高等有机化学人名反应
100种有机化学人名反应

100种有机化学人名反应1. Arndt-Eistert反应醛、酮与重氮甲烷反应,失去氮并重排成多一个CH2基的相应羰基化合物,这个反应对于环酮的扩环反应很重要。
O+CH2N22. Baeyer-Villiger氧化由樟脑生成内酯:-OO+-N2CH2NN重排应用过氧酸使酮氧化成酯。
反应中在酮的羰基和相邻的碳原子之间引人一个氧原子。
如OCH3CH3CH3 OOCH3CH3H2SO5有时反应能生成二或多过氧化物,但环状酮转变为内酯能得到单一的预期产物。
合适的酸为过硫酸(Caro’s 酸)、过氧苯甲酸、三氟过氧乙酸。
除环酮外,无环的脂肪、芳香酮也可发生此反应。
二酮生成酸酐类、α、β-不饱和酮得到烯醇酯类。
3.Bechamp还原(可用于工业制备)在铁、亚铁盐和稀酸的作用下,芳香族硝基化合物能还原成相应的芳香胺。
C6H5-NO2 + 2Fe + 6HClC6H5-NH2 + 2FeCl3 + 2H2O。
当某些盐(FeCl2、FeCl3、FeSO4、CaCl2等)存在时,所用酸无论是过量还是少量,甚至在中性溶液中都能够进行这种还原。
此方法适用于绝大部分各种不同结构的芳香族化合物,有时也用来还原脂肪族硝基化合物。
4.Beckmann重排醛肟、酮肟用酸或路易斯酸处理后,最终产物得酰胺类。
单酮肟重排仅得一种酰胺,混酮肟重排得两种混合酰胺。
但一般质子化羟基的裂解和基团R的转移是从相反的位置同OR OH ORR'NRN时进行的。
R'NHR' R' OH NHR无论酯酮肟和芳酮肟都会发生此反应。
环酮肟重排得内酰胺,这在工业生产上很重要,利用此反应可帮助决定异构酮肟的结构。
5.Beyer喹啉类合成法芳香伯胺与一分子醛及一分子甲基酮在浓盐酸或ZnCl2存在下,反应生成喹啉类化合物。
HHNN R'HClNH2R'-H2+ R'CHO+RCOCH3RR这是对Doebner-Miller喹啉合成法的改进。
大学有机化学人名反应机理汇总

3.Baeyer----Villiger 反应拜耳-维立格氧化重排反应过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:4.Beckmann 重排%肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变。
7.Cannizzaro 反应凡α位碳原子上无活泼氢的醛类和浓NaOH或KOH水或醇溶液作用时,不发生醇醛缩合或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物。
此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇:!脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛类与强碱液,作用发生醇醛缩合或进一步变成树脂状物质。
醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上。
9.Claisen 酯缩合反应含有α-氢的酯在醇钠等碱性缩合剂作用下发生缩合作用,失去一分子醇得到β-酮酸酯。
如2分子乙酸乙酯在金属钠和少量乙醇作用下发生缩合得到乙酰乙酸乙酯。
乙酸乙酯的α-氢酸性很弱(,而乙醇钠又是一个相对较弱的碱(乙醇的pK a~),因此,乙酸乙酯与乙醇钠作用所形成的负离子在平衡体系是很少的。
但由于最后产物乙酰乙酸乙酯是一个比较强的酸,能与乙醇钠作用形成稳定的负离子,从而使平衡朝产物方向移动。
大学有机化学人名反应总结

⼤学有机化学⼈名反应总结有机化学⼀、烯烃1、卤化氢加成(1)【马⽒规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】【本质】不对称烯烃的亲电加成总是⽣成较稳定的碳正离⼦中间体。
【注】碳正离⼦的重排(2)【特点】反马⽒规则【机理】⾃由基机理(略)【注】过氧化物效应仅限于HBr 、对HCl 、HI ⽆效。
【本质】不对称烯烃加成时⽣成稳定的⾃由基中间体。
【例】2、硼氢化—氧化【特点】不对称烯烃经硼氢化—氧化得⼀反马⽒加成的醇,加成是顺式的,并且不重排。
【机理】【例】 3、X 2加成【机理】【注】通过机理可以看出,反应先形成三元环的溴鎓正离⼦,然后亲和试剂进攻从背⾯进攻,不难看出是反式加成。
不对称的烯烃,亲核试剂进攻主要取决于空间效应。
【特点】反式加成 4、烯烃的氧化1)稀冷⾼锰酸钾氧化成邻⼆醇。
3H 33H3稀冷KMnO433M n OOH 2O 3H 33H 3 2)热浓酸性⾼锰酸钾氧化3)臭氧氧化 4)过氧酸氧化 5、烯烃的复分解反应【例】 6、共轭⼆烯烃 1)卤化氢加成2)狄尔斯-阿德尔(Diels-Alder )反应【描述】共轭⼆烯烃和烯烃在加热的条件下很容易⽣成环状的1,4加成产物。
【例】⼆、脂环烃1、环丙烷的化学反应【描述】三元环由于张⼒⽽不稳定,易发⽣加成反应开环,类似碳碳双键。
【特点】环烷烃都有抗氧化性,可⽤于区分不饱和化合物。
【注】遵循马⽒规则【例】2、环烷烃制备1)武兹(Wurtz)反应【描述】通过碱⾦属脱去卤素,制备环烷烃。
【例】2)卡宾①卡宾的⽣成A、多卤代物的α消除B、由某些双键化合物的分解②卡宾与烯烃的加成反应【特点】顺式加成,构型保持【例】③类卡宾【描述】类卡宾是⼀类在反应中能起到卡宾作⽤的⾮卡宾类化合物,最常⽤的类卡宾是ICH2ZnI。
【特点】顺式加成,构型保持【例】三、炔烃1、还原成烯烃1)、顺式加成2)、反式加成2、亲电加成1)、加X2【机理】中间体Br+R2 R1【特点】反式加成2)、加HXRRHBr R Br(⼀摩尔的卤化氢主要为反式加成)3)、加H2O【机理】【特点】炔烃⽔合符合马式规则。
大学有机化学人名反应总结

有机化学一、烯烃1、卤化氢加成 (1)CHCH 2RHXCH 3RX【马氏规则】在不对称烯烃加成中,氢总就是加在含碳较多的碳上。
【机理】CH 2CH 3+CH 3CH 3X +CH 3CH 3+H +CH 2+C3X +CH 3X主次【本质】不对称烯烃的亲电加成总就是生成较稳定的碳正离子中间体。
【注】碳正离子的重排 (2)CHCH 2RCH 2CH 2R BrHBrROOR【特点】反马氏规则 【机理】 自由基机理(略)【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】CH 2CH3BrCH CH 2BrC H 3CH +CH 3C H 3HBrBrCH 3CH 2CH 2BrCH CH 3C H 32、硼氢化—氧化CHCH 2R CH 2CH 2R OH1)B 2H 62)H 2O 2/OH-【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成就是顺式的,并且不重排。
【机理】2CH33H323H32 CH CH2CH3H BH2CH CH=CH(CH3CH2CH2)3-H3CH2CH2C22CH3CH2B OCH2CH2CH3H3CH2CH2C2CH2CH3+O H-OHB-OC H2CH2CH3CH2CH2CH3H3CH2CH2B OC H2CH2CH3CH2CH2CH3H2CH2CH3HOO-B(OCH2CH2CH3)3 B(OCH2CH2CH3)3+3NaOH3NaOH3HOC H2CH2CH33+Na3BO32【例】CH31)BH32)H2O2/OH-CH3HHOH3、X2加成C CBr/CClC CBr【机理】CCC CBrBr CBr+C CBrOH2+-H+C CBrOH【注】通过机理可以瞧出,反应先形成三元环的溴鎓正离子,然后亲与试剂进攻从背面进攻,不难瞧出就是反式加成。
不对称的烯烃,亲核试剂进攻主要取决于空间效应。
大学有机化学人名反应机理汇总
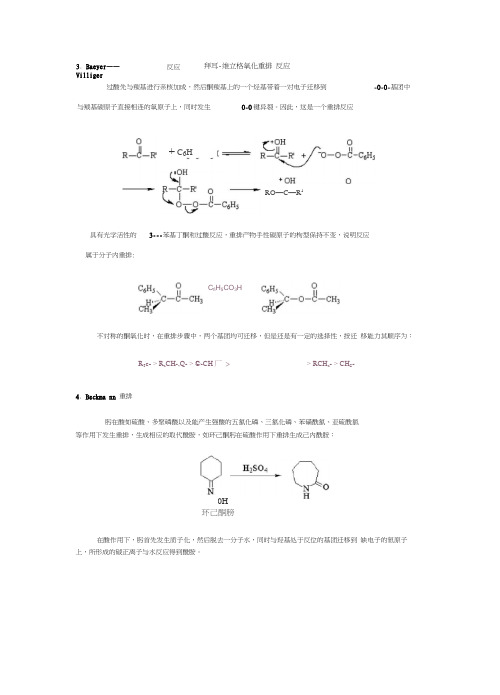
过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-0-0-基团中与羰基碳原子直接相连的氧原子上,同时发生0-0键异裂。
因此,这是一个重排反应具有光学活性的 3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁 移能力其顺序为:R 3c- > R a CH-,Q- > ©-CH 厂 > > RCH a - > CH S -4. Beckma nn 重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:环己酮膀在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到 缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
3. Baeyer ——Villiger反应拜耳-维立格氧化重排 反应0H+ C 6H A ;O HRO —C —R 1C fi H 5CO 3H迁移基团如果是手性碳原子,则在迁移前后其构型不变。
7. Cannizzaro 反应凡a 位碳原子上无活泼氢的醛类和浓NaOH 或KOH 水或醇溶液作用时,不发生醇醛缩合 或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物。
此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇:脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛 类与强碱液,作用发生醇醛缩合或进一步变成树脂状物质。
HCHO + C 6H 5CHO醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上。
眷£6+ C^CHQH9. Claisen 酯缩合反应HQR'-N=「一R + ---------- ■ R'—N=C-R甕一KHC —RHCHONaOH||C s Hj —C —H十0HII0H含有a -氢的酯在醇钠等碱性缩合剂作用下发生缩合作用,失去一分子醇得到B -酮酸酯。
(完整版)经典有机人名反应

有机化学人名反应1.拜耳维利格Baeyer----Villiger 反应(p317)反应机理(不要求)过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例2.康尼查罗Cannizzaro 反应(p321)凡α位碳原子上无活泼氢的醛类和浓NaOH或KOH水或醇溶液作用时,不发生醇醛缩合或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物。
此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇:脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛类与强碱液,作用发生醇醛缩合或进一步变成树脂状物质。
具有α-活泼氢原子的醛和甲醛首先发生羟醛缩合反应,得到无α-活泼氢原子的β-羟基醛,然后再与甲醛进行交叉Cannizzaro反应,如乙醛和甲醛反应得到季戊四醇:反应机理醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上。
反应实例3.克莱森许密特Claisen—Schmidt 反应(交叉羟醛缩合)(p314)一个无氢原子的醛与一个带有氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇溶液存在下发生缩合反应,并失水得到不饱和醛或酮:反应机理反应实例3.Claisen 重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R′I >R′Br >R′Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按 S2 进行的分子重排反应:N反应实例二、Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer----Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
有机化学人名反应

取代反应:1,加特曼反应:加特曼(Gattermann L)发现:用催化量的金属铜代替氯化亚铜或溴化亚铜作催化剂,也可使重氮盐与盐酸或氢溴酸反应制得芳香氯化物或溴化物。
这样进行的反应叫做加特曼反应。
2,加特曼-科赫反应:苯、一氧化碳和氯化氢反应生成苯甲醛,此反应称为加特曼-科赫反应。
3,傅-克反应:芳香化合物芳环上的氢被烷基取代的反应称为傅-克烷基化反应;芳香化合物芳环上的氢被酰基取代的反应称为傅-克酰基化反应;统称傅-克反应。
4,布赫尔反应:萘酚在亚硫酸氢钠存在下与氨作用,转变成相应萘胺的反应称为布赫尔反应。
5,齐齐巴宾反应:吡啶与氨基钠反应,生成α-氨基吡啶,如果α位已被占据,则得γ-氨基吡啶,但产率很低。
这个反应称为齐齐巴宾(Chichibabin)反应。
6,刚穆伯—巴赫曼反应:芳香重氮盐中的芳基在碱性条件下与其它芳香族化合物偶联成联苯或联苯衍生物的反应称为刚穆伯(Gomberg)—巴赫曼(Bachmann)反应。
7,柯尔伯—施密特反应:干燥的酚钠或酚钾与二氧化碳在加温加压下生成羟基苯甲酸的反应称为柯尔伯—施密特(Kolbe-Schmitt)反应。
8,威廉森合成法:在无水条件下,醇钠和卤代烷作用生成醚的反应称为威廉森(Williamson A W)合成法。
9,席曼反应:芳香重氮盐和氟硼酸反应,生成溶解度较小的氟硼酸盐,后者加热分解产生氟苯,这称为席曼(Schiemann)反应。
10,桑德迈耳反应:1884年,桑德迈耳(Sandmeyer T)发现:在氯化亚铜或溴化亚铜的催化下,重氮盐在氢卤酸溶液中加热,重氮基可分别被氯或溴原子取代,生成芳香氯化物或溴化物。
这一反应称为桑德迈耳反应。
11,普塑尔反应:一些重氮盐在碱性条件下或稀酸的条件下可以发生分子内的偶联反应。
这个反应是普塑尔(Pschorr R)在寻找合成菲环的新方法中首先发现的,故称为普塑尔反应。
12,瑞穆尔—悌曼反应:酚与氯仿在碱性溶液中加热生成邻位及对位羟基醛的反应称为瑞穆尔—悌曼(Reimer —Tiemann)反应。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯与一个新得卤代烷:卤代烷反应时,其活性次序为:R′I >R′Br 〉R′Cl.除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a—或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总就是先脱除含碳原子数最少得基团。
本反应就是由醇制备卤代烷得很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用得卤代烷R’X得烷基与亚磷酸三烷基酯(RO)3P得烷基相同(即R'=R),则Arbuzov反应如下:这就是制备烷基膦酸酯得常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯RP(OR')2与次亚膦酸酯R2POR'也能发生该类反应,例如:反应机理一般认为就是按SN2 进行得分子内重排反应:反应实例二、Arndt—Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer——--Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上得一个烃基带着一对电子迁移到—O-O-基团中与羰基碳原子直接相连得氧原子上,同时发生O-O键异裂.因此,这就是一个重排反应具有光学活性得3-—-苯基丁酮与过酸反应,重排产物手性碳原子得枸型保持不变,说明反应属于分子内重排:不对称得酮氧化时,在重排步骤中,两个基团均可迁移,但就是还就是有一定得选择性,按迁移能力其顺序为:醛氧化得机理与此相似,但迁移得就是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应得酯,其中三氟过氧乙酸就是最好得氧化剂。
这类氧化剂得特点就是反应速率快,反应温度一般在10~40℃之间,产率高。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R′I 〉R′Br 〉R′Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a—或b—卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷R’X 的烷基和亚磷酸三烷基酯(RO)3P 的烷基相同(即R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯RP(OR')2和次亚膦酸酯R2POR' 也能发生该类反应,例如:反应机理一般认为是按S N2 进行的分子内重排反应:反应实例二、Arndt—Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸.反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺.反应实例三、Baeyer—--—Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到—O—O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂.因此,这是一个重排反应具有光学活性的3—-—苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂.这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R′I >R′Br >R′Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理2 进行的分子内重排反应:一般认为是按 SN反应实例二、Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer----Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R′I 〉R′Br〉R′Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a—卤代醚、a- 或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应.当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷R’X的烷基和亚磷酸三烷基酯(RO)3P 的烷基相同(即R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯RP(OR')2和次亚膦酸酯R2POR' 也能发生该类反应,例如:反应机理一般认为是按S N2 进行的分子内重排反应:反应实例二、Arndt-Eister反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸.反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer--—-Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O—O键异裂.因此,这是一个重排反应具有光学活性的3-—-苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸.反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。
100种有机化学人名反应

120~200℃时热解,顺利产生烯烃,相应醇和氧硫
化碳。黄原酸酯在热解前制备不须离析除黄原酸酯外,其他的酯氨基甲酸酯,碳
酸酯和羧酸酯热解。特别是使用大分子量的羧酸酯(棕榈酸酯)的烯烃提供了有
利条件。因为这些酯本身沸点高,而热解温度较低(约
300℃),在液相中简单加热即可。
250℃
Cl OCH2CH=CH2
;
Cl 醚分子中,
如临位未被取代则不起重排反应,产生复杂的热分解作用。此反应是在苯环引入丙基的
简易方法,因为烯丙基可还原成丙基。
16. Claisen缩合反应(P352~354)
17. Claisen-Schmidt反应(P287)
N+
*
H CH3
CH3 △
H3C
*
CH3
CH2 +(CH3)2NOH
O
H5C6
C6H5
20. Criegee氧化法
乙二醇类在稀醋酸或苯溶液中,室温时用四乙酸铅进行很温和的氧化,两个相连的各带
有游离羟基的碳原子之间的碳链就断裂,得到定量的醛酮类。此法用于研究醇类结构及
此法用来合成呋喃类化合物,在吡啶或氨存在下,α
-氯化羰基化合物或α
,β-氯醚类与
1,3-二羰基化合物发生缩合反应,生成呋喃类化合物。
R C O H2C COOR'
O
COOR'
R
吡啶或氨
-H2O, -HCl
CH2
+
OC
Cl CH3
CH3
解,可得醛类化合物,
R’MgX中
大学有机化学人名命名的部分反应

傅克反应Friedel-Craafts在无水AlCl3、AlFe3、ZnCl2、BF3和硫酸等无机酸存在下烷基化苯和卤代烃、烯烃、醇反义生成烷基苯酰基化苯与酰氯或酸酐生成芳基酮拜尔-魏立格反应Baeyer_Villiger过氧酸氧化可使酮分子中插入一个氧成为酯康尼扎罗反应Cannizzaro无α氢的醛在浓碱作用下,一分子氧化为酸,另一分子还原为醇克莱门森还原Clemmensen醛酮与锌汞齐和浓盐酸反应,羰基被还原成亚甲基麦尔外因-彭道夫反应Meerwein-Ponndorf异丙醇铝可在异丙醇溶剂中将羰基还原为醇羟基,不影响其他不饱和基团Collins试剂——三氧化铬与吡啶将伯醇的氧化停留在醛的阶段瑞穆尔-梯曼反应Reimer-Tiemann苯酚、氯仿、NaOH共热,得到邻羟基苯甲醛——水杨醛柯尔贝反应Kolbe苯酚的钠盐在高温高压下和二氧化碳反应,得到水杨酸钠盐。
酸化后得到邻羟基苯甲酸——水杨酸克莱森-施密特反应Claisen-Schmidt脂肪烃与含有α氢的酮或醛在NaOH或者乙醇水溶液中进行交叉羟醛缩合克内费纳格尔缩合反应Knoevenagel含有活泼亚甲基的化合物与芳醛在碱作用下发生缩合,形成不饱和酯书199佩金反应Perkin芳香醛与酸酐在相应的钠或钾盐存在下缩合达森反应Darzens醛酮在强碱作用下和α卤代醛酸酯反应,生产环氧醛酸酯克莱森缩合Claisenα氢的酯自身缩合,形成β羰基酸酯狄克曼缩合Dieckmann中间相隔4、5个碳的二元羧酸酯,在NaOH的醇溶液下发生内酯缩合,生成五、六元环酮酯罗森孟还原法Rosenmund用二甲苯或硫喹啉降低钯催化剂的活性,可使酰卤还原成醛(LiAlH4还原到醛后进一步变成伯醇)霍夫曼降解Hofmann酰胺与溴或氯在碱性水溶液中反应,生成比原料少一个碳原子的伯胺盖布瑞尔反应GabrielN-烷基的酰胺水解得到高纯度的伯胺。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R′I >R′Br >R′Cl。
除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。
本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按 S2 进行的分子内重排反应:N反应实例二、Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer----Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。
因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排:不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。
100种有机化学人名反应(有机化学)

1. ArndtEistert反应醛、酮与重氮甲烷反应失去氮并重排成多一个CH2基的相应羰基化合物这个反应对于环酮的扩环反应很重要。
OCH2N2O-CH2NNN2重排O2. BaeyerVilliger氧化应用过氧酸使酮氧化成酯。
反应中在酮的羰基和相邻的碳原子之间引人一个氧原子。
如由樟脑生成内酯OCH3CH3CH3OOCH3CH3H2SO5有时反应能生成二或多过氧化物但环状酮转变为内酯能得到单一的预期产物。
合适的酸为过硫酸Caro’s 酸、过氧苯甲酸、三氟过氧乙酸。
除环酮外无环的脂肪、芳香酮也可发生此反应。
二酮生成酸酐类、α、β不饱和酮得到烯醇酯类。
3. Bechamp还原可用于工业制备在铁、亚铁盐和稀酸的作用下芳香族硝基化合物能还原成相应的芳香胺。
C6H5-NO2 2Fe 6HCl C6H5-NH2 2FeCl3 2H2O。
当某些盐FeCl2、FeCl3、FeSO4、CaCl2等存在时所用酸无论是过量还是少量甚至在中性溶液中都能够进行这种还原。
此方法适用于绝大部分各种不同结构的芳香族化合物有时也用来还原脂肪族硝基化合物。
4. Beckmann重排醛肟、酮肟用酸或路易斯酸处理后最终产物得酰胺类。
单酮肟重排仅得一种酰胺混酮肟重排得两种混合酰胺。
但一般质子化羟基的裂解和基团R的转移是从相反的位置同时进行的。
NOHRRRNHRONRROHRNHRO 无论酯酮肟和芳酮肟都会发生此反应。
环酮肟重排得内酰胺这在工业生产上很重要利用此反应可帮助决定异构酮肟的结构。
5. Beyer喹啉类合成法芳香伯胺与一分子醛及一分子甲基酮在浓盐酸或ZnCl2存在下反应生成喹啉类化合物。
NH2NHRRHNRRRCHORCOCH3HCl H2这是对Doebner-Miller喹啉合成法的改进。
Doebner-Miller合成法由芳胺和不饱和醛或酮反应得到喹啉衍生物。
NH2NHCH3HNCH3 H2CH3O2CH3CHO 6. Blanc氯甲基化反应芳香族化合物苯、萘、蒽、菲、联苯及衍生物在ZnCl2或NH4Cl、AlCl3、SnCl4、H2SO4、H3PO4 存在下用甲醛和极浓盐酸处理发生芳香化合物的氯甲基化反应。
有机化学人名反应

有机化学人名反应取代反应:1,加特曼反应:加特曼(GattermannL)发现:用催化量的金属铜代替氯化亚铜或溴化亚铜作催化剂,也可使重氮盐与盐酸或氢溴酸反应制得芳香氯化物或溴化物。
这样进行的反应叫做加特曼反应。
2,加特曼-科赫反应:苯、一氧化碳和氯化氢反应生成苯甲醛,此反应称为加特曼-科赫反应。
3,傅-克反应:芳香化合物芳环上的氢被烷基取代的反应称为傅-克烷基化反应;芳香化合物芳环上的氢被酰基取代的反应称为傅-克酰基化反应;统称傅-克反应。
4,布赫尔反应:萘酚在亚硫酸氢钠存在下与氨作用,转变成相应萘胺的反应称为布赫尔反应。
5,齐齐巴宾反应:吡啶与氨基钠反应,生成-氨基吡啶,如果位已被占据,则得-氨基吡啶,但产率很低。
这个反应称为齐齐巴宾(Chichibabin)反应。
6,刚穆伯—巴赫曼反应:芳香重氮盐中的芳基在碱性条件下与其它芳香族化合物偶联成联苯或联苯衍生物的反应称为刚穆伯(Gomberg)—巴赫曼(Bachmann)反应。
7,柯尔伯—施密特反应:干燥的酚钠或酚钾与二氧化碳在加温加压下生成羟基苯甲酸的反应称为柯尔伯—施密特(Kolbe-Schmitt)反应。
8,威廉森合成法:在无水条件下,醇钠和卤代烷作用生成醚的反应称为威廉森(WilliamonAW)合成法。
9,席曼反应:芳香重氮盐和氟硼酸反应,生成溶解度较小的氟硼酸盐,后者加热分解产生氟苯,这称为席曼(Schiemann)反应。
10,桑德迈耳反应:1884年,桑德迈耳(SandmeyerT)发现:在氯化亚铜或溴化亚铜的催化下,重氮盐在氢卤酸溶液中加热,重氮基可分别被氯或溴原子取代,生成芳香氯化物或溴化物。
这一反应称为桑德迈耳反应。
11,普塑尔反应:一些重氮盐在碱性条件下或稀酸的条件下可以发生分子内的偶联反应。
这个反应是普塑尔(PchorrR)在寻找合成菲环的新方法中首先发现的,故称为普塑尔反应。
12,瑞穆尔—悌曼反应:酚与氯仿在碱性溶液中加热生成邻位及对位羟基醛的反应称为瑞穆尔—悌曼(Reimer—Tiemann)反应。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
The palladium-catalyzed C-C coupling between aryl halides or vinyl halides and activated alkenes in the presence of a base is referred as the "Heckp yReaction". Recent developments in the catalysts and reaction conditions have resulted in a much broader range of donors and acceptors being amenable to the Heck Reaction.g y One of the benefits of the Heck Reaction is its outstanding trans selectivity.Mechanism of the Heak ReactionRecent Literature●Trifunctional N,N,O-terdentate amido/pyridyl carboxylate Pd(II) complexes were highly active and stable phosphine-free catalysts for Heck and room-temperature Suzuki reactions with high turnover numbers.M. L. Kantam, P. Srinivas, J. Yadav, P. R. Likhar, S. Bhargava,J. Org. Chem.,2009,74, 4882-4885.●New N-Heterocyclic Carbene Palladium Complex/Ionic Liquid Matrix Immobilized on Silica: Application as Recoverable Catalyst for the Heck Reactionpp yB. Karimi, D. Enders,Org. Lett.,2006,8, 1237-1240.●Pd(quinoline-8-carboxylate)2as a Low-Priced, Phosphine-Free Catalyst for Heck and SuzukiReactionsX. Cui, J. Li, Z.-P. Zhang, Y. Fu, L. Liu, Q.-X. Guo,J. Org. Chem.,2007,72, 9342-9345.Recent Literature●Triethanolamine as an Efficient and Reusable Base, Ligand and Reaction Medium for Phosphane-Free Palladium-Catalyzed Heck ReactionsH. J. Li, L. Wang,Eur. J. Org. Chem.,2006, 5101-5102.●Triaryl phosphine-functionalized N-heterocyclic carbene ligands for Heck reactionA.-E. Wang, J.-H. Xie, L.-X. Wang, Q.-L. Zhou,Tetrahedron,2005,61, 259-266.A E Wang J H Xie L X Wang Q L Zhou259266p g p q●Heck Couplings at Room Temperature in Nanometer Aqueous MicellesB. H. Lipshutz, B. R. Taft,Org. Lett.,2008,10, 1329-1332.Recent Literature●Efficient Aqueous-Phase Heck and Suzuki Couplings of Aryl Bromides Using Tri(4,6-dimethyl-3-sulfonatophenyl)phosphine Trisodium Salt (TXPTS)L. R. Moore, K. H. Shaughnessy,Org. Lett.,2004,6, 225-228.●Poly(ethylene glycol) (PEG) as a Reusable Solvent Medium for Organic Synthesis. Application in the Heck ReactionS. Chandrasekhar, C. Narsihmulu, S. S. Sultana, N. R. Reddy,Org. Lett.,2002,4, 4399-4401.●Functionalized Ionic Liquid as an Efficient and Recyclable Reaction Medium for Phosphine-Free Palladium-Catalyzed Heck ReactionPalladium Catalyzed Heck ReactionL. Zhou, L. Wang,Synthesis,2006, 2649-2652.Recent Literature●Brønsted Guanidine Acid-Base Ionic Liquids: Novel Reaction Media for the Palladium-Catalyzed Heck ReactionS. Li, Y. Lin, H. Xie, S. Zhang, J. Xu,Org. Lett.,2006,8, 391-394.●Pd-m BDPP-Catalyzed Regioselective Internal Arylation of Electron-Rich Olefins by Aryl Halidesy y gS. Liu, N. Berry, N. Thomson, A. Pettman, Z. Hyder, J. Mo, J. Xiao,J. Org. Chem.,2006,71, 7467-7470.●Regioselective Heck Vinylation of Electron-Rich Olefins with Vinyl Halides: Is the Neutral Pathway in Operation?in Operation?M. McConville, O. Saidi, J. Blacker, J. Xiao,J. Org. Chem.,2009,74, 2692-2698.Recent Literature●The Heck Reaction of Electron-Rich Olefins with Regiocontrol by Hydrogen-Bond DonorsJ. Mo, J. Xiao,Angew. Chem. Int. Ed.,2006,45, 4152-4157.J Mo J Xiao Angew Chem Int Ed41524157●Palladium-Tetraphosphine Complex Catalysed Heck Reaction of Vinyl Bromides with Alkenes: Aj gPowerful Access to Conjugated DienesM. Lemhadri, A. Battace, F. Berthiol, T. Zair, H. Doucet, M. Santelli,Synthesis,2008, 1142-1152.●Heck Coupling with Nonactivated Alkenyl Tosylates and Phosphates: Examples of Effective 1,2-g y()Migrations of the Alkenyl Palladium(II) IntermediatesA. L. Hansen, J.-P. Ebran, M. Ahlquist, P.-O. Norrby, T. Skydstrup,Angew. Chem. Int. Ed.,2006,45, 3349-3353.Recent Literature●Synthesis of 2-Vinylic Indoles and Derivatives via a Pd-Catalyzed Tandem CouplingReactionA. Fayol, Y.-Q. Fang, M. Lautens,Org. Lett.,2006,8, 4203-4206.●Heck Vinylations Using Vinyl Sulfide, Vinyl Sulfoxide, Vinyl Sulfone, or Vinyl Sulfonate Derivativesy y y p p pand Aryl Bromides Catalyzed by a Palladium Complex Derived from a TetraphosphineA. Battace, T. Zair, H. Doucet, M. Santelli,Synthesis,2006, 3495-3505.●Direct Acylation of Aryl Bromides with Aldehydes by Palladium CatalysisJ. Ruan, O. Saidi, J. A. Iggo, J. Xiao,J. Am. Chem. Soc.,2008,130, 10510-10511.Protective Groups & StabilitiesFmoc‐NR29‐Fluorenylmethyl carbamate, FMOC amino, FMOC amine, FMOC amideH2O:pH < 1,100°CpH = 1, RT pH = 4, RT pH = 9, RT pH = 12, RTpH > 12,100°CBases:LDA NEt3, Py t‐BuOK Others:DCC SOCl2Nucleophiles:RLi RMgX RCuLi Enolates NH3, RNH2NaOCH3 Electrophiles:RCOCl RCHO CH3I Others::CCl2Bu3SnH Reduction:H2/ Ni H2/ Rh Zn / HCl Na / NH3LiAlH4NaBH4 Oxidation:KMnO4OsO4CrO3/ Py RCOOOH I2, Br2, Cl2MnO2/CH2Cl2 T. W. Green, P. G. M. Wuts,Protective Groups in Organic Synthesis,Wiley Interscience New York503507736739Wiley-Interscience, New York,1999, 503-507, 736-739.Protective Groups & StabilitiesProtective Groups & Stabilities BOC‐NR2t‐Butyl carbamate, BOC amine, BOC amino, BOC amideH2O:pH < 1,100°CpH = 1, RT pH = 4, RT pH = 9, RT pH = 12, RTpH > 12,100°CBases:LDA NEt3, Py t‐BuOK Others:DCC SOCl2 Nucleophiles:RLi RMgX RCuLi Enolates NH3, RNH2NaOCH3Electrophiles:RCOCl RCHO CH3I Others::CCl2Bu3SnH Reduction:H2/ Ni H2/ Rh Zn / HCl Na / NH3LiAlH4NaBH4Oxidation:KMnO4OsO4CrO3/ Py RCOOOH I2, Br2, Cl2MnO2/ CH2Cl2T.W.Green,P.G.M.Wuts,Protective Groups in Organic Synthesis, T. W. Green, P. G. M. Wuts,Protective Groups in Organic Synthesis, Wiley-Interscience, New York,1999, 518-525, 736-739.Protective Groups & StabilitiesProtective Groups & Stabilities Cbz‐NR2/ Z‐NR2Benzyl carbamateH2O:pH < 1,100°CpH = 1, RT pH = 4, RT pH = 9, RT pH = 12, RTpH > 12,100°CBases:LDA NEt3, Py t‐BuOK Others:DCC SOCl2 NucleophileRLi RMgX RCuLi Enolates NH3, RNH2NaOCH3 s:Electrophiles:RCOCl RCHO CH3I Others::CCl2Bu3SnH Reduction:H2/ Ni H2/ Rh Zn / HCl Na / NH3LiAlH4NaBH4Oxidation:KMnO4OsO4CrO3/ Py RCOOOH I2, Br2, Cl2MnO2/ CH2Cl2T. W. Green, P. G. M. Wuts,Protective Groups in Organic Synthesis, Wiley-Interscience, New York,1999, 518-525, 736-739.Protective Groups & StabilitiesProtective Groups & Stabilities PhthalimideH2O:pH < 1,100°CpH = 1, RT pH = 4, RT pH = 9, RT pH = 12, RTpH > 12,100°CBases:LDA NEt3, Py t‐BuOK Others:DCC SOCl2Nucleophiles:RLi RMgX RCuLi Enolates NH3, RNH2NaOCH3Electrophiles:RCOCl RCHO CH3I Others::CCl2Bu3SnH/Reduction:H2/ Ni H2/ Rh Zn / HCl Na NH3LiAlH4NaBH4Oxidation:KMnO4OsO4CrO3/ Py RCOOOH I2, Br2, Cl2MnO2/ CH2Cl2T. W. Green, P. G. M. Wuts,Protective Groups in Organic Synthesis, Wiley-Interscience, New York,1999, 518-525, 736-739.Protective Groups & StabilitiesProtective Groups & Stabilities Bn‐NR2BenzylamineH2O:pH < 1, 100°C pH = 1, RT pH = 4, RT pH = 9, RT pH = 12, RT pH > 12, 100°CBases:LDA NEt3, Py t‐BuOK Others:DCC SOCl2Nucleophiles:RLi RMgX RCuLi Enolates NH3, RNH2NaOCH3Electrophiles:RCOCl RCHO CH3I Others::CCl2Bu3SnH Zn / HCl LiAlH NaBH Reduction:H2/Ni H2/Rh n Na/NH3iAlH4Na H4Oxidation:KMnO4OsO4CrO3/ Py RCOOOH I2,Br2, Cl2MnO2/ CH2Cl2T. W. Green, P. G. M. Wuts,Protective Groups in Organic Synthesis, Wiley-Interscience, New York,1999, 518-525, 736-739.R H'Aldol'is an abbreviation of aldehyde and alcohol.When the enolate of an aldehyde or a ketone reacts at theα‐h l f ld h d k hcarbon with the carbonyl of another molecule under basicor acidic conditions to obtainβ‐hydroxy aldehyde or ketone,ketonethis reaction is called Aldol Reaction.Mechanism of the Aldol AdditionMechanism of the Aldol AdditionMechanism of the Aldol AdditionUnder conditions of kinetic control,the mixed Aldol Addition can be used to prepare adducts that are otherwise difficult to obtain selectively.This process begins with the irreversible generation of the kinetic enolate,e.g.by employing a sterically hindered lithium amide base such as LDA(lithium diisopropylamide). With an unsymmetrically substituted ketone,such a non‐nucleophilic,t i ll b tit t d k t h l hili sterically‐demanding,strong base will abstract a proton from the least hindered side.Proton transfer is avoided with lithium enolates at low temperatures in ethereal solvents,so that addition of a second carbonylsolventspartner(ketone or aldehyde)will produce the desired aldol product.Recent Literature●Highly Enantioselective Organocatalytic Direct Aldol Reaction in an Aqueous MediumV. Maya, M. Raj, V. K. Singh,Org. Lett.,2007,9, 2593-2595.V Maya M Raj V K Singh Org Lett25932595●Chiral Amine-Polyoxometalate Hybrids as Highly Efficient and Recoverable Asymmetric Enamine Catalysts. S. Luo, J. Li, H. Xu, L. Zhang, J.-P. Cheng,Org. Lett.,2007,9, 3675-3678.Catalysts S Luo J Li H Xu L Zhang J P Cheng Org Lett36753678●A Highly Efficient Organocatalyst for Direct Aldol Reactions of Ketones with AldedydesZ. Tang, Z.-H. Yang, X.-H. Chen, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang, L.-Z. Gong,J. Am. Chem.Z Tang Z H Yang X H Chen L F Cun A Q Mi Y Z Jiang L Z Gong J Am ChemSoc.,2005,127, 9285-9289.Recent Literature●The First Direct and Enantioselective Cross-Aldol Reaction of AldehydesA. B. Northtrup, D. W. C. MacMillan,J. Am. Chem. Soc.,2002,124, 6798-6799.●Enantioselective organocatalytic aldehyde–aldehyde cross-aldol couplings. The broad utility of α-thioacetal aldehydes。