第三章自由基聚合反应.ppt
第三章 自由基聚合反应

[M]0 ln = kp'[I]1/2t [M] fkd 1/2 1/2 [I] t = kp kt
3. 引发效率较低时的聚合反应动力学方程 对于引发剂效率较低的聚合反应,链引发速率不仅
取决于引发剂浓度,而且与单体浓度有关。
vi=2fkd[I][M]
O O Ph C O O C Ph 过氧化苯甲酰(BPO) O Ph C O Ph + CO2 O 2 Ph C O
白色吸潮性粉末,稍 有气味,易溶于大多数有 机溶剂,微溶于水。同时 也是一种易燃、易爆炸的 危险化学品,必须在含湿 状态下保存。
容易发生诱导分解从而使引发效率降低。此外,引发合成 的聚合物会慢慢变黄,因此在合成光学性能要求较高的聚合物 时应避免选用。
2. 笼蔽效应
在溶液聚合反应中,浓度较低的引发剂分子及其分解出 的初级自由基始终处于含大量溶剂分子的高粘度聚合物溶液 的包围之中,一部分自由基无法与单体分子接触而更容易发 生向引发剂或溶剂的转移反应,从而使引发效率降低。 3. 初级自由基的副反应 如初级自由基的双基偶合反应、向溶剂分子的转移反应 等都可能消耗自由基,从而使引发效率降低。
3.3.2 引发剂分解反应动力学
R O O R' R N N R' RO + R'O R + R' +N2
单分子一级反应 故引发剂分解速率 vd=-d[I]/dt=kd[I]
t=0时引发剂浓度为[I]0,上式积分得 ln([I]0/[I])=kdt 引发剂分解50%所需的时间定义为引发剂半衰期t1/2来表达一 级反应的速率常数。 t1/2 = 1 [I] 0.693 ln 0 = kd [I]0/2 kd
+ H2 C CH X
高分子化学-第3章 自由基聚合
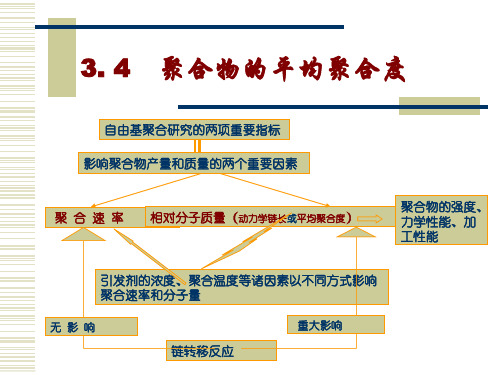
3. 4
聚合物的平均聚合度
1、动力学链长和聚合度
(1)动力学链长υ (kinetic chain length)的定义
每个活性种从引发阶段到终止阶段所消耗单体分子数。无 链转移时,动力学链长为增长速率和引发速率的比。 依据稳态时引发速率等于终止速率,则动力学链长可表 示为增长速率与终止速率的比: 即为单体消耗速率与
自由基产生(或消失) 速率之比
3. 4
聚合物的平均聚合度
如将稳态时的自由基浓度 入上式,可得下式:
,代
3. 4
聚合物的平均聚合度
若自由基聚合反应由引发剂引发时,
引发速率Ri = 2 f kd[I],则:
3. 4
聚合物的平均聚合度
可知动力学链长与引发速率存在以下关系:
1) 动力学链长与单体浓度的一次方成正比,与 引发剂浓度平方根成反比。 2) 说明了在自由基聚合体系中,增加引发剂用 量虽然可以提高聚合速率,但又使聚合物相对分子 质量降低。由此说明引发剂在自由基聚合中的重要
(1)温度对聚合速率的影响
总聚合速率常数k与温度T(K)遵循Arrhenius经验公式: 由前面推导可知: k=Ae-E/RT
k=kp(kd/kt)1/2
因此:
3.5 影响自由基聚合反应的因素
从而可知,总活化能E=(Ep-Et/2)+Ed/2
由Ep、 Et和Ed的大小可以得到总活化能E约为83 kJ/mol,为正值,表明温度升高,速率常数增大k增大。
3.5 影响自由基聚合反应的因素
1. 链自由基的双基终止过程的三步曲:
1) 链自由基的平移;
2) 链段重排,使活性中心靠近;
3) 双基相互反应而使链终止。
第二步(链段重排)是 控制步骤,受体系粘度 影响显著。
高分子化学课件第三章 自由基共聚合

m1= d[M1] = k11[M1*][M1] + k21[M2*][M1] (i)
m2 d[M2]
k12[M1*][M2] + k22[M2*][M2]
第三章 自由基共聚合
(3)假设共聚反应是一个稳态过程,即总的活性中心的浓 度[M1*+M2*]恒定,[M1*]和[M2*]的消耗速率等于[M1*]和 [M2*]的生成速率,并且 M1* 转变为M2*的速率等于M2*转 变为M1*的速率;
二元共聚合的理论研究较系统深入,而三元及三元以上共 聚合复杂,理论研究很少,但实际应用的例子颇多。ABS, SBS
三元以上聚合,一般以两种单体确定主要性质,另外单体 改性。
二元共聚物根据两单体单元在分子链上的排列方式可分四 类:
第三章 自由基共聚合
(1)无规共聚物(random copolymer) 两种单体单元的排列没有一定顺序,A单体单元相邻的单
第三章 自由基共聚合
四种竞争链增长反应:
k11 M1* + M1
k12 M1* + M2
k21 M2* + M1
k22 M2* + M2
M1* R11 = k11[M1*][M1]
M2* R12 = k12[M1*][M2]
M1*
R21 = k21[M2*][M1]
M2* R22 = k22[M2*][M2]
若含一段A链与一段B链,如~AAAAAAA-BBBBBBBBBB~, 称AB型二嵌段共聚物;如果是由一段A链接一段B链再届一 段A链,如~AAAAAA-BB~BBB-AAAAAAA~,则称ABA型 三嵌段共聚物;若由多段A链和多段B链组成,则称(AB)n型 多嵌段共聚物。
第三章 自由基共聚合
高分子化学第三章 自由基聚合

• 链转移反应前后,自由基的数目未变。
35
1. 向单体转移
· ~~CH2-CH + CH2=CH Cl Cl
· ~~CH=CH + CH3-CH Cl Cl
• 注意CH2=CHCl单体
36
2. 向溶剂或链转移剂转移
X ~~CH2CH · + YS X ~~CH2CHY + S ·
• 溶剂:
• 链转移剂:有较强的链转移能力的化合
1 2
[I ]
1
2
[M ] (3—35式)
注意本方程的适用范围
73
二、温度对聚合速率的影响
• 阿累尼乌斯公式:K=Ae–Ea/RT
其中:K=kp(kd/kt)½ 则:Ea=Ep+Ed/2–Et/2
74
一般情况下: Ep≈29kJ•mol–1, Ed≈126kJ•mol–1 Et≈17kJ•mol–1
10
一、 聚合的可能性
• 主要取决于双键上取代基的空间 效应
11
1.烯类单体: CXY=CMN
(1)一取代( CH2=CHX)
可均聚合
12
(2)二取代
(CH2=CXY、CHX=CHY) (a)1,1——二取代:一般不考虑空 间位阻效应,可均聚合。
注意:CH2=C(Ar)2只能形成二聚体
13
(b)1,2——二取代
54
2.半衰期
[I] ln = Kd t [I0]
• 60℃
ln2 t½ = K d
(3—17)
t½ >6h,低活性引发剂 1h< t½ <6h,中活性引发剂 t½ <1h,高活性引发剂
55
3. 引发效率
第三章自由基共聚合

无规共聚物名称中,放在前面的单体为主单体,后为第二单体 如:氯乙烯-co-醋酸乙烯酯共聚物 嵌段共聚物名称中的前后单体代表聚合的次序 接枝共聚物名称中,前面的单体为主链,后面的单体为支链 如:聚丙烯-g-丙烯酸
三 、研究共聚反应的意义 ⒈ 对聚合物进行改性
通过共聚,可以改善聚合物的许多性能,如机械性能、 弹性塑性、柔顺性、玻璃化温度、塑化温度、熔点、 溶解性能、染色性能和表面性能等等。性能改变的程 度与第二、第三单体的种类、数量以及单体单元的排 布方式有关。
M1代表丁二烯单体单元,M2代表苯乙烯单体单元。
(2)大分子主链上含M1单体单元(或也含M2单体单元),支链上含M2,M3两种单 单元。如ABS树脂
M1M1M1 M1M1M1M1 M2M2M3 M1M1M1M1M1 M3M3M2M2
M1代表丁二烯(B)单体单元,M2代表丙烯腈(A)单体单元, M3代表苯乙烯 (S)单体单元。
共聚物的命名:
聚- 两单体名称以短线相连,前面加“聚”字 如聚丁二烯-苯乙烯 -共聚物 两单体名称以短线相连,后面加“共聚物” 如乙烯-丙烯 共聚物、氯乙烯-醋酸乙烯共聚物
在两单体间插入符号表明共聚物的类型 co alt b g copolymer 无规 alternating 交替(alternate交替的, 轮流的) block 嵌段(block [blCk] 木块, 石块, 块, 街区) graft 接枝(graft 嫁接, (接技用的)嫩枝)
主单体 乙烯 乙烯 异丁烯 丁二烯 丁二烯 苯乙烯 氯乙烯 MMA 丙烯腈
第二单体 丙烯 苯乙烯 丙烯腈 丙烯腈
改进的性能和主要用途 破坏结晶,增加柔性和弹性。其为乙丙橡胶。 增加强度。其为通用橡胶。 增加耐油性。其为丁-苯橡胶。 提高抗冲性能。其为增韧塑料。
《材化高分子化学》第3章 自由基聚合
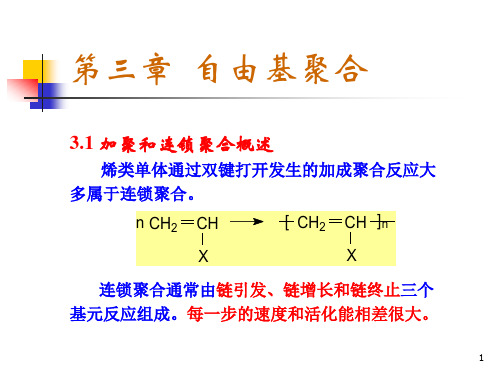
E = 105~150 kJ/mol (3—1)
kd = 10-4~10-6 s-1
(3—2)
19
第三章 自由基聚合
b. 单体自由基的形成
R + CH2 CH X
RCH2 CH X
由初级自由基与单体加成产生,为放热反应, 活
化能低,反应速度快。
E = 20 ~ 34 kJ/mol
(3—3)
20
第三章 自由基聚合
(CH3)2C N N C (CH3)2
2 (CH3)2C + N2
CN
CN
CN
优点:
(1)分解只形成一种自由基,无诱导分解。 (2)常温下稳定。80℃以上剧烈分解。
35
第三章 自由基聚合
(2)有机过氧化类引发剂
最简单的过氧化物:过氧化氢。活化能较高, 220kJ/mol,一般不单独用作引发剂。
HO OH 2HO
7
第三章 自由基聚合
分子中含有推电子基团,如烷基、烷氧基、苯基、乙 烯基等,碳=碳双键上电子云增加,有利于阳离子聚合进 行。
δ
CH2 CH Y
注意:丙烯分子上有一个甲基,具有推电子性和超共轭双 重效应,但都较弱,不足以引起阳离子聚合,也不能进行 自由基聚合。只能在配位聚合引发体系引发下进行配位聚 合。
30
第三章 自由基聚合
3.4.2 自由基聚合反应的特征
(1)可分为链引发、链增长、链终止等基元反应。 各基元反应活化能相差很大。其中链引发反应速率 最小,是控制聚合过程的关键。
慢引发、快增长、有转移,速终止。
与逐步缩聚机理特征比较见p75表3-6。
31
第三章 自由基聚合
(2)只有链增长反应使聚合度增加。从单体转化为 大分子的时间极短,瞬间完成。体系中不存在聚合 度递增的中间状态(p75图3-2)。聚合度与聚合时间 基本无关。
第三章自由基共聚合反应

知识目标 樉学习掌握自由基共聚合反应的基本概念、基本计算; 樉学习掌握自由基共聚合反应的机理、影响因素。
能力目标 樉能初步运用自由基共聚合的原理对高聚物进行改性; 樉能初步运用自由基共聚合的规律指导双组分共聚物的合成。
第一节 自由基共聚合反应的意义与类型一、自由基共聚合反应的意义 由两种或多种单体共同参加的自由基聚合反应称为自由基共聚合反应,简称共聚反应。
其产物中含有两种或两种以上不同单体链节的聚合物称为共聚物。
如nM1+mM2→~M1M2M2M1M2M2M2M1M1M2M2M1~ 通过自由基共聚合反应,可以改变均聚物的组成和结构,进而改变均聚物的使用性能。
如聚苯乙烯是抗冲击强度和抗溶剂性能都很差的易碎性塑料,因此实际使用受到很大限制。
而将苯乙烯与少量丁二烯共聚,就可以得到高抗冲击聚苯乙烯;将苯乙烯与丙烯腈、丁二烯共聚,就可得到广泛应用的ABS工程塑料;又如通过共聚改善材料的染色性能,黏合性能等。
通过自由基共聚合反应,还可以使本身不能均聚的单体如顺丁烯二酸酐、反丁烯二酸酐、顺丁烯二酸酯、1,2二苯基乙烯等参加共聚反应,扩大了单体范围。
通过自由基共聚合反应,能够测定单体和自由基的相对活性,设计、预测共聚物的性能、组成与结构。
自由基共聚合反应应用非常广泛,产品非常多。
如丁苯橡胶、丁腈橡胶、乙丙橡胶、丙烯酸酯类共聚物、ABS树脂、含氟共聚物塑料、氯乙烯乙烯醋酸乙烯酯三元共聚物等等,都是由自由基共聚合反应合成的。
二、共聚反应的类型 根据参加共聚反应单体的种类多少可以分为:只有两种单体共同参加的二元共聚反应和两种以上单体共同参加的多元共聚反应。
如果按聚合反应的活性中心不同,可以分为自由基型共聚和离子型共聚。
由于多元共聚反应非常复杂,这里着重介绍自由基型二元共聚反应。
离子型共聚在其他章节介绍。
三、共聚物的类型 由二种单体共同参加共聚反应所形成的共聚物,根据两种结构单元在共聚物大分子链的排列方式不同,可以分为以下四种类型。
第3章 自由基共聚

k 12 [M1 ][ M 2 ] [M ] 2 k 21 [M1 ]
代入
d[M1 ] k11 M1 M1 k 21 M M1 2 d[M 2 ] k 22 M 2 M 2 k12 M1 M 2
得:
d[M 1 ] k 11k 21[M 1 ] k 12 k 21[M 1 ][ M 2 ] 2 d[M 2 ] k 22 k 12 [M 2 ] k 12 k 21[M 1 ][ M 2 ]
n CH2=CH + mCH2=CH Cl
共聚
[CH2 CH]n [CH2 CH ]m Cl OCOCH 3
OCOCH 3
根据参加反应单体的单元数,共聚反应可分为:
二元共聚: 三元共聚: 多元共聚: 两种单体 三种单体 3种或3种以上单体 共同进行反应
3.1 引言
二、 共聚物类型
共聚物按大分子链中单体链节的排列方式可分为下列四种: (1)无规共聚物(random copolymer)
链引发(initiation): 2个引发反应。
I R
R M1 RM
ki,1 1
Ri,1 ki ,1
R M
1
R M 2 RM
ki, 2
2
Ri,2 ki , 2 R M 2
3.2 二元共聚物组成
链增长(propagation):4个增长反应
3.2 二元共聚物组成
链终止(termination): 3个终止反应
M M1
M M2
2
1
死大分子
kt,11
Rt ,11 2kt ,11 M
第3章自由基聚合反应

tr,I Mx I Mx R
k
Rtr,I ktr,I [M ][I]
Rtr,S ktr,S[M ][S]
Rp Rt Rtr Rp C ( D) Rt Rtr 2
tr,S Mx S Mx S
k
聚合度的表达式应是:
高分子化学
3.5 聚合中期聚合反应速率
聚合初期的恒速阶段一般可持续到转化率达10~15%。
fkd 1/2 Rp k p [M ] [I ] kt
1/2
自动加速效应(autoacceleration)——聚合反应速率 自动加快的现象称为自动加速现象。
一般单体的聚合体系都会存在自动加速现象,差别是 体系不同,出现的早晚和程度不同。 主要原因 3.5.3.1 凝胶效应 ——由于反应过程体系粘度的增加引起的自动加速现象称为 凝胶效应(gel effect)。
Rp k p [M ][M ]
Rt 2kt [M ]2
ktr , M ktr , I [ I ] ktr , S [ S ] ktr , P [ P ] 2 kt R p 1 C ( D ) 2 2 2 k p [M ] kp k p [M ] k p [M ] k p [M ] Xn
凝胶效应的主要原因在于链终止反应是受扩散控制的反应。
高分子化学
第3章 自由基聚合反应
3.5-3.9
对于正常的双基终止,链自由基双基终止过程可以分为 三步:链自由基质心的平移、链段重排(控制步骤)和双基 碰撞发生反应。体系黏度是影响的主要因素。
fk R p k p [ M ] d [ I ]1/2 kt
k p [M ] (2kt )1/2 Ri1/2
高分子化学第三章_自由基聚合
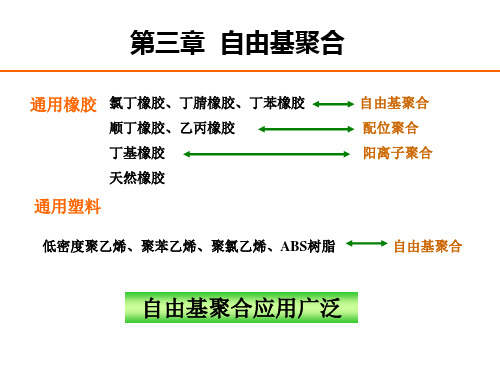
(1)取代基吸电子能力较弱,如偏氯乙烯中的氯,两个氯 吸电子 作用的叠加,使单体更易聚合。 ( 2 )取代基吸电子能力强,如偏二腈乙烯,两个腈基强吸 电子作 用使双键上电荷密度降低太多,从而使双键失去了与 自由基加成的能力,只能阴离子聚合,而难自由基聚合。 (3)两个取代基都是给电子性,如异丁烯中的两个甲基, 给电子作用的叠加,使异丁烯不能发生自由基聚合,而易 于阳离子 聚合。 ( 4 )两个取代基中,一个是弱给电子性,另一个是强吸 电子性,如甲基丙烯酸酯类,这类单体易发生自由基聚合 反应。
组成
连 锁 聚 合
具有活性中 心,聚合过 程由链引发 ,链增长, 链终止等三 个基元反应 组成。自由 基聚合反应 是连锁聚合 反应的一种
聚合过程中也可能存在另一个基元反应 —链转移反应(chain transfer reaction);链转移反应对聚合物的分子量、结构和聚 合速率产生影响。
7
连锁聚合反应过程
分解 I 引发剂 R* + H2C CH X 或离解
(以乙烯基单体聚合为例)
引发活性种(中心) R* 链增长活性中心 R CH2 CH* X
R CH2 CH* + H2C CH X X
CH2 CH* X 增长链
增长链
终止反应
聚合物链
连锁聚合反应的基本特征
a. 连锁聚合反应是合成碳链聚合物的聚合反应; b. 由多个机理不同基元反应组成,反应速率和活化能差别 大; c. 单体只能与活性中心反应生成新的活性中心,单体之间 不能反应; d. 反应体系始终是由单体、聚合产物和微量引发剂及含活 性中心的 增长链所组成; e. 聚合产物的相对分子质量一般不随单体转化率而变。
CH B X
3.1.3 连锁聚合的单体
第三章自由基聚合

进行聚合的判据吉布斯自由能ΔG: 如果ΔG<0,单体才有聚合的可能; 如果ΔG>0,则聚合物将发生解聚; 若ΔG=0,则单体聚合与聚合物解聚处于可逆平衡状态。
第三章 自由基聚合
ΔG=ΔH – TΔS 根据焓变和熵变的符合变化,有四种组合: ΔH<0和ΔS<0,这是最普通的组合。 因为一般聚合是放热或减焓反应,故ΔH<0;另一方面,单 体聚合成大分子聚合物后,无序性减小,是减熵过程,故 ΔS<0。因此,只有焓变大于TΔS的时候,ΔG<0,聚合才能 发生。 在某一临界温度下,ΔG=0,则ΔH =TΔS,聚合和解聚处 于平衡状态,这一临界温度称为聚合上限温度Tc,可以简 单计算如下:
无取代基,结构对称,无诱导效应和共轭效应,较难 聚合,只能高温高压的苛刻条件下进行自由基聚合, 或者在特殊催化剂下进行配位聚合。
带供电基团的乙烯类单体:
如带有烷氧基、烷基、苯基、乙烯基等单体。
供电子基团使碳碳双键的电子云密度增加,有利于阳 离子的进攻,进行阳离子聚合。
δ
CH2
CH2 H C O R
> RCHCOR > > C6H5CH2
RCHCN >
> (C6H5)2CH
前两个过于活泼,引起爆聚;最后五种则是稳定的自由基; 三苯甲基自由基非常稳定,无引发能力,为阻聚剂。
第三章 自由基聚合
自由基聚合机理 自由基聚合机理是由单体分子转变成大分子的微观历程, 由链引发、链增长、链终止、链转移等基元反应组成。
CH2=CHCH2CH3 CH2=C(CH3)2 CH2=CHCH=CH2 CH2=C(CH3)CH=CH2 CH2=CClCH=CH2 CH2=CHC6H5 ⊕ + ⊕ ⊕ + + + ⊕ ⊕ + ⊕
第3章 自由基聚合

(2)偶氮类引发剂 带吸电子取代基的偶氮化合物,分对称和不对称两大类:
R
1
R2 R2 1 C N N C R X X 对称
R R2 R C N N C R1 X X 不对称
X为吸电子取代基:-NO2, -COOR, -COOH, -CN等
CH3 CH3 H3C C N N C CH3 CN CN 偶氮二异丁腈(AIBN) CH3 2 H3C C CN + N2
n = kp[M]/2(fktkd[I])1/2
动力学链长与平均聚合度的关系
无链转移反应时,每一条增长链都是由一个初级自由基引 发而成,因而ν=平均每条增长链所含的单体单元数: 当发生歧化终止时, 两条增长链生成两个聚合物分子,因而 聚合产物的 Xn = n; 当发生偶合终止时,两条增长链结合生成一个聚合物分子, 因而聚合产物的Xn = 2n 。
四、温度对聚合反应速率的影响 聚合反应速率常数与温度的关系遵守Arrhenius方程: k = Ae-E/RT Rp ∝ kp(kd/kt)1/2
表观聚合速率常数 k =kp(kd/kt)1/2 k = Ap(Ad/At)1/2exp{-[Ep - Et/2 + Ed/2]/RT} 式中:Ed 引发剂分解活化能;Ep 链增长活化能;Et 链终止 活化能; 总活化能 E= Ep -Et/2 + Ed/2 一般Ed = 126 kJ/mol, Ep =29 kJ/mol, Et = 17 kJ/mol,即 E = 84 kJ/mol, 聚合反应速率随温度升高而加快。
其中加入的能产生聚合反应活性中心的化合物常称为引发剂。 引发剂(或其一部分)在反应后成为所得聚合物分子的组成 部分。
(以乙烯基单体聚合为例)
分解 I 引发剂 R* + H2C CH X R CH2 CH* X 或离解 引发活性种(中心) R* 链增长活性中心
高分子化学课件-自由基连锁聚合反应

(1)自由基的活性
自由基的活性与其结构有关
共轭效应较强的自由基具有较大的稳定性
极性基团使自由基活性降低 体积较大的基团可妨碍反应物的靠近,将
使反应活性降低
各种自由基的相对活性顺序
体积较大
3.1.3 自由基聚合的基元反应
自由基聚合是链式聚合 至少由三个基元反应组成 链引发 链增长 链温度Te下,有对应的平衡单体浓度。 若平衡单体浓度非常低,就可看成是聚合完全。 25℃时,酯酸乙烯[M]e=1.4×10-11mol/L, 苯乙烯为 2×10-8, 甲基丙烯酸甲酯 2.86×l0-5, α-甲基苯乙烯 [M]e=2.6mol/L, 130℃聚合时, MMA [M]e=0.5mol/L,
例如
CH3 n CH2=C
CH3 -[-CH2-C-]n-
α-甲基苯乙烯在 萘钠或烷基锂存 在下,在苯溶液中聚合,温度反复冷却至70℃和加热到+40℃,体系粘度反复增减,表 明聚合和解聚可逆地进行。
低温聚合多一些,温度高时,会解聚出更多的 单体。
链增长和解聚是一对可逆反应
如下式所示,
链增长和解聚
高 分 子 化 学
第 三 章 自由基连锁聚合反应
1
自由基聚合是高分子化学中极重要的反应
工业上处于领先地位: 自由基聚合产物占总 聚合物的60%以上, 约占热塑性树脂的80 %。 理论上也较完善:
自由基活性中心的产生和性质,各基元反应和
反应机理、聚合反应动力学、分子量及影响分 子量的因素, 聚合反应热力学理论, 都较为成熟。 这些理论常作为研究离子型及配位聚合的基础 。
22
3. 多取代烯烃
一般不能聚合,除对于半径很小的取代基(如:F), 不管几取代,均能聚合。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3.1 概述 烯类单体在聚合条件下,碳碳双键被打开,通过链式聚合反应,
生成乙烯基聚合物:
n H2C CH X
CH2 CH Xn
X: H, R,
, Cl, CN, OR , -COOR
乙烯基聚合物在高分子合成工业上占据及其重要的地位,其主 要品种如聚乙烯、聚氯乙稀、聚苯乙烯等的产量遥遥领先,主宰整 个合成聚合物的市场。
(2)电子效应决定聚合机理的选择性
乙烯基单体(CH2=CH-X)对聚合机理的选择性,即是否能进行 自由基、阴离子、阳离子聚合,取决于取代基-X的诱导效应和共轭 效应(合称为电子效应),取代基电子效应的影响主要表现在它们 对单体双键的电子云密度的改变,以及对形成活性种的稳定能力的 影响。
(a) X为给(推)电子基团
与乙烯相比,一取代乙烯中的取代基往往在降低双键对称性的同 时会改变其极性,从而聚合活性增加。1,1-二取代烯烃,由于同一 碳原子上两个取代基的存在而具有一定程度的以双键为对称轴的对 称性,从而使聚合活性与单取代乙烯相比,稍有降低。
1,2-二取代以及三、四取代烯烃原则上都不能聚合,其原因是这 三类取代烯烃的α-和β-碳原子都带有取代基,活性中心不论是从α位还是β-进攻单体时都存在空间障碍,从而无聚合活性。唯一例外 的是当取代基为F时,它的一、二、三、四取代乙烯都可以聚合, 这时因为F原子半径小,与H非常接近,从而无空阻效应。
H2C CH X
增大电子云密度,易 与阳离子活性种结合
H R CH 2 C
X
分散正电性,稳定阳离子
因此带给电子基团的烯类单体易进行阳离子聚合, 如X = -R,-OR,-SR,-NR2等。
(b) X为吸电子基团
H2C CH X
降低电子云密度,易 与富电性活性种结合
H R CH 2 C
X
分散负电性,稳定活性中心
羰基化合物和一些杂环化合物,其中以烯烃最具实际应用意义。 评价一个单体的聚合反应性能,应从两个方面考虑:首先是其
聚合能力大小,然后是它对不同聚合机理如自由基、阳离子、阴离 子聚合的选择性。
(1)位阻效应决定单体聚合能力
H2C CH X
一取代烯烃
Y H2C C
X
1,1-二取代烯烃
一取代烯烃和1,1-二取代烯烃原则上都能进行聚合,原因是活性 中心可从无取代基的β-碳原子上进攻单体。除非取代基体积太大, 如带三元环以上的稠环芳烃取代基的乙烯不能聚合,1,1-二苯基乙 烯也只能聚合生成二聚体而得不到高聚物。
H2C CH NO2
偏二腈乙烯
硝基乙烯
只能进行阴离子聚合
CH3 H 2C C
CH3
H2C CH OR
异丁烯 乙烯基醚
只能进行阳离子聚合
(d)X 取代基为吸电子基团,但同时又具有 p-p 给电子共轭效 应
由于其给电子效应部分地抵消了吸电子效应,使其吸电子效应 减弱,该类单体一般难以进行阴离子聚合,而只能进行自由基聚合, 如:
根据引发活性种与链增长活性中心的不同,链式聚合反应可 分为自由基聚合、阳离子聚合、阴离子聚合和配位聚合等。
自由基: AA 阳离子: AB 阴离子: AB
CH2=CHX 2A
CH2=CHX A+B-
A CH2 CH X
A CH2
HC+
B
X
CH2=CHX A-B+
A
CH2
HC-
+ B
X
3.1.2 链式聚合单体 能进行链式聚合的单体主要有烯烃(包括共轭二烯烃)、炔烃、
HO OH
2HO.
O
O
KO S O O S OK
O
O
. O
_
2KO S O (SO4)
链式聚合的反应历程根据反应活性中心的性质,可分为自由 基聚合、阳离子聚合、阴离子聚合和配位聚合等。其中以自由基聚 合不论是在理论研究上还是应用上占主导地位,是整个链式聚合的 基础。
3.3.1 链式聚合反应的一般特征 逐步聚合反应是由单体及不同聚合度中间产物之间,通过功能基
反应来进行的。
链式聚合反应则是通过单体和反应活性中心之间的反应来进行, 这些活性中心通常并不能由单体直接产生,而需要在聚合体系中加 入某种化合物,该化合物在一定条件下生成聚合反应活性中心,再 通过反应活性中心与单体加成生成新的反应活性中心,如此反复生 成聚合物链。
其中加入的能产生聚合反应活性中心的化合物常称为引发剂。引 发剂(或其一部分)在反应后成为所得聚合物分子的组成部分。
链式聚合反应的基本特征总结如下:
➢ 聚合过程一般由多个基元反应组成; ➢ 各基元反应机理不同,反应速率和活化能差别大; ➢ 单体只能与活性中心反应生成新的活性中心,单体之 间不能反应; ➢ 反应体系始终是由单体、聚合产物和微量引发剂及含 活性中心的增长链所组成; ➢ 聚合产物的分子量一般不随单体转化率而变。(活性 聚合除外)。
H2C CH Cl
氯乙烯
H2C CH
O C CH3 O
乙酸乙烯酯
(e) 具有共轭体系的烯类单体
p电子云流动性大,易诱导极化,可随进攻试剂性质的不同而取 不同的电子云流向,因此视引发条件不同而可进行阴离子型、阳离 子型、自由基型等各种链式聚合反应。如苯乙烯、丁二烯等:
R+
H2C CH
+
R-
+ H 2C CH
3.2 自由基聚合引发剂和链引发反应 3.2.1 引发剂种类
自由基聚合引发剂通常是一些在一定条件下(加热或光照) 可分 解生成自由基,并能引发单体聚合的化合物。
链引发反应包括两个基本反应:引发剂分解产生初级自由基; 初级自由基与单体加成生成单体自由基。
kd
I
பைடு நூலகம்2R
初级自由基
R + H2C CH
ki
R CH2 CH 单体自由基
因此带吸电子基团的烯类单体易进行阴离子聚合与自由基, 如X = -CN,-COOR,-NO2等。
(c)自由基聚合对单体的选择性低
许多带吸电子基团的烃类单体,如丙烯腈,丙烯酸酯类等既可 以进行阴离子聚合,也可以进行自由基聚合。只是在取代基的电子 效应太强时,才不能进自由基聚合,如
CN H2C C
CN
X
X
常用的自由基聚合引发剂可分为四大类:过氧化物、偶氮类化 合物、氧化-还原体系以及某些在光作用下产生自由基的物质。
(1)过氧化物引发剂
分子结构中含有过氧基团,由过氧基团在受热或光照条件下均 裂产生引发自由基。包括无机过氧化物和有机过氧化物。
无机过氧化物包括过氧化氢、过硫酸钾、过硫酸铵等,其分解反 应机理如下: