欧洲药典中文翻译
欧洲药典 10.0 EP 10.0 长春西汀 中文翻译

01/2008:2139修订:7.3长春西汀Vinpocetine欧洲药典10.0Ph.Eur. 10.0EP 10.0C22H26N2O2Mr 350.5[42971-09-5]定义乙基(13as,13bs)13α-乙基-2, 3 ,5 ,6-13α 13b六氢-1H-吲哚3, 2, 1-d吡啶3, 2, 1-ij,l, 5-二痰杂萘-12-羧酸。
(Ethyl (13aS,13bS)-13a-ethyl-2,3,5,6,13a,13b-hexahydro- 1H-indolo[3,2,1-de]pyrido[3,2,1-ij][1,5]naphthyridine-12-carboxylate.)含量:98.5%- 101.5%(干品)。
特征外观:白色或微黄色结晶性粉末。
溶解性:几乎不溶于水,可溶于二氯甲烷,微溶于无水乙醇。
鉴别A.比旋度(见检测项)。
B.红外吸收光谱(2.2.24)。
对比:长春西汀CRS。
检测比旋光度(2.2.7):+127到+134(干品)。
取0.25 g溶于二甲基甲酰胺R,并用相同的溶剂稀释至25.0 ml。
有关物质。
液相色谱(2.2.29).供试溶液。
取50.0mg供试品溶于流动相并用流动相稀释至50.0ml。
对照溶液(a).取1.0ml 供试品溶液用流动相稀释至50.0ml。
对照溶液(b).取5.0mg 长春西汀杂质B CRS,6.0mg长春西汀杂质A CRS,5.0mg 长春西汀杂质C CRS 5.0mg长春西汀杂质D CRS,溶于流动相,并用流动相稀释至50.0ml。
对照溶液(c).取1.0ml 对照溶液(a)和1.0 ml对照溶液(b)用流动相稀释至20.0ml。
色谱柱:-尺寸:l = 0.25m, Ø = 4.6mm-固定相:色谱用末端封尾的十八烷基硅烷键和硅胶R(5μm)。
流动相:15.4g/l 的醋酸铵R溶液,乙腈R(45:55 V/V)。
流速:1.0ml/min。
欧洲药典中英文翻译 EP8.0干燥失重

2.2.32. LOSS ON DRYING 干燥失重Loss on drying is the loss of mass expressed as per cent m/m.干燥失重指重量损失,表述为% 重量/重量Method. Place the prescribed quantity of the substance to be examined in a weighing bottle previously dried under the conditions prescribed for the substance to be examined. Dry the substance to constant mass or for the prescribed time by one of the following procedures. Where the drying temperature is indicated by a single value rather than a range, drying is carried out at the prescribed temperature +/- 2?C.方法:将要求数量的待检样品放置于预先干燥的称量瓶中,按要求条件进行干燥,直至样品干至恒重或下述程序指定的时长。
如果干燥温度给定的是一个值而不是一个范围,则在指定温度+/- 2?C进行干燥。
a) “in a desiccator”: the drying is carried out over diphosphorus pentoxide R at atmospheric atmostpheric pressure and at room temperature;“在干燥器中”:指在室温常压下,用五氧化二磷试剂,进行干燥b) “in vacuo”: the drying is carried out over diphosphorus pentoxide R, at a pressure of 1.5 kPa at room temperature;“真空”:在室温下,真空1.5kPa下,用五氧化二磷试剂进行干燥c) “in vacuo within a specified temperature range”: the drying is carried out over diphosphorus pentoxide R, at a pressure of 1.5kPa to 2.5kPa within the temperature range prescribed in the monograph;“在指定温度范围内真空下”:真空1.5kPa至2.5kPa下,各论要求的温度范围内,用五氧化二磷进行干燥d) “in an oven within a specified temperature range”: the drying is carrie d out in an oven within the temperature range prescribed in the monograph;“在烘箱里指定温度下”:在各论要求的温度范围内,用烘箱进行干燥e) “under high vacuum”: the drying is carried out over diphosphorus pentoxide R at a pressure not exceeding 0.1kPa, at the temperature prescribed in the monograph.“在高真空下”:在各论要求的温度下,不超过0.1kPa的真空下用五氧化二磷进行干燥If other conditions are prescribed, the procedure to be used is described in full in the monograph.如果需要采用其它条件,则在各论中应进行详细描述。
欧洲药典翻译一部分

5.4 残留溶剂在活性物质、辅料和医药产品中限定残留溶剂水平ICH采取了关于残留溶剂杂质指导原则,这个指导原则描述了在活性物质、辅料和医药产品中的溶剂浓度限定。
指导原则排除了已存在的市场产品。
EP采用了和这项指导原则同样的原则,不管存在的活性物质、辅料和医药产品是否是药典专论的内容。
所有的物质和产品都要测定它们中可能出现的溶剂浓度。
其中限额适用遵从以下的,溶剂残留量测试在特定专论一般不提及,因为从一个制造商到另一个采用的溶剂可能会有所不同,这一总章的要求通过制药用物质(2034)适用。
在产品生产过程中所采用的溶剂应告知主管,该信息也列于为EP专论的合格证而递交的卷宗,在证书中也提及。
其中只有使用第三类溶剂时,采用干燥损失测定或者进行溶剂的具体测定。
如果采用了一种被确认可行的、权威的第三类溶剂,但高于限度0.5%,溶剂的具体测定是需要的。
当使用第一类或第二类溶剂(或第三类溶剂超过限度0.5%)时,用在一般方法中描述的方法,否则采用经证实的合适的方法。
当进行残留溶剂的定量测定时,在计算物质含量时,这个结果被考虑进去,除了干燥检验。
杂质:残留溶剂的指导原则1.前言该指导原则的目的是建议为了病人安全在药物中可允许的残留溶剂的量。
该指导原则建议少使用有毒溶剂,描述了一些残留溶剂的毒性允许水平。
在这医药产品中的残留溶剂被定义为在活性物质或辅料的制造或医药产品的制剂中使用的或产生的有机挥发性物质。
这些溶剂没有通过实用生产技术完全清除,在活性物质合成中合适地选择溶剂会提高产量,决定一些晶体的结构、纯度、溶解性等特征,因此有时溶剂会成为合成过程中的重要参数,该指导原则并没有指出辅料中使用的溶剂和溶剂化物,但是,这些产品中的溶剂含量应该评估并说明理由。
由于残留溶剂没有疗效,所有残留溶剂的去除都应该达到产品的标准或药品生产和管理规范或其他质量要求,医药产品不应包含比安全系数更高的水平的残留溶剂,一些溶剂会造成不允许的毒性,应该在生产中避免,除非他们的使用在风险效益评估中有强烈的理由。
欧洲药典EP8.0 2.6.1无菌检验 sterility中英文翻译

2.6.1. STERILITY2.6.1 无菌检查法The test is applied to substances, preparations or articles which, according to the Pharmacopoeia, are required to be sterile. However, a satisfactory result only indicates that no contaminating micro-organism has been found in the sample examined in the conditions of the test.本检查方法适用于按照药典要求应当无菌的原料、制剂或其他物质。
但是,如果按照本无菌检查法的结果符合要求,仅表明在该检查条件下未发现微生物污染。
PRECAUTIONS AGAINST MICROBIAL CONTAMINATION微生物污染防范The test for sterility is carried out under aseptic conditions. In order to achieve such conditions, the test environment has to be adapted to the way in which the sterility test is performed. The precautions taken to avoid contamination are such that they do not affect any micro-organisms which are to be revealed in the test. The working conditions in which the tests are performed are monitored regularly by appropriate sampling of the working area and by carrying out appropriate controls.无菌检测试验应在无菌的条件下进行。
欧洲药典附录中文版.

第二部分、附录附录1 溶液的澄清度 (2)附录2 溶液颜色检查 (3)附录3 旋光度 (7)附录4 铵盐检查法 (9)附录5 氯化物检查法 (11)附录6 硫酸盐灰分 (13)附录7 铁 (14)附录8 重金属 (16)附录9 干燥失重 (21)附录10 硫酸盐检查法 (23)附录11 红外吸收分光光度法 (25)附录12 pH测定 (29)附录13 滴定 (34)附录14 氯化物鉴别反应 (37)附录15 指示剂颜色与溶液pH 的关系 (38)附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
欧洲药典附录中文版

欧洲药典附录中文版第二部分、附录附录 1 溶液的澄清度.................................. 2附录 2 溶液颜色检查.................................. 3附录 3 旋光度 ....................................... 6附录 4 铵盐检查法.................................... 8附录 5 氯化物检查法.................................. 9附录 6 硫酸盐灰分................................... 10附录 7铁 .......................................... 11附录 8 重金属 ...................................... 12附录 9 干燥失重..................................... 15附录 10 硫酸盐检查法................................. 16附录 11 红外吸收分光光度法 (17)附录 12 pH 测定 ...................................... 20附录 13 滴定 ........................................22附录 14 氯化物鉴别反应............................... 24附录 15指示剂颜色与溶液 pH 的关系 ................... 25 附录 1 溶液的澄清度在内径 15,25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为 40mm,按如下所述方法进行比较。
浊度标准液制备 5 分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
间苯三酚欧洲药典翻译

间苯三酚二水合物分子式:C6H6O3·2H2O摩尔质量:162.1CAS Number:6099-90-7用途:肌肉解挛剂系统命名:1,3,5-三羟基苯二水合物含量:99.0 % ~ 101.0 %(以无水间苯三酚计算)理化性质:外观:白色或类白色粉末溶解性:难溶于水,易溶于乙醇(96 %),部分溶于二氯甲烷鉴别:A.红外光谱(2.2.24)样品制备:事先将供试品在105 °C烘箱中干燥对比:将供试品和无水间苯三酚标准品进行图谱对比B. 薄层色谱(2.2.27)供试溶液:将0.2g供试品溶解在甲醇中,并稀释至10mL参比溶液:将0.2g无水间苯三酚标准品溶解在甲醇中,并稀释至10mL薄层板:TCL F254硅胶板流动相:无水甲酸:正己烷:乙酸乙酯= 2:37.5:62.5(体积比)用量:10 µL爬板高度:超过2/3板长检测条件:在254 nm紫外光照射下检测结果:供试溶液和参比溶液的样品点在TCL板上,应大小相似、位置一致C.干燥失重(见测试)溶解:将2.5g供试品溶解在乙醇(96 %)中,并稀释至25mL外观:溶液应澄清,并不得比参比溶液BY5(方法二)颜色更深pH:将10mL溶液用去除二氧化碳的蒸馏水稀释至100mL,测得pH应在4.0 ~ 6.0之间有关物质:液相色谱:使用前现配溶液,避光保存混合溶液:流动相B:流动相A = 10:90(体积比)供试溶液:将50mg供试品溶解在混合溶液中,并稀释至10mL参比溶液(a):取1mL供试溶液用混合溶液稀释至100mL,后取1mL此溶液用混合溶液稀释至10mL参比溶液(b):分别取6mg的间苯二酚(杂质B),2,3',4,5',6-五羟基联苯(杂质D),邻苯三酚(杂质A)溶解于10mL的混合溶液中,加入2mL的供试溶液并用混合溶液稀释至20mL,后取1mL此溶液用混合溶液稀释至50mL参比溶液(c):分别取4mg的邻苯三酚(杂质A),2,3',4,5',6-五羟基联苯(杂质D),杂质E,杂质I,杂质K,杂质L溶解于10mL的混合溶液中,并用混合溶液稀释至20mL参比溶液(d):取10mg的供试品溶解于10mL的混合溶液,加入1mL的参比溶液(c)并用混合溶液稀释至20mL色谱柱:柱长:0.25m直径:4.0mm固定相:末端修饰十八烷基硅羟基耦合极性集团R(5µm)流动相:流动相A:1.36 g/L的磷酸二氢钾并用磷酸调节pH至3.0流动相B:乙腈程序洗脱流速:1.0 mL/min检测波长:265nm注射量:供试溶液,参比溶液(a),(b),(d)各20µL杂质鉴别:对比参比溶液(d)色谱图定位鉴别杂质A, D, E, I, K and L相对保留时间:以间苯三酚为参照点(保留时间大约为12min):impurity E = about 0.7; impurity A = about 0.9; impurity D = about 1.3; impurity B = about 1.35; impurity K = about 1.5; impurity I = about 1.8; impurity L = about 2.0系统适用性:分离度:杂质A和间苯三酚大于2.5,杂质B和D大于4.0限度:校正因子:对于计算含量,各杂质的峰面积应乘以如下的校正因子:impurity A = 0.6; impurity D = 0.2; impurity E = 0.7; impurity I = 0.6; impurity K = 0.6; impurity L = 0.4且对于杂质A, D, E, I, K, L,其峰面积不应大于参比溶液(a)中主峰峰面积的1.5倍(0.15 %)非特定杂质:对于非特定杂质,其面积不应大于参比溶液(a)中主峰峰面积(0.10 %)总杂质:面积不应大于参比溶液(a)中主峰峰面积的3.0倍(0.30 %)报告限:面积不应大于参比溶液(a)中主峰峰面积的0.5倍(0.05 %)氯含量:不应大于200 ppm将2.5mL的溶液用水稀释至15mL硫酸盐:不应大于500ppm将3mL的溶液用蒸馏水稀释至15mL干燥失重:小于1.0 %将1.000g无水间苯三酚在105 °C烘箱中干燥灼烧残渣:小于0.1 %样品检测量1.0g滴定:将0.500g供试品溶解在50mL水中,用1M氢氧化钠溶液滴定,计算计量终点1mL的1M氢氧化钠相当于63.05mg的C6H6O3保存:避光保存可能存在杂质:A. benzene-1,2,3-triol (pyrogallol),B. benzene-1,3-diol (resorcinol),D. 2,3′,4,5′,6-biphenylpentol (phloroglucide),E. benzene-1,2,4-triol,I. 2,6-dichlorophenol,K. 4-chlorobenzene-1,3-diol (4-chlororesorcinol),L. 3,5-dichloroaniline,O. 4,6-dichlorobenzene-1,3-diol (4,6-dichlororesorcinol).。
欧洲药典-凡例(中英文对照)

欧洲药典-凡例1.1. GENERAL STATEMENTSThe General Notices apply to all monographs and other texts of the European Pharmacopoeia.总论的内容适用于各论和欧洲药典中的其它章节。
The official texts of the European Pharmacopoeia are published in English and French. Translations in other languages may be prepared by the signatory States of the European Pharmacopoeia Convention. In case of doubt or dispute, the English and French versions are alone authoritative.欧洲药典以英语和法语形式发行,欧洲药典委员会的签署国可将药典内容译成其它语言,但若发生争议,应以英语和法语版为权威。
In the texts of the European Pharmacopoeia, the word "Pharmacopoeia" without qualification means the European Pharmacopoeia. The official abbreviation Ph. Eur. may be used to indicate the European Pharmacopoeia.在欧洲药典中,如无特殊规定,“药典”是指欧洲药典,缩写Ph. Eur.也指欧洲药典。
The use of the title or the subtitle of a monograph implies that the article complies with the requirements of the relevant monograph. Such references to monographs in the texts of the Pharmacopoeia are shown using the monograph title and reference number in italics.文章中如果引用了各论中的标题和副标题意味着文章内容符合相关各论的要求。
欧洲药典译文(药品玻璃容器)

药品玻璃容器药品玻璃容器用于直接与药品接触.现有的几种药品玻璃容器例如:安瓿: 这是一种薄壁玻璃容器,在被填装后,以熔合的方式式封装.容器中的药物在容器破碎后被抽取.针剂瓶仅为一次性使用各种规格的玻璃瓶:这是一些规格不一的厚壁玻璃容器,以玻璃或其他材料封装,例如塑料,人造橡胶等.容器中的药物可以被一次或多次按任意的量取用.盛装人类血液或人类血液中的提取物的容器:这是一种圆柱形的壁厚,容积不一的厚壁玻璃容器,由无色,透明的中性玻璃制成。
玻璃的质量:无色玻璃指人类肉眼看上去高度透明的玻璃。
有色玻璃是指添加了少量氧化金属物质的玻璃,根据需要的吸光率选材制成.中性玻璃指一种硼硅酸盐玻璃,其中含有大量硅氧化物,铝或碱土金属氧化物. 因为中性玻璃上述的成分构成,它具有很高的抗热冲击性和抗水解性.钠钙硅玻璃指一种硅玻璃含有碱金属氧化物--主要是钠氧化物;和碱土金属氧化物--主要是钙氧化物. 钠钙硅玻璃的化学成份决定它只具有中等的抗水解性. 药品玻璃容器的化学稳定性是由它的抗水解性表现出来的. 比如在一个特定的水和容器内表面接触的条件下或粉末状玻璃和水接触时,玻璃所表现出来的抗溶解性. 抗水解性以滴定法被测溶液碱度来衡量. 以下是根据其抗水解性对玻璃容器的分类以下斜体字部分是对不同的药品所应采用的玻璃容器类型的推荐. 药品生产厂家必须负责保证选用合适的容器来包装药品.一类玻璃容器通常适用于所有的药品(口服或非口服药品及人类血液或人类血液的提取物). 二类玻璃容器通常适用于酸性和中性及水性药品(非口服).三类玻璃容器通常适用于:非水性的药品(非口服)、粉末剂(非口服)以及口服类药品.四类玻璃容器通常适用于一些固体药品(口服)和一些液体或半固体药品(口服)某些特殊药品的玻璃容器的抗水解性可能高于以上介绍的玻璃容器的水解性.对于口服类药品,有色和无色玻璃容器都适用,非口服类药品通常用无色玻璃容器包装,但有色玻璃容器亦可用于某些对光不敏感的药品.建议所有包装液体和粉末状(非口服)药品的玻璃容器可以让人凭肉眼就能观察其盛装的物品.可以对玻璃容器的内表面进行特殊处理以获得或提高抗水解性. 玻璃容器的外表面也可以被处理以减少磨擦和提高抗磨损性. 对玻璃容器的外表面处理必须不能污染其内表面.除了一类玻璃容器, 药品玻璃容器不可被重复使用. 盛装人类血液或人类血液提取物的玻璃容器不可被重复使用.药品玻璃容器必须符合关于抗水解性的测试标准或其他相关标准. 对于有非玻璃部件的玻璃容器, 上述测试标准只适用于玻璃部件.测试以下是必须的测试用以评定不同用途的玻璃容器的质量.抗水解性设备和试剂—研钵、杵(如图3.2.1.-1)、锤子、磁铁—一套不锈钢方网格筛子,装在不锈钢的框架上,筛子的规格号码为:a) 筛子no. 710b) 筛子no. 425c) 筛子no. 250—一块永久磁铁—一个旧的中性玻璃制成的烧瓶和盖子(即曾被使用于同样试验的烧瓶和盖子或者是一个烧瓶被装入水试剂后保持在121°C的高压下至少一个小时)—一片惰性金属(例如铝)的箔.—一个可以使温度保持在121 °C ± 1 °C的高压锅,这个高压锅必须装有压力计、温度计、旋钮气阀和一个托盘,高压锅的水平面以上必须有足够的空间来容纳用被测试的容器。
纯化水英汉对照-欧洲药典8.0

WATER,PURIFIED纯化水H2O M r18.12 DEFINITIONWater for the preparation of medicines other than those that are required to be both sterile and apyrogenic,unless otherwise justified and authorized.定义制药用水不同于其它用水,要求它是无菌的、无热源的,除非另有调整或授权。
Purified water in bulk散装纯化水PRODUCTIONPurified water in bulk is prepared by distillation,by ion exchange,by reverse osmosis or by any other suitable method from water that complies with the regulations on water intended for human consumption laid down by the competent authority.Purified water in bulk is stored and distributed in conditions designed to prevent growth of micro-organisms and to avoid any other contamination.生产:散装纯化水是经合格的当局规定的适宜人类使用的水经蒸馏、离子交换、反渗透膜或其他任何适合的方法制备。
散装纯化水存储和分配于可防止微生物生长和可避免其他任何污染的条件下。
Microbiological monitoring During production and subsequent storage, appropriate measures are taken to ensure that the microbial count is adequately controlled and monitored.Appropriate alert and action levels are set so as to detect adverse trends.Under normal conditions,an appropriate action level is a microbial count of100CFU/mL,determined by filtration through a membrane with a nominal pore size not greater than0.45μm,using R2A agar and incubating at30-35°C for not less than5days.The size of the sample is to be chosen in relation to the expected result.微生物监测在生产和其后的存储过程中,采取适当的方式以确保水的微生物数受到足够的控制和监测。
欧洲药典EP8.0 滴位测定 中文翻译

2.2.20. POTENTIOMETRIC TITRATION 电位滴定In a potentiometric titration the end-point of the titration isdetermined by following the variation of the potential difference between 2electrodes (either one indicator electrode and one reference electrode or 2indicator electrodes) immersed in the solution to be examined as a function ofthe quantity of titrant added.在一个电位滴定中,滴定终点判定是依据浸入被测液中两支电极(或者是一支指示电极一支参比电极,或者是两支指示电极)之间的电位差,对应加入滴定液的数量。
The potential is usually measured at zero or practically zerocurrent.电位一般在零点测量,或在零点电流。
Apparatus. The apparatus used (a simple potentiometer or electronicdevice) comprises a voltmeter allowing reading to the nearest millivolt.仪器:所用仪器(一个简单的电位计或电极装置)包括一个可以准确读至毫伏的电位计。
The indicator electrode to be used depends on the substance to bedetermined and may be a glass or metal electrode (for example, platinum, gold,silver or mercury). The reference electrod is generally a calomel or asilver-silver chloride electrode.所用的指示电极依赖于所检测的物质,可以是玻璃电极或金属电极(例如,铂电极、金电极、银或水银电极)。
欧洲药典 10.0 EP 10.0 长春胺 中文翻译

01/2020:1800长春胺(VINCAMINE)(Vincaminum)欧洲药典10.0Ph.Eur. 10.0EP 10.0C21H26N2O3[1617-90-9]定义Methyl 14-hydroxyvincane-14β-carvoxylate。
含量:99.0 %到101.0% (干燥物质)。
性状外观:白色或近白色,结晶性粉末。
溶解性:几乎不溶于水,溶于二氯甲烷,微溶于无水乙醇。
鉴别A.比旋度(见检测)。
B.红外吸收光谱(2.2.24)。
参比标准品:长春胺CRS。
检测比旋度(2.2.7):+44.3°~+49.0° (以干计)。
将0.1g样品溶解在二甲基甲酰胺R(dimethyformamide R) 并用相同的试剂稀释至20.0 mL。
有关物质。
液相色谱(2.2.29)。
溶液临用现配。
使用超声溶解样品,避免任何高温。
供试溶液. 将50.0mg被测物质溶解于10 mL四氢呋喃R中,并用流动相稀释至100.0mL。
对照溶液(a). 取1.0mL供试溶液用流动相稀释至200.0mL。
对照溶液(b). 将5 mg系统适用性用长春胺CRS (含有杂质A, B和C)溶解于流动相中并用流动相稀释至10.0mL。
色谱柱:-尺寸:l=0.25m,φ=4.6mm;-固定相:末端封口的用于嵌入极性基团色谱分析的十八烷基硅烷硅胶R(end-capped octadecylsilyl silica gel for chromatography with embedded polar groups R)(5μm);流动相:四氢呋喃R,乙腈R,15.4 g/L的乙酸铵R溶液(17:18:65 V/V/V);流速:1.0 mL/min。
检测器:分光光度计272nm。
进样体积:20ul。
运行时间:长春胺保留时间的3.5倍。
杂质鉴别:用系统适用性用长春胺CRS提供的图谱以及对照溶液(b)所得的图谱来鉴别杂质A, B和C。
EP7.0 欧洲药典 2.9.38. Particle-size distribution 粒度分布_中文_翻译

2.9.38. 筛分法评估颗粒度大小分布(17)PARTICLE-SIZE DISTRIBUTION ESTIMATION BY ANALYTICAL SIEVING(17)筛分是对粉末和颗粒通过颗粒大小分布进行分级的最老的方法之一。
当使用织物筛布时,筛分基本上依据筛网的中间尺寸大小(即幅度或宽度)对颗粒进行分类。
如果大部分的颗粒大于75微米左右,采用机筛法是最合适的。
对于更小的颗粒,由于质量轻,在筛分过程中,它们不能克服表面凝聚力和粘附力,使颗粒相互粘在一起或粘在筛网上,从而导致那些本该能通过筛孔的颗粒而未能通过。
对于这样的材料,其它的振动方式,如喷气筛分或声波筛分器筛分可能更为合适。
然而,筛分有时可被用于一些中值颗粒尺寸小于75微米的粉末或颗粒,只要该方法经过验证。
在制药行业,对单一粉末或颗粒进行粗略等级分类时,筛分通常是被选择的方法。
这是一个尤其引人注目的方法,因为该方法仅依据粒径大小对粉末和颗粒进行颗粒大小分级,在大多数情况下,干粉状态下即可进行检测。
Sieving is one of the oldest methods of classifying powders and granules by particle-size distribution. When using a woven sieve cloth, the sieving will essentially sort the particles by their intermediate size dimension (i.e. breadth or width). Mechanical sieving is most suitable where the majority of the particles are larger than about 75 µm. For smaller particles, their light weight provides insufficient force during sieving to overcome the surface forces of cohesion and adhesion that cause the particles to stick to each other and to the sieve, and thus cause particles that would be expected to pass through the sieve to be retained. For such materials other means of agitation such as air-jet sieving or sonic-sifter sieving may be more appropriate. Nevertheless, sieving can sometimes be used for some powders or granules having median particle sizes smaller than 75 µm where the method can be validated. In pharmaceutical terms, sieving is usually the method of choice for classification of the coarser grades of single powders or granules. It is a particularly attractive method in that powders and granules are classified only on the basis of particle size, and in most cases the analysis can be carried out in the dry state.筛分方法的局限性包括:需要可观质量的样品(通常至少为25克,这取决于粉末或颗粒的密度和试验筛的直径);以及在筛分那些容易堵塞筛孔的油性或有粘着力的粉末或颗粒时存在困难。
欧洲药典标准

欧洲药典标准欧洲药典(European Pharmacopoeia, EP)是欧洲药草准则的官方法典,目的是确保世界范围内的药品质量和治疗有效性,以保护公众健康。
它提供了一套标准和指南,以确保药品的安全性、质量和有效性,是药品配方和生产的准则。
欧洲药典的历史可以追溯到1964年,当时的欧洲各国决定合作制定共同的药品标准。
从那时起,药典每年定期发布,并随着科学和技术的进步而不断更新。
药典由名为欧洲药典总局(EDQM)的组织负责管理和发布。
欧洲药典的标准涵盖了许多方面,包括活性成分、辅助成分、生产过程和质量控制方法等。
每个标准都被认为是行业内识别药品质量和治疗有效性的基准。
它们是通过利用最新的科学知识、专家的经验和立法要求来确定的。
欧洲药典还包括有关药品质量控制的方法和程序的指南。
这些指南旨在帮助药品制造商和监管机构确保药物的质量和安全性。
它们涵盖了药品的制造、合规性评估、质量控制和批准过程等方面。
欧洲药典的标准是在专家小组的指导下制定的。
这些专家来自各个领域,包括医学、药学、化学和法律。
他们的目标是确保药物是安全和有效的,以及可追溯的,从而保护公众的健康和安全。
欧洲药典的标准在欧洲范围内具有法律效力。
成员国在国家层面上有责任确保药品符合这些标准。
根据欧洲药典的要求,药品必须通过一系列的质量控制测试和临床试验,以确保其在安全和有效性方面符合标准。
除了欧洲范围内的应用,许多国家和地区也使用欧洲药典的标准作为其国家药典的基础。
这些国家和地区包括非欧洲国家,如中国、日本和澳大利亚。
总之,欧洲药典是一本由欧洲各国联合制定的药品标准的权威法典。
它的目标是确保药品在全球范围内的质量和治疗有效性,并保护公众的健康和安全。
通过提供一套统一的标准和指南,欧洲药典在药品配方、生产和质量控制方面发挥了重要的作用。
它对全球医药行业的发展起着积极的推动作用。
欧洲药典附录中文版

欧洲药典附录中文版————————————————————————————————作者:————————————————————————————————日期:第二部分、附录附录1 溶液的澄清度 (1)附录2 溶液颜色检查 (2)附录3 旋光度 (6)附录4 铵盐检查法 (8)附录5 氯化物检查法 (9)附录6 硫酸盐灰分 (10)附录7 铁 (11)附录8 重金属 (12)附录9 干燥失重 (15)附录10 硫酸盐检查法 (16)附录11 红外吸收分光光度法 (17)附录12 pH测定 (20)附录13 滴定 (22)附录14 氯化物鉴别反应 (24)附录15 指示剂颜色与溶液pH 的关系 (25)附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1ⅠⅡⅢⅣ浊度标准液 5.0ml 10.0ml 30.0ml 50.0ml水95.0ml 90.0ml 70.0ml 50.0ml附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
欧洲药典附录中文版

第二部分、附录溶液的澄清度 ............................. 2 溶液颜色检查 ............................. 3 旋光度 ................................... 7 铵盐检查法 ............................... 9 氯化物检查法........................................ 1..1..硫酸盐灰分 ........................................ 1..3..铁 ........................................ 1...4附录 1 附录 2 附录 3 附录 4 附录 5 附录 6 附录 7 附录 8 附录 9 附录 10 附录 11 附录 12 附录 13 附录 14附录 15...重金属........................................ 1..6. ..干燥失重........................................ 2 (1)..硫酸盐检查法........................................ 2..3. .红外吸收分光光度法..................... 2..5.pH 测定........................................ 2 (9)..滴定........................................ 3..4. ..氯化物鉴别反应........................................ 3..7. .指示剂颜色与溶液 pH 的关系............. 3..8附录 1 溶液的澄清度在内径15 ~25mm ,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm ,按如下所述方法进行比较。
氯化钾《欧洲药典》9.0中英对照
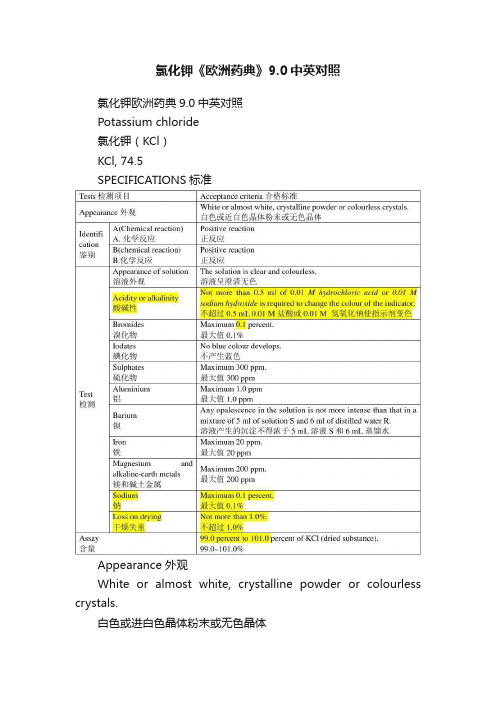
氯化钾《欧洲药典》9.0中英对照氯化钾欧洲药典9.0中英对照Potassium chloride氯化钾(KCl)KCl, 74.5SPECIFICATIONS标准Appearance 外观White or almost white, crystalline powder or colourless crystals.白色或进白色晶体粉末或无色晶体IDENTIFICATION鉴别A.It gives the reactions of chlorides (2.3.1).和氯化物反应B.Solution S (see T ests) gives the reactions of potassium.溶液和钾反应TESTS检测Solution S. Dissolve 10.0g in carbon dioxide-free water R prepared from water R and dilute to 100 ml with the same solvent.溶液S. 将10.0g 溶于脱二氧化碳水并稀释至100ml。
Appearance of solution. Solution S is clear (2.2.1) and colourless (2.2.2, Method Ⅱ).溶液外观:溶液呈无色澄清Acidity or alkalinity.To 50 ml of solution S add 0.1 ml of bromothymol blue solution R1. Not more than 0.5 ml of 0.01 M hydrochloric acid or 0.01 M sodium hydroxide is required to change the colour of the indicator.酸碱性:取50ml溶液S加入0.1ml酚蓝溶液,不超过0.5ml 0.01M盐酸或0.01M氢氧化钠使指示剂变色。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附录1溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取硫酸肼溶于水,加水稀释至,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以水溶解乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容。
滴定并以上述盐酸溶液调整,使黄色液每毫升含 FeCl3﹒6H2O。
避光保存。
滴定在一个配有磨口塞的250ml锥形瓶内,加入黄色液,15ml 水,5ml浓盐酸和4g碘化钾,塞上瓶塞,在暗处放置15分钟,再加100ml 水。
用的硫代硫酸钠标准溶液滴定游离的碘,在滴定接近终点时加淀粉试液作指示剂。
1ml 的硫代硫酸钠标准溶液相当于 FeCl3﹒6H2O。
红色液称取60克氯化钴,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容。
滴定并以上述盐酸溶液调整,使红色液每毫升含 CoCl2﹒6H2O。
滴定在一个配有磨口塞的250ml锥形瓶内,加入红色液,5ml稀过氧化氢溶液和10ml 300g/l的氢氧化钠溶液,缓慢煮沸10分钟,冷却后,加60ml稀硫酸和2g碘化钾,塞上瓶塞,缓慢摇动锥形瓶,使沉淀溶解完全。
用的硫代硫酸钠标准溶液滴定游离的碘,在滴定接近终点时加入淀粉试液作为指示剂。
溶液变成粉红色时到达滴定终点。
的硫代硫酸钠标准溶液相当于 CoCl2﹒6H2O。
蓝色液称取63克硫酸铜加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容。
滴定并以上述盐酸溶液调整,使蓝色液每毫升含 CuSO4﹒5H2O。
滴定在一个配有磨口塞的250ml锥形瓶内,加入蓝色液,50ml水,12ml稀醋酸和3g碘化钾。
用的硫代硫酸钠标准溶液滴定游离碘,在滴定接近终点时加入淀粉试液作为指示剂。
当溶液变为轻微的淡褐色时到达滴定终点。
的硫代硫酸钠标准溶液相当于 CuSO4﹒5H2O。
颜色标准溶液用3种贮备液制备5种颜色标准液。
如表2-1。
表2-1,颜色标准液方法I和方法Ⅱ的标准比色液用5种颜色标准溶液,制备以下各种颜色的标准比色液。
表2-2,标准比色液B表2-3,标准比色液BY表2-4,标准比色液Y表2-5,标准比色液GY表2-6,标准比色液R储存对于方法I,标准比色液在外径为12mm的无色透明中性封口的玻璃管中储存,避光。
对于方法Ⅱ,使用前直接从颜色标准液制备标准比色液。
仪器和试剂:附录3 旋光度旋光是手性物质的特性,即能使偏振光的平面旋转。
右旋物质的旋光度为正的(+),即右旋物质可以使偏振光平面顺时针方向旋转;左旋物质的旋光度为负的(-)。
精确的旋光度是指,在温度t下,波长为λ的光透过长1m或含1kg/m3旋光活性物质的液体,所发生的旋转,用弧度(rad)表示。
实际操作中,旋光度常用mrad ﹒m2﹒kg-1表示。
本药典采用以下常规定义纯液体旋光度:旋光度以角度(°)表示,即20℃下,1dm长测定管的纯液体使钠光谱D线(λ=)的偏振光平面所旋转的角度;对于溶液,按专论规定方法制备。
液体旋光度:测定即20℃下,1dm长的测定管的含待测液体的溶液使钠光谱D线(λ=)的偏振光平面所旋转的角度(°),即溶液旋光度。
溶液中液体旋光度,由溶液旋光度除以溶液中被检测液体的密度(g/cm3)计算得出。
固体物质的旋光度:测定20℃下,1dm长测定管的含被测物质1g/ml的溶液使钠光谱D线(λ=)的偏振光平面所旋转的角度,即溶液旋光度。
溶液中固体物质的旋光度由溶液的旋光度计算得出。
溶液中某物质的旋光度与溶剂和浓度有关。
按本药典采用的惯例,旋光度不标注单位;它的实际单位为(°)﹒ml﹒dm-1﹒g-1。
本药典的旋光度同国际标准单位旋光度的换算关系如下:如果专论有特别要求,按要求选择温度(可能不是20℃)和波长。
旋光计的读数必须精确到°。
测量范围通常由鉴定用石英片检查;测量范围内线性由蔗糖溶液检查。
方法20±℃下,旋光计调零,用钠光谱的D线(λ=)测定,或者按专论要求的温度测定旋光度。
测定液体的旋光度,测定前放入封闭的空测定管,调零;测定固体的旋光度,测定前放入盛有所用溶剂测定管,调零。
按下式计算旋光度:纯液体旋光度:溶液中物质的旋光度:c为浓度,单位g/l。
按下式计算以g/l为单位的溶解物质的浓度c,或以m/m百分比为单位的浓度c′:= 20 ± °C下,旋光度读数,单位度(°)l = 测定管长度,单位dm。
20 °C 下溶液密度,单位g/cm3,本药典在节中以相对密度代替密度。
20 =c = 溶解物质的浓度,单位g/l。
c′= 溶解物质的浓度,单位g/l。
附录4 铵盐检查法除非另有规定,通常用方法A。
方法A供试溶液:在比色管中用14ml水溶解规定质量的供试品,必要时加入稀释的氢氧化钠溶液使溶解,用水稀释至15ml。
再加碱性碘化汞钾试液。
标准溶液:取10ml的标准铵溶液(1ppmNH4),加5ml水和碱性碘化汞钾试液。
两溶液摇匀后分别用塞子塞住比色管。
5分钟后,供试溶液中的黄色不得比标准溶液中的颜色更深。
方法B在25ml有盖子2的广口瓶中,加入规定数量的供试品细粉,使其溶解或悬浮在1ml的水中,加重氧化镁。
取一片5mm的正方形银锰纸,滴几滴水使其湿润,铺在瓶口,然后立即盖上聚乙烯瓶盖。
漩涡混和,防止液体溅出,在40℃下放置30分钟。
如银锰纸显示灰色,其颜色不得比,规定量的标准铵溶液(1ppmNH4),加1ml 的水,克氧化镁制成的标准溶液的银锰纸的颜色更深。
附录5 氯化物检查法供试溶液:15ml待测溶液,加1ml稀硝酸于测试管中,混合,然后将混合溶液倒入装有1ml硝酸银溶液的比色管中。
标准溶液:10ml的氯化物标准液(5ppm Cl),加5ml水,加1ml稀硝酸,混合,然后将混合溶液倒入装有1ml硝酸银溶液的比色管中。
黑色背景下对比两份溶液的颜色。
避光放置5分钟后,供试溶液中的乳白色不得比标准溶液更深。
附录6 硫酸盐灰分将坩埚(由铂、瓷或石英制成)在600±50℃灼烧30分钟,取出放入已放置硅胶的干燥器内,冷却后称重。
将规定量的供试品置于上述坩埚内,称重。
加少量硫酸(通常1ml)湿润供试品,按要求温度缓慢加热,直至供试品完全炭化。
冷却后,加少量硫酸润湿残渣,继续加热到没有白烟冒出。
再在600±50℃灼烧至完全灰化。
操作过程中应避免燃着。
取出坩埚置于已放置硅胶的干燥器内冷却,冷却后称重,计算残渣的重量。
如果残渣超过规定,除有其他规定,重复以上操作,直至恒重。
附录7 铁供试溶液:将规定数量的供试品溶于水中,并用水稀释至10ml,或者直接用10ml 规定溶液。
加2ml 200g/l的柠檬酸溶液和的硫醇基乙酸(硫乙醇酸),混合,加氨水使偏碱性,再用水稀释至20ml。
供试溶液:10ml的标准铁溶液(1ppm Fe)按供试溶液的方法制备成20ml标准溶液,5分钟后,供试溶液中的粉红色不得比标准溶液深。
附录8 重金属方法A供试溶液:12ml待测水溶液,2ml pH为的缓冲溶液,混合后加的硫代乙酰胺试液,立即混合。
对照溶液:10ml的标准铅溶液(1ppm or 2ppm Pb), 2ml pH为的缓冲溶液, 2ml 的待测液,混合后加的硫代乙酰胺试液,立即混合。
空白溶液:10ml的水,2ml pH为的缓冲溶液,2ml的测试溶液。
混合后加的硫代乙酰胺试液,立即混合,同空白溶液比较,对照溶液显浅棕色。
2分钟后,供试的溶液颜色不得比对照溶液深。
方法B用含最少量水的溶剂(例如含15%水的二氧杂环乙烷或含15%水的丙酮)溶解规定量的供试品,制成待测液供试溶液:12ml待测液, 2ml pH为的缓冲溶液,混合后加的硫代乙酰胺试液,立即混合。
对照溶液:10ml的标准铅溶液(1ppm or 2ppm Pb), 2ml pH为的缓冲溶液,2ml的待测液,混合后加的硫代乙酰胺试液,立即混合。
空白溶液:10ml的水,2ml pH为的缓冲溶液,2ml的测试溶液。
混合后加的硫代乙酰胺试液,立即混合,同空白溶液比较,对照溶液显浅棕色。
2分钟后,供试溶液的颜色不得比对照溶液深。
方法C供试溶液:规定量(不超过2g)的待检测物质置于坩埚内,加4ml的250g/l 的硫酸镁溶液(稀硫酸溶解硫酸镁),玻璃棒搅拌混和,小心加热。
如果混合物还是液体,则在水浴中蒸发使其干燥。
连续加热灼烧,灼烧温度不超过800℃,直到获得白色或灰白色的残渣。
取出,冷却后以稀硫酸润湿残渣,加热蒸发后继续灼烧,灼烧的总时间不能超过2小时。
取出,冷却。
如法制取2份残渣,分别加入5ml稀盐酸,的酚酞试液,然后滴加氨水,直到粉红色出现。
冷却,滴加冰醋酸至颜色消失,颜色消失后再多加冰醋酸。
必要时过滤,并洗涤残渣。
加水稀释至20ml,制成待测液。
取12ml该待测液,加2ml 的缓冲溶液,混和,加硫代乙酰胺试液,立即混合,制成供试溶液。
对照溶液:4ml 250g/l的硫酸镁溶液(稀硫酸溶解硫酸镁),规定量的标准铅溶液(10ppmPb)。