GaussView基础教程
高斯view基础
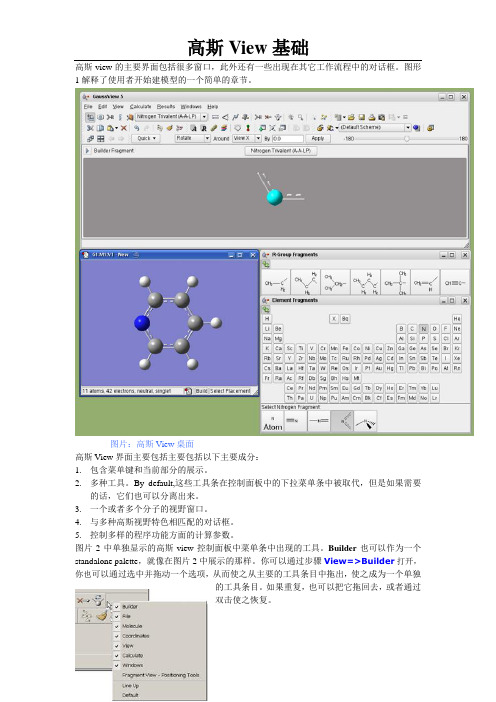
高斯view的主要界面包括很多窗口,此外还有一些出现在其它工作流程中的对话框。
图形1解释了使用者开始建模型的一个简单的章节。
图片:高斯View桌面高斯View界面主要包括主要包括以下主要成分:1.包含菜单键和当前部分的展示。
2.多种工具。
By default,这些工具条在控制面板中的下拉菜单条中被取代,但是如果需要的话,它们也可以分离出来。
3.一个或者多个分子的视野窗口。
4.与多种高斯视野特色相匹配的对话框。
5.控制多样的程序功能方面的计算参数。
图片2中单独显示的高斯view控制面板中菜单条中出现的工具。
Builder也可以作为一个standalone palette,就像在图片2中展示的那样。
你可以通过步骤View=>Builder打开,你也可以通过选中并拖动一个选项,从而使之从主要的工具条目中拖出,使之成为一个单独的工具条目。
如果重复,也可以把它拖回去,或者通过双击使之恢复。
如果鼠标右击任何工具条中的灰色区域,你将看到工具条中的下拉菜单。
列表的第一部分包括在窗口中可利用的工具条的名称。
从列表中选择一个工具条名称能够在它的可视或者隐藏的窗口中toggle它。
这个Line Up选项能够使工具条转移到它们之间排除空缺的空间处,以及Default选项可以在合适的时候重建工具条中default的配置。
Figure 3. GaussView Toolbars in Standalone ModeMolecules, Molecule Groups and Views 高斯View这些构图来组建分子。
1.一个分子在使用时通常是一个独立的分子,与打开的分子隔离。
通常而言,分子与磁盘总初春的文件一致。
但也有的时候不一致,如涉及模型的时候。
注意:这个概念上的分子可能包括不止一中的化学种类。
2.如果同样的分子不只有一个窗口打开,那么它们将以不同的方式展示于窗口。
而且,一个View窗口只能展示一个分子,不同的View和不同的窗口可以展示相同分子不同的方向和不同的性能。
GaussView教程

所需软件:GaussView
步骤:
1.从GaussView打开目标体系的。
chk文件,如下图
图1
2逐步点选Results; surface,出现如下图对话框
图2
3。
其中Cube Actions…一项,选择New Cub,跳出下图对话框,kind一栏选择Total Density,然后点击OK
图3
4。
此时返回图2,稍等待空白栏中出现计算结果.Surface Actions…一项选择New Mapped Surface,跳出下图对话框,点击OK
图4
5.稍等,便出现下图漂亮彩色静电势图
图5
怎么样,简简单单就作出了漂亮的静电势图^—^
可能你作出来的和我的不一样,这是因为透明度不同,显示的效果不一样。
编辑显示效果,依次请点击File, Preferences,如下图示:点击Surface选项卡,Transparent就是用来调节透明度的。
图6
编辑制作:赵继阳,参考GaussView《help》。
高斯软件-基础教程
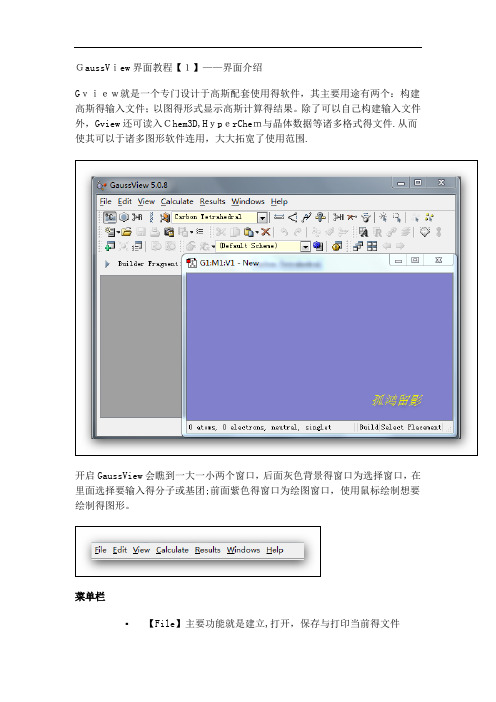
GaussView界面教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件.从而使其可以于诸多图形软件连用,大大拓宽了使用范围.开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
GaussView基础教程
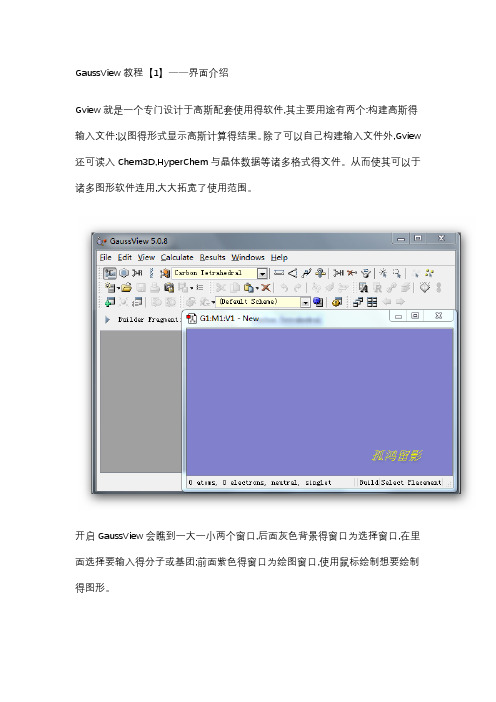
GaussView教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview 还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
GaussView-画图技巧

• 一般可以用迪卡尔坐标,也可以用Z-矩阵(ZMatrix)
• 二,Gaussian 计算实例(进行几何优化,在起始 结构的附近寻找具有最低的能量的结构.)
• 计算优化乙烯结构:
• 第一步,写好高斯输入文件:
• 1、(画出乙烯分子结构并保存文件)
• 2、点击菜单上的Calculate,选中Gaussian..,打 开高斯计算设置窗口。
• 3、设定工作类别optimization,
• 4、设定计算方法,Ground State 表示计算基态 ,DFT…表示用密度泛函理论,Unrestricted表示 用非限制性的,即分别计算α电子和β电子; B3LYP 表示用的函数为B3LYP 函数。下一行为 计算所用基组设置,本计算采用的基组为6-31G ,后面的是加极化函数和弥散函数,我们这儿暂 时不考虑。然后再一下行第一个Charge 表示计算 分子所带电荷量,这里设为0,表示是中性分子; 然后是自旋多重度spin,singlet 表示自旋多重度 为1,分子中没有单电子。
尔坐标轴的定向将按照分子的对称轴进行选取,
如下图所示。
•
如果再选中下方的“Always track point
group symmetry”选项,则后面当我们利用变形工
具改变某个几何参数(键长或键角或二面角)时
或者删除工具删除某个原子时,分子将按照对称
性的要求自动改变其他对称相关的几何参数(键
长或键角或二面角)或删除其他对称相关的原子
• 后面添加其他基团做准备。用基团工具点击氢原 子将氢原子取代掉后即可达到在一定的成键方向 添加某个基团的目的。
• 例2.1:构造出环戊二烯,用删除工具删除 CH2的两个氢原子。再用加氢工具加一个氢原子 即可得到正确取向的氢。
三章:GaussView使用

第三章:GaussView使用:(3.1)用高斯进行几何优化后,在GaussView中发现断了根键(或者成键方式变化了),这是什么原因啊?(3.2)想固定某部分原子(键长、键角、二面角、相对位置、相对方向。
)怎么做?(3.3)GaussView怎么打不开输出文件?后续问题:我明明放在桌面上了啊,怎么会有中文路径呢?后续问题:不好意思,楼上说的我还是不太明白。
请问什么是路径啊?把对话框中的Library 改成你的GV 的安装位置比如,我的安装位置D:\ProgramFiles\gview4,那我就把CostomFragment中的Library改成“D:\ProgramFiles\gview4”(3.4)原子可以改变编号吗?怎么改?Quote:原子当然可以改变编号了,这在寻找过渡态的时候有大用处。
在GV 工具栏上有个A的按钮,【图10】打开它可以改变原子编号,还可以插入虚原子,改变原子坐标等等。
注意改变原子编号的时候要按顺序,比如从小到大,不要乱改。
论坛附件上传下载教程阅尽铅华,不再吱声回复引用报告道具 TOPyjcmwgk版主帖子179 精华3 积分57 威望5楼发表于 2010-2-18 11:17 | 只看该作者踩窝窝送礼物问候Ta第四章:其它问题(4.1)要查某个杂志属于哪个数据库,怎么查呢?后续问题:不能下载啊,听说是权限问题,怎么解决呢?Quote:对于大部分的大学和科研单位,由于资本有限,不可能购买很多数据库。
只能利用别人帮忙下载一些期刊文献,但总18 点金币331 枚贡献0 点讨论指数21 点麻烦朋友总不太好。
熟话说,求人不如求己。
下面我把我怎么获得免费文献的经验告诉大家,希望有所帮助。
首先我们要知道我们要查的期刊文献属于哪个数据库。
至于哪些文献属于哪个数据库,例如化学期刊所属数据库列表:/blog/user _content.aspx?id=10282接下来我们就要知道各数据库的地址,我在这里列一些比较大的数据库:SD: WILEY:http://www3.interscience.wil /browse/?type=JOURNAL ACS:/about.html RSC:/Publishing/Journals/ SPRINGERLINK:http://www.springerli/journals/ THIEME:http://www.thieme.de/connec t/en/product-type/journals.html等等......知道这些数据库,我们就可以搜索到具体文献的下载地址,当然绝大部分文献下载要权限.你采用下面方式快速的获取你所需要的文献。
GaussView高斯软件教程

(4).双击Gview界面上的 图标。出现以下窗口点击氟的元素符号“F”,就选中氟原子。
-
(5).回到工作窗口在苯环上的任一个H上单击左键,将H置换成F。
但要注意此时仅是元素符号发生了改变,C-F间的距离仍是C-H键的距离。需要用建 长工具进行调整。单击Gview界面上 图标。 然后再点击工作窗口中的C原子和F原 子,看到被选中的两个原子于周围的原子再亮度上有差异此时会出现下面的窗口
-
(2).双击窗口中 图标,得到如下窗口里面有常用的环状官能团。选中苯环(单 击即可选中)
-
(3).在当前工作窗口(打开Gview时程序自动打开一个工作窗口,如下图)也可通过 File-new 路径 新建一个工作窗口
-
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上, 前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子 当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下 命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离 可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。 Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调 节分子的角度]这个功能连用。
-
根据C-F键的长度在 0.675和2.700之间的方框内进行C-F键的调整。完毕后点击OK即可。
-
(6).双击Gview界面上的 图标,出现以下窗口这是Gview里内置的链烃库,选中乙 烷
-
GaussView讲解

Z表示时内坐标,C表示直角坐标。可以在里面对坐标做适当的调整
(12).向Gauss递交计算。点Gview界面上Calculation 会出来一个递交计算的对话框。从所给的对话框中可以选择工作类型Job Type(如优化, 能量或频率等);计算方法Method(如半经验方法,HF方法,DFT方法,MP方法等, 还可以选定基组);Title(对所要做的计算给一个说明,以备以后的查看) Link 0(给检查 点文件命名,还可以在此用RWF命令设置临时数据交换文件的大小); General, Guess,(这两个选项主要是给出体系中各原子的连接关系及如何给出初始猜测); NBO(可在此设定NBO计算),PBC(可在此设定晶体的有关计算), Solvation(可在 此设定溶液中的计算,除了选择溶剂外,还要选择模拟溶剂的理论模型)的Gaussian
错误的判断。
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
gassianview使用技巧

gassianview使用技巧GaussView是一款常用的计算化学软件,可以用于分子结构的绘制、气体分子动力学模拟、振动频谱计算等。
下面是几个使用GaussView的技巧:1. 分子结构绘制:GaussView提供了用户友好的界面,可以通过“Build”工具绘制分子结构。
可以选择不同的元素、键类型以及连接方式来构建分子的结构。
在绘制分子时,可以使用鼠标拖动和旋转分子来获得最佳的显示视角。
2. 动画和振动频谱:GaussView可以用于分子的动力学模拟,并可生成动画。
可以通过“Job Control”面板设置分子的初始状态、步长和模拟的时间等参数,然后点击“Run”按钮开始模拟。
模拟完成后,可以通过“Results”面板查看动画和振动频谱。
3. 能量优化:GaussView可以进行能量优化计算,从而确定分子的最稳定构型。
可以通过“Job Control”面板选择能量优化计算类型和优化方法,然后点击“Run”按钮开始计算。
计算完成后,可以在“Results”面板中查看优化后的分子结构和相关能量信息。
4. 离子与溶剂模型:GaussView支持模拟溶剂中的分子,可以选择不同的离子和溶剂模型,并通过“Job Control”面板设置溶剂分子的初始位置和计算参数。
可以将溶剂中的分子视为盒状结构,通过手动添加溶剂分子或者选择预定义的盒状结构。
5. 输入文件生成:GaussView可以自动生成Gaussian计算所需的输入文件(通常为`.com`或`.gjf`格式),可以通过“File”菜单下的“Export Gaussian Input”选项来生成输入文件。
可以通过该选项设置计算所需的方法、基组、计算类型等参数,并指定输入文件的保存路径。
这些是使用GaussView的一些常见技巧,希望能对你有帮助!。
GaussView高斯软件教程
根据C-F键的长度在 0.675和2.700之间的方框内进行C-F键的调整。完毕后点击OK即可。
-
(6).双击Gview界面上的 图标,出现以下窗口这是Gview里内置的链烃库,选中乙 烷
-
(7).在工作窗口内空处点左键,用我们前面讲过的命令调节苯和乙烷间的距离和角度
-
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度;将鼠标放在分 子上,前后移动,可以将分子放大或缩小。Shift+Alt+鼠标左键组合可以在窗口内平 移分子当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可 用以下命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间 的距离。可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子 间的角度。Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移 动,可以调节分子的角度]这个功能连用。具体的好处需要自己去试
-
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
-
至此分子构建已完成。
-
9).Gview存储个人常用分子的功能。Gview中只有常用的一些环状分子和链状分子, 远不能满足研究特定体系的特殊需要。不过Gview有一个功能可以弥补这个缺憾:可 以把常用的分子或官能团存在制定的文件夹内。在需要时可以直接调用。双击view上 的 图标,在下面的对话框中键入相关项目,保存即可
-
(2).双击窗口中 图标,得到如下窗口里面有常用的环状官能团。选中苯环(单 击即可选中)
gaussview使用教程1

• 元素板块
• 在周期表中选择元素种类,以及原子的成键 方式
• 端基板块提供了一些列已经过AM1方法优化的端 基碎片,默认端基为羰基。
• 环形板块提 供了一系列 AM1方方优 化后的环形
结构,默认 环为苯。
• 生物分子下拉菜单可选择所需的是氨基酸或核苷。
• 当选择氨基酸时,左边下拉菜单可选择氨基酸种 类,右边下拉菜单选择分子片断作为中间部分、 氨基端或酰基端。这样就制定了氨基酸的种类。
• Always track point group symmetry:当结构变化时,不断计 算分子所属点群,此选项与对称性限制为None联合使用,当 改变键长的结构参数时,可以看到对称性的变化,通常来说, 只有参数的改变完成后,对称性才会发生改变,对一些大的 体系,此选项可能导致反应时间明显变长。
Bond SmartSlide对话框也可以在没有化学键相连的原子间添 加化学键。当对话框打开时,可通过单击选择所需要的化学 键类型。同样也可以取消原子间的化学键。
• Displacement选项指认当键长改变时,原子所连接的基团的 变化。
• Translate Atom:只移动原子,所连接基团的位置不变。 • Translate Group:原子所连接基团的位置随原子改变(即看
• Rebonding Structures • Rebonding按钮和Edit-Rebond选项:以间距算法为
基础确认原子间距离,Gaussian不用屏幕所显示的 键长信息来计算,这些信息是为了便于我们观看。
Imposing Symmetry对话框
• Imposing Symmetry
• Enable point group symmetry:使Gaussview图形具有对称 性。
GaussView讲解
Gview是一个专门设计于高斯配套使用的软件,其 主要用途有两个构建高斯的输入文件以图的形式显示 高斯计算的结果除了可以自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式的文件。从而使其可以于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
(2).双击窗口中 击即可选中)
图标,得到如下窗口里面有常用的环状官能团。选中苯环(单
(3).在当前工作窗口(打开Gview时程序自动打开一个工作窗口,如下图)也可通过 File-new 路径 新建一个工作窗口
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上, 前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子 当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下 命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离 可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。 Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调 节分子的角度]这个功能连用。
Result :显示计算的结果,包括电荷,静电势表示的表面,振动,频率,核磁,势能面 扫描,优化等。注意有些结果只能 用检查点文件才能显示。
常用功能键的介绍
以构建一个间氟苯乙烷分子并从 Gview里递交计算为例来说明
(1)双击Gview图标, 打开Gview ,下图就是Gview打开后的窗口 打开Gview ,下图就是Gview打开后的窗口
Gaussview软件使用手册

Gaussview软件使用手册GaussView是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView是不在高斯软件的计算模块中,而是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译和放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其他的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1.经过优化的分子结构;2.分子轨道3.任何估算密度的电子密度表面;4.静电潜在表面;5.原子价;6.与振动频率相符的一般模型;7.IR,Raman,NMR,VCD以及其他的频谱;8.结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP和BOMD轨道的模拟;9.整体能量的小部分以及其他来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目是按照逻辑结构来排列的,开始于最简单的项目,然后是引用最广泛的项目,最后是更加复杂和不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。
GaussView高斯软件教程
Gview是一个专门设计于高斯配套使用的软件,其 主要用途有两个构建高斯的输入文件以图的形式显示 高斯计算的结果除了可以自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式的文件。从而使其可以于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
-
(10).查看分子的对称性 从Edit-Point group路径可以查看所构建分子的点群。点击Point group后,出现如下 窗口:为C1点群,其下拉菜单中的为可能的点群(改变Tolerance,也可帮助我们判断 所构建体系可能有的点群)
-
(11).查看分子坐标。单击Gview界面上的 图标。出现下面窗口
在构建分子前,应对分子的键长,键角和点群等有一个详细的了解。这样在构建过程 中才能更好的把握细节,如构建大分子时,各个集团间的键长,键角和二面角一定要 给的准确。相当一部分的错误(如SCF不收敛,1502报错等)都是分子建模不合理 造成的。分子模型不合理导致程序根据分子模型所得的薛定鄂方程不合理,也就导致 了对方程的解不能顺利完成,所以报错。对于大分子,不会出现分子直线排步的情况, 链烃通常为锯齿型的排列,环之间总是有一定的夹角,共面情况虽有但不多见,这些 都是在构建分子模型的过程中需要注意的。再者,分子建模是量化计算的一个难点, 特别是对金属配合物,块状体系和纳米体系的构建。只有给出合理的结构,程序才能 给出合理的解。如果不能体现出分子的对称性,则程序不能正确判断分子轨道的对称 性,这在涉计到反应机理和轨道相互作用的研究中可能会导致错误的判断。
-
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
gaussview使用简介PPT课件

2021/6/4
16
(3).在当前工作窗口(打开Gview时程序自动打开一个工作窗口,如下图)也可通过 File-new 路径 新建一个工作窗口
2021/6/4
17
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上, 前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子
2021/6/4
24
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
2021/6/4
25
至此分子构建已完成。
2021/6/4
26
9).Gview存储个人常用分子的功能。Gview中只有常用的一些环状分子和链状分子, 远不能满足研究特定体系的特殊需要。不过Gview有一个功能可以弥补这个缺憾:可 以把常用的分子或官能团存在制定的文件夹内。在需要时可以直接调用。双击view上 的 图标,在下面的对话框中键入相关项目,保存即可
2021/6/4
7
Result :显示计算的结果,包括电荷,静电势表示的表面,振动,频率,核磁,势能面 扫描,优化等。注意有些结果只能 用检查点文件才能显示。
2021/6/4
8
常用功能键的介绍
2021/6/4
9
2021/6/4
10
2021/6/4
11
2021/6/4
12
2021/6/4
13
GaussView使用简介
Gview是一个专门设计于高斯配套使用的软件,其 主要用途有两个构建高斯的输入文件以图的形式显示 高斯计算的结果除了可以自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式的文件。从而使其可以于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
Gauss画图技巧

4. 显示坐标轴
点击右键菜单可以选择显示笛卡尔坐标 轴。显示坐标轴可以更准确地添加原子或虚 原子。 例子4.1:接上例,添加一个笛卡尔坐标 轴
5. 添加虚原子(构建晶体结构)
点击上面图标“Atom List Editor”,点击 symbol选择一个任意重复的原子为虚原子。
6. 删除虚原子
点击上图,在弹出的“Atom List Editor”窗 口中选中要删除的原子,然后选择菜单 Edit >Delete Atoms > Selected。
例6.1:接上例添加虚原子
7. 利用晶体工具在一定方向复制分子
晶体工具也可以通过选择菜单Edit > PBC...调 出。晶体工具除了构造一维至三维晶体结构外, 还可以实现分子的平移复制。方法是:在弹出的 PBC 对话框中选择Symmetry选项卡中的“Lattice Dimension”中的“Three”,即构造三维晶体结构 (一般来说三个晶轴的方向中的一个是沿着分子 主轴的方向)。我们可以通过调节Alpha 至 Gamma 角来得到我们想要的一个晶轴取向,然后
8、点击Edit,提示保存作业,如下图所示:
9、出现保存页面,输入文件名ethylene.gjf, 点击Save 保存,
10、出现优化后的键长,键角,二面角 的优化结果。
制作一维聚乙炔与三维二 茂铁
1.2 化学模型(Model Chemistries)
Gaussian包含多种化学模型,比如 计算方 法 Gaussian关键词 方法 HF Hartree-Fock 自恰场模型 B3LYP Becke型3参数密度泛函 模型,采用Lee-Yang-Parr泛函 MP2 二级 Moller-Plesset微扰理论 MP4 四级MollerPlesset微扰理论 QCISD(T) 二次CI
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
GaussView教程【1】——界面介绍
Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview 还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏
▪【File】主要功能就是建立,打开,保存与打印当前得文件
▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等
▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等
▪【Calculate】直接向Gaussian提交计算
▪【Results】接收并显示Gaussian计算后得结果
▪【Windows】控制窗体,如关闭、恢复等
▪【Help】帮助
快速工具栏
【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;
【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等
【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸
【左面第五个】用户自定义基团,您可以将常用得基团存放到此处
这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】
这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏
这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子
这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
1、开启Gview
2、双击按钮,选择苯基(第一个),会瞧到主程序框体中出现苯环
在工作窗口单击,可以瞧到工作窗口也出现了一个苯环。
单击左键,可以将主程序框体中得分子或基团加入到工作窗口;
按上下左右键,或者左键单击不放移动鼠标,可以调节分子得角度;
滚动鼠标滚轮,可以放大缩小分子;
按住shift键,左键单击不放移动鼠标,可以移动分子;
当工作窗口内有多个分子时(在构建大得分子时,这种情况很容易出现)
用Shift+Alt+鼠标左键组合,移动想要移动得分子;
用Ctrl+Alt+鼠标左键组合调节其中一个分子得角度;
双击图标(或者左键不放),在元素周期表中选择【F】元素,回到工作窗口在苯得任意一个【H】上单击,使之变成【F】
双击图标,从链烃库中选择【乙基】(第一个),然后点击间位上得【H】即可
至此,分子式已经构建完成,Gview存储个人常用分子得功能,双击view上得图标,在下面得对话框中键入相关项目,保存即可。
(注:我得程序此项功能不行,用户自定义基团)
查瞧分子坐标:单击Gview界面得图标,查瞧分子得坐标。
至此,我们保存一下我们构建好得间氟苯乙烷分子,在绘图界面右键单击【File】——【Save】然后选择保存路径即可。
GaussView教程【3】——向Gaussian提交计算
上一讲我们已经构建好了一个间氟苯乙烷分子,现在打开间氟苯乙烷分子。
点击Gview界面上得【Calculate】——【Gaussian Calculate Setup】
从对话框中我们可以选择许多参数,下面依次选择一下
【Job Type】工作类型:
【Energy能量】|【Optimization优化】|【Frequency频率】|【Opt+Freq 优化+频率】|【Scan扫描】|【Stability稳定性】|【NMR核磁】,
这里我们选择【NMR】核磁
【Method】方法:每种计算模式都提供了若干种方法,这里选择默认值即可【Title】题目:这项可以根据自己得项目自行命名
【Link 0】给检查点文件命名
其它选择默认即可
选好后点击【Submit】提交至Gaussian,并保存。
(最好存到安装文件所在磁盘)
系统会询问就是否提交至Gaussian,选择【OK】,Gaussian会自动开启并计算,计算时间因硬件配置而异
计算完毕后系统会提示关闭Gaussian,点击【就是】
计算完毕后会生成两个文件,、log得就是系统日志,便于查瞧计算结果。
这里我们选择、chk文件【Ok】
接下来我们又一次瞧到了刚才绘制得那个分子,表面上没什么不同,但这次我们可以预言它得许多性质了,在GView得主界面点击【Results】——【Summary】可以瞧到刚才得计算总结,点击【View File】可以瞧到日志文件。
因为我们刚才选择计算模式就是【NMR】核磁,所以我们点击【Results】——【NMR】,随即我们瞧到了很熟悉得核磁图谱,但似乎很乱,因为这就是所有元素得核磁谱图,在左下方【Element】右方选择【H】,即H谱,旁边【Reference】选择【TMS HF/6-31G(d)】,这次我们就可以瞧到间氟苯乙烷分子理论计算得核磁谱图了。
在绘图界面右击【View】——【Labels】可以瞧到每个H原子对应场中得位置。