世界上最完善最详细的神经元原代培养完全黄金版资料
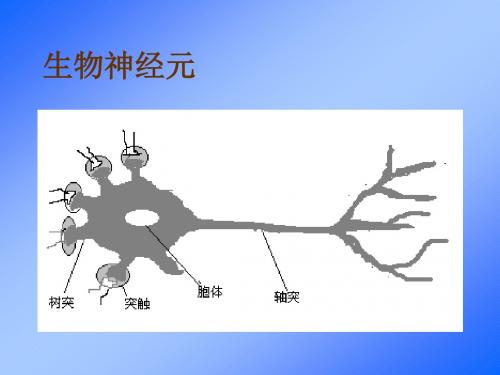
生物神经元资料
人工神经元的网络输入
1、设n个输入分别为x1 , x2 , … , xn,对应的权分别为 w1, w2, …, wn,即有输入向量和权向量:
X=(x1 , x2 , … , xn)
W=(w1, w2, …, wn) 神经元网络输入: net=xiwi net=XW
xx2 1w2w1 xnwn
net=XW
目前还很难确定它们与问题类型和规模的关系,
隐藏层数及其神经元个数的增加不一定能够提高网络 精度和表达能力。BP网一般都选二级网络。
BP网络训练过程
1、样本集 (输入向量,理想输出向量)--实际系统采集 2、向前传播阶段
(1) 从样本集中取一个样本(Xp,Yp),将Xp输入网络; (2) 计算相应的实际输出Op; 3、向后传播阶段 (1) 计算实际输出Op与理想输出Yp的差; (2) 根据这个误差,按极小化误差方式调整权矩阵;
生物神经网络工作机理1
• 在突触的接受侧,信号被送入胞体,在胞体内 进行综合,有的信息起刺激作用,有的起抑制 作用,当胞体中接受的累加刺激超过一个阈值 时,胞体被激发,此时它沿轴突通过树突向其 它神经元发出信号。
生物神经网络工作机理2
• 一个神经元沿轴突通过树突发出的信号是相同 的,而这个信号可能对接受它的不同神经元有 不同的效果,这一效果主要有突触决定:突触 的“联结强度”越大,接受的信号就越强,反 之,就越弱。
构造人工神经元的要求
神经元是构成神经网络的最基本单元 要构造神经网络必须先构造人工神经元 人工神经元应是简单实现的数学模型 人工神经元应该能模拟生物神经元的工作机理
人工神经元的构造方法
对于每个神经元,它可以接受来自系统其他 神经元的输入信号
每个输入信号对应一个权,相当于突触的“联结强度”
神经元_精品文档

神经元神经元是神经系统中最基本的功能单位。
作为神经系统的基本结构单元,神经元在神经信号的传递和处理中发挥着重要的作用。
本文将介绍神经元的结构和功能,并探讨其在神经系统中的作用。
一、神经元的结构神经元由多个组成部分构成,包括细胞体(soma)、树突(dendrites)、轴突(axon)和终末突触(synaptic terminals)。
细胞体是神经元的主体部分,其中包含细胞核和细胞质。
树突是从细胞体伸出的分支,用于接收来自其他神经元的输入信号。
轴突是神经元中的长突起,负责将神经信号传递到其他神经元。
终末突触位于轴突的末端,与其他神经元的树突或细胞体相连接,通过化学或电信号将信息传递给下一个神经元。
二、神经元的功能神经元通过电化学信号传递信息,从而实现神经系统中的信息处理和传递。
当神经元受到足够的刺激时,会产生电势差,即动作电位。
动作电位沿着神经元的轴突传递,通过终末突触的释放神经递质传递给下一个神经元或肌肉细胞。
这种信号传递的方式被称为突触传递。
神经元之间的连接形成了复杂的神经网络,这些网络在神经系统中起着关键的作用。
神经元的树突接收来自其他神经元的输入信号,并将这些信号集成起来。
通过集成和处理各种输入信号,神经元可以产生适当的输出信号作出反应。
这使得神经系统能够有效地感知外界环境和调控机体内部的生理过程。
三、神经元的类型根据功能和结构的不同,神经元可以分为多种类型。
最常见的神经元类型是感觉神经元、运动神经元和中间神经元。
感觉神经元负责接收和传递感觉刺激的信息,将其传递到中枢神经系统。
这些神经元分布在身体的各个感官器官中,如皮肤、眼睛和耳朵等。
运动神经元负责控制机体的运动活动。
它们接收中枢神经系统发出的指令,将其传递给肌肉细胞以产生相应的运动。
中间神经元是位于中枢神经系统中的神经元。
它们在大脑和脊髓中起着连接不同神经元之间的桥梁作用。
中间神经元参与了许多复杂的信息处理和调控过程。
四、神经元在神经系统中的作用神经元是神经系统中信息传递和处理的基本单元。
原代神经元培养

神经细胞原代培养从动物(大鼠或小鼠等)的胚胎或新生动物的脑组织取下某一局部区域,分离细胞,培养在培养容器后不再移植,常称为原代神经细胞培养。
一、培养前准备1. 器械和器皿器械:外科剪、镊子、虹膜剪等、小剪刀、细镊子和虹膜小刀等各种金属器械如解剖器械,使用后及时刷洗干净,用酒精棉球擦拭后晾干,或经60℃烘干,以防生锈。
由于湿热消毒对手术器械容易可用70-80%酒精浸泡1小时以上消毒。
器皿:1)玻璃瓶及相应的胶塞或胶木螺旋盖,2)用于分装血清、多聚赖氨酸、解剖液、各种盐溶液和培养液等。
3)培养瓶、盖玻片4)培养用的盖玻片必须不含铅、不发霉,可用0.2N盐酸浸泡10分钟,用蒸馏水洗2次,每次10分钟,然后移入丙酮或乙醇浸泡10分钟,再用双蒸水洗2次,每次10分钟,烘干待消毒。
凡组织学用过的旧盖玻片一般不用。
5)移液管、吸管、烧杯、离心管和培养皿。
6)塑料培养皿(35mm)、塑料培养板(6、24、96孔)。
辅助工具:放置试管、吸管和玻璃瓶等的架子。
2. 培养基和培养用液的配制1) 培养基质:有多聚赖氨酸、牛皮胶原和鼠尾胶原。
本室目前常用分子量为7-140000多聚赖氨酸(Poly-L-lysine,浓度为0.1mg/ml。
2) 平衡盐溶液:主要以无机盐和葡萄糖配制而成。
各种平衡盐溶液主要的不同点在于NaCl的浓度、离子浓度及缓冲系统的不同,可根据实际需要选用适当的平衡盐溶液。
3) 培养液:目前已有现成的干粉出售,按说明书进行配制即可。
神经细胞培养需高糖,培养液应含有或加葡萄糖至6g/L。
目前培养海马神经元可选用B27无血清培养液。
4) 血清:有胎牛血清、小牛血清和马血清等。
分装成小瓶,4℃保存备用(分装前需56℃灭活30分钟)。
5) 胰蛋白酶溶液:用于分离细胞。
先用少量灭菌三蒸水将胰蛋白酶粉末溶成糊状,然后补水至2.5%浓度,振荡摇匀,4℃过夜,待完全溶解后用滤器过滤灭菌,并分装成1-2ml冻存。
用前以D-Hanks平衡盐水或其他无钙镁离子溶液溶解稀释至0.25%或0.125%。
神经元原代培养实验报告

一、实验背景神经元是神经系统中最基本的结构和功能单位,研究神经元生物学特性对于理解神经系统疾病和开发新的治疗方法具有重要意义。
神经元原代培养是研究神经元生物学特性的重要方法之一,它能够模拟体内神经元的生长和发育过程,为神经元生物学研究提供了一种有效的手段。
二、实验目的1. 掌握神经元原代培养的方法和步骤。
2. 观察神经元原代培养过程中的形态学变化。
3. 分析神经元原代培养过程中细胞生长、增殖和分化情况。
三、实验材料1. 实验动物:E17-18d孕大鼠或E14-16d孕小鼠。
2. 试剂:含10%胎牛血清的DMEM培养基、神经元维持培养基(NeurobasalB27谷氨酰胺)、胰蛋白酶、抗神经丝抗体、荧光染料等。
3. 仪器:细胞培养箱、倒置显微镜、离心机、移液器等。
四、实验方法1. 取E17-18d孕大鼠或E14-16d孕小鼠大脑皮质,用胰蛋白酶消化,制成细胞悬液。
2. 将细胞悬液接种于培养皿中,置于37℃、5%CO2的培养箱中培养。
3. 每天观察细胞生长情况,记录细胞形态学变化。
4. 培养第3天,进行细胞计数,观察细胞生长速率。
5. 培养第5天,进行免疫荧光染色,观察神经元纯度。
6. 培养第10天,进行神经元分化实验,观察神经元功能。
五、实验结果1. 细胞形态学观察:神经元原代培养过程中,细胞从圆形逐渐转变为梭形,并逐渐出现突起,形态逐渐成熟。
2. 细胞生长速率:培养第3天,细胞生长速率为(2-4)×10^4个/皿;培养第5天,细胞生长速率为(4-8)×10^4个/皿。
3. 神经元纯度:免疫荧光染色结果显示,神经元纯度达到90%以上。
4. 神经元分化实验:神经元分化实验结果显示,神经元在培养过程中逐渐分化为不同类型的神经元,如运动神经元、感觉神经元等。
六、实验讨论1. 神经元原代培养过程中,细胞形态学变化与体内神经元生长和发育过程相似,为研究神经元生物学特性提供了有力手段。
2. 细胞生长速率受多种因素影响,如培养基、温度、CO2浓度等。
神经元原代培养_

神经元原代培养_
神经元原代培养
从孕17-18天的雌鼠的胎儿分离神经元细胞。
孕雌鼠麻醉然后解剖,胎儿收集到HBSS-1中然后快速断头。
剥离脑膜和白质后,大脑皮质收集入HBSS-2 液中机械磨碎。
皮质碎片移到有0.025%胰酶的H BSS-2液中37°C消化15分钟。
胰酶消化后,细胞用含有10%胎牛血清的HBSS-2液冲洗两次,用神经基础培养液重悬细胞(培养液添加了0.5mM左旋-谷氨酰胺,25μM左旋-谷氨酸,2%B27和0.12mg/ mL庆大霉素)。
以1×105cell/cm2接种到事先用多聚赖氨酸包被的培养皿上,放到37°C,5%CO2湿温培养箱里进行培养。
每3天用吸管换液,一次换0.5mL。
体外培养8天细胞就能用于实验。
选用17-18天的胎鼠能够提高神经元培养的效率,因为与乳鼠相比胚胎组织的细胞连接还很少。
因此,用乳鼠会使神经元的分离更加困难,会造成细胞连接不可逆的损伤,细胞之间的共同的轴突和树突更容易发生损伤。
另外,用出生后1天内的大鼠皮质培养容易有胶质细胞污染,而用胎鼠则可以避免这种情况。
如果用的是乳鼠,一般是在培养36个小时后加入阿糖胞苷,抑制胶质细胞生长。
另外,如果把分化的影响看成是考虑的重要因素,可以用培养4天的、8天的、18天的细胞。
其中一个例子可以是观察毒性物质对小孩和成年的不同效应。
原代神经元培养培训课件

精品文档神经细胞原代培养的胚胎或新生动物的脑组织取下某一局部区域,分离细胞,培养在从动物(大鼠或小鼠等)培养容器后不再移植,常称为原代神经细胞培养。
一、培养前准备1. 器械和器皿器械:外科剪、镊子、虹膜剪等、小剪刀、细镊子和虹膜小刀等℃烘干,各种金属器械如解剖器械,使用后及时刷洗干净,60用酒精棉球擦拭后晾干,或经以防生锈。
由于湿热消毒对手术器械容易可用70-小时以上消毒。
80%酒精浸泡1 器皿:1) 2) 3)玻璃瓶及相应的胶塞或胶木螺旋盖,用于分装血清、多聚赖氨酸、解剖液、各种盐溶液和培养液等。
5)培养瓶、盖玻片次,盐酸浸泡可用0.2N10分钟,用蒸馏水洗2培养用的盖玻片必须不含铅、不发霉,分钟,10分钟,再用双蒸水洗2次,每次1010每次分钟,然后移入丙酮或乙醇浸泡烘干待消毒。
凡组织学用过的旧盖玻片一般不用。
6)7)移液管、吸管、烧杯、离心管和培养皿。
8)。
孔)、、、塑料培养板(35mm9)塑料培养皿()62496精品文档.精品文档辅助工具:放置试管、吸管和玻璃瓶等的架子。
2. 培养基和培养用液的配制多7-1400001) 培养基质:有多聚赖氨酸、牛皮胶原和鼠尾胶原。
本室目前常用分子量为0.1mg/ml,浓度为。
聚赖氨酸(Poly-L-lysineNaCl主要以无机盐和葡萄糖配制而成。
各种平衡盐溶液主要的不同点在于2) 平衡盐溶液:的浓度、离子浓度及缓冲系统的不同,可根据实际需要选用适当的平衡盐溶液。
培养液:目前已有现成的干粉出售,按说明书进行配制即可。
神经细胞培养需高糖,培3)无血清培养液。
养液应含有或加葡萄糖至6g/L。
目前培养海马神经元可选用B27℃564) 血清:有胎牛血清、小牛血清和马血清等。
分装成小瓶,4℃保存备用(分装前需。
灭活30分钟)胰蛋白酶溶液:用于分离细胞。
先用少量灭菌三蒸水将胰蛋白酶粉末溶成糊状,然后补5) 冻℃过夜,待完全溶解后用滤器过滤灭菌,并分装成1-2ml水至2.5%浓度,振荡摇匀,4。
世界上最完善最详细的神经元原代培养完全黄金版

世界上最完善最详细的神经元原代培养完全黄金版世界上最完善最详细的神经元原代培养完全黄金版[精华]序言:国内外关于原代培养有很多文献,不幸的是,没有一篇是详细的,没有一篇解答过why. 为了让大家少走弯路,根据我杀过3000只老鼠的经验,我把我这几年摸索的经验和大家分享,请求多投几票得几个叮当。
相信你follow我的这篇文章一定会做的很好。
我的标题说的很像吹牛,但是我是严肃认真的说的,I earn it.note:这篇文章全是我个人的经验,99。
9%原创,经过无数失败的到的最完美的原代神经元培养法,除了最后那一副图是来自下面第2行那篇文献,所以我才说是99.9%原创。
(注:附录的图是常用的消化酶对实验的存活率影响,来自下面链接里的文献)另外,成年鼠的原代培养我没有摸索过,推荐一篇文献在我以前的帖子(无需叮当)(/bbs/post/view?bid=156&id=17651341&sty=3)本篇专门讨论胎鼠和新生鼠神经元的原代培养。
1.材料选择。
一定要严格按照国际上,NCBI数据库,其他文献的材料一致。
胎鼠就要胎鼠,新生鼠就要新生鼠,不能混淆。
因为新生鼠有一些受体,在培养的工作中失去功能,会不能再生,比如NMDA受体。
和文献不同会导致错误结论,甚至困惑。
很多人写信问我新生鼠的培养问题,我回答用新生鼠的实验很少,八成是你自己搞错了实验对象,请确认国际上的相关研究文献所用老鼠,再来问我下面的问题。
2,培养基选择(neurbasal/neurobasal-a,invitrogen Co.ltd)。
我强烈不建议用血清培养。
原因是:首先,血清刺激胶质细胞和杂细胞分裂,最后非常影响神经元产量;其次,为了抑制胶质生长,往往要加入阿糖孢苷。
严重的毒性会影响许多灵敏实验;再次,最重要的,血清培养的细胞状态很不均一,从正常到凋亡都有,严重影响试验准确,而无血清专用培养基所有的正常细胞都处于一样的状态,要么都好,要么都差,高度一致。
世界上最完善最详细的神经元原代培养完全黄金版

世界上最完善最详细的神经元原代培养完全黄金版精华序言:国内外关于原代培养有很多文献;不幸的是;没有一篇是详细的;没有一篇解答过why.为了让大家少走弯路;根据我杀过3000只老鼠的经验;我把我这几年摸索的经验和大家分享;请求多投几票得几个叮当..相信你follow我的这篇文章一定会做的很好..我的标题说的很像吹牛;但是我是严肃认真的说的;I earn it.note:这篇文章全是我个人的经验;99..9%原创;经过无数失败的到的最完美的原代神经元培养法;除了最后那一副图是来自下面第2行那篇文献;所以我才说是99.9%原创..1.材料选择..一定要严格按照国际上;NCBI数据库;其他文献的材料一致..胎鼠就要胎鼠;新生鼠就要新生鼠;不能混淆..因为新生鼠有一些受体;在培养的工作中失去功能;会不能再生;比如NMDA受体..和文献不同会导致错误结论;甚至困惑..很多人写信问我新生鼠的培养问题;我回答用新生鼠的实验很少;八成是你自己搞错了实验对象;请确认国际上的相关研究文献所用老鼠;再来问我下面的问题..2;培养基选择neurbasal/neurobasal-a;invitrogen Co.ltd..我强烈不建议用血清培养..原因是:首先;血清刺激胶质细胞和杂细胞分裂;最后非常影响神经元产量;其次;为了抑制胶质生长;往往要加入阿糖孢苷..严重的毒性会影响许多灵敏实验;再次;最重要的;血清培养的细胞状态很不均一;从正常到凋亡都有;严重影响试验准确;而无血清专用培养基所有的正常细胞都处于一样的状态;要么都好;要么都差;高度一致..Invitrogen/GIBCO 无血清培养基neurobasalneurbasal-A被认为是最标准;最好的培养基..需要的添加剂为B27和谷氨酰胺..培养出的细胞状态均一;胶质细胞几乎不分裂..其中;neurobasal是胎鼠培养基;而-A为新生鼠培养基..不过根据笔者实验;没什么太大区别;都能很轻松培养成功;但是;切忌中途换培养基;要从一而终..很多实验室是不加抗生素的;也有的实验室加..由于很多初学者操作不熟练;不注意;很多人会污染;所以我还是建议加双抗..注意由于无血清培养基添加剂不是血清;而是人工合成的B27也有用N2的;但是推荐B27;只差10美金;血清有复杂的解毒作用而B27没有;双抗对细胞的干扰;无血清培养基要大的多..所以;如果你在neurobasal里添加抗生素;记得用量只要标准量的一半..另一个添加成分谷氨酰胺溶液不稳定;所以每次用时候要现配现加..大概是0.5-2mmol/l大部分人用0.5mmol/l.虽然笔者试过;没加这个也正常生长;但是几乎所有文献都添加;we just simply follow.3.解剖过程一定要全程在冰上预冷..这就意味着;从你处死母鼠后;胎鼠的大脑的温度要最快的降低到0度..另外;在解剖过程中;胎鼠直到皮层或海马被剪碎的过程;有时候可能需要2小时;如果样品多..一定要注意多准备冰袋子..确保你的任何过程都在冰浴的培养液中进行消化步骤除外..这样可以大大提高存活率..在解剖大脑时候;有人用hank's;在其中解剖..根据我的经验;即使是0度;神经元还是在进行着很大程度代谢..所以;hanks的无糖环境很不利..我个人建议;用DMEM-高糖或DMEM-F12+马血清来浸泡解剖过程中的大脑;来供给大脑的代谢;血清可以抑制神经凋亡..这其中还有一个问题;就是DMEM培养液会在空气中变碱性..国外有的实验室用L-15培养液来浸泡解剖过程中的脑;因为L-15不会再空气中变碱性..没有L-15的国内几乎没有;可以全程用DMEM-HG或者DMEM-F12+血清浸泡解剖过程中的脑..注意注意:一切操作都在冰浴的液体中;所以要多准备冰袋..中途冰袋没了的是猪头4.关于培养皿的问题..一般来说;6孔板是最佳选择..神经元让人苦恼的问题是;神经元发育的情况和密度相关..大于6孔板比如6CM DISH会使得中间很难和周围的细胞密度一样;造成板中间和边缘成熟度不一样;而这会给实验带来很大干扰..而太小的话96孔或128孔细胞会倾向于向中间集中也会照成发育不均匀..所以笔者经验是6孔板是最佳选择..神经原代培养一定是要包被;用POLY-LYS或者胶原..关于POLY-LYS包被;我们是包被30分钟;吸干晾过夜;然后洗两次;或者只洗一次但是要30分钟接着晾干..由于neurobasal培养法相当成熟;所以没必要用难固定的胶原..5.处死母鼠并把胎鼠解剖时候;一定要注意母鼠状态;是否有死胎;胎儿胎盘是不是正常;有多少胎;母鼠是多少天的一般是E16-E18这些信息也要严格记录..处死孕鼠后;要立即取出子宫放入冰冷培养液中..注意孕鼠处死后要短暂的浸泡酒精消毒;防止污染..一般来说;冰袋笔者选择一面蓝一面白的..蓝的朝上;可以很清晰看到海马结构..而去除血管膜的时候;白的那面朝上;这样可以很清楚看到血管膜..从处死老鼠到大脑被剥离出这段是人就会做;我就不多说了..不过有一点要注意;不管你是用皮层还是海马;不要取一个分离一个;而是要快速把所有胎鼠大脑都迅速取出放到冰预冷的培养液中..我建议胎鼠取出来的时候胎鼠就放在冰冷的缓冲液中..就是说;处死后要迅速把大脑的温度降到0.6:最影响原代培养的成败因素之一:这小段请大家一定注意看..无论皮层还是海马;上面都是有一层血管膜..注意:一定要尽可能的去掉所有的血管和血管膜..可以用解剖显微镜..胎鼠很好剥离;而新生鼠则比较难..这里千万不能粗心大意血管膜没剥离干净的后果是:1 吹打时候很难吹打下来神经元;被迫多次吹打;造成神经元大量死亡;2 血管膜一部分细胞会混入原代培养中;这些细胞往往会分裂;导致你的培养成果根本不能用..可以说;剥离的不好不干净;实验就是失败的;不可能得到高质量的神经元..注意的事情二:如果是要的是皮层的神经元原代培养;而皮层的细胞很多;切忌不要放太多皮层消化吹打过多的细胞反而会导致神经元大量死亡..另外;请仔细注意文献中要的是皮层哪一部位..没有详细要求的话一般是取顶端..从大脑到单个海马的剥离我不细说了..我现在以海马神经元为例..在所有海马都成功剥离后;请仔细的去除血管膜;原因上面说过..最后;请再仔细检查一下是不是都去掉了..确认后;海马片段被移入一个小烧杯里;加少量培养液;用剪刀剪成0.5-1立方毫米的小块;放置冰上准备消化;7:实验成败的关键之二:消化..一般来说;我看很多人都用0.125%的胰酶消化..实话说;胰酶消化非常的难以掌握速度;而且消化效果真的是--很烂稍微不注意;就会消化过头;细胞都消化光了..而胰酶消化的快;还会出现细胞快速破裂释放出DNA;和其他蛋白缠结成絮状包裹在组织块外面;使得块里面的不到应有的消化..所以我个人不推荐胰酶..想做一个完美成功的消化;我强烈推荐木瓜酶+DNA酶..木瓜酶是消化过后;存活率最高的酶见附图..笔者是配成2mg/ml的木瓜酶..另外要用的是DNA 酶..DNA酶的作用是;消化掉破裂细胞的DNA;避免了释放的DNA和蛋白缠结在组织块表面而阻碍进一步消化..DNA酶由于各个公司的活性不一样;很难有标准浓度;需要摸条件;不过不是很严格;只要能防止DNA缠结就好..木瓜酶的特点是不会消化过头;非常温和..缺点是一定要现配请直接用无血清的培养液配来保证神经元的营养代谢;而且一定要现用现配..消化的技巧:消化时候;很多人有不良习惯;喜欢用个50ml试管装着所有东西..结果是组织都沉底;下面消化不足;上面消化的没细胞..笔者的技巧是;用一个6或10CM 培养皿来消化..这样消化液在培养皿里形成浅而广阔的一层;均匀分布着组织块;从而彻底解决了上述问题..消化酶是2mg/ml木瓜蛋白酶+适量DNA酶..木瓜蛋白酶很便宜请放心..消化过程是20-30分钟..由于木瓜蛋白酶温和;因此不会消化过头;使得你的实验消化的很稳定..消化时候请在37度温箱里;每5分钟稍微摇动一下..注意:1木瓜酶不要用巯基化合物激活;激活后消化好很多但是成活率坏很多;2尽量用不含血清的培养液来配置木瓜酶而不是用hanks;使得神经元保持营养;3木瓜酶一定要现用现配..再次重申:除了消化过程中要37度;其余任何时刻都是要浸泡在0度冰浴的液体中..消化过后可以用1ml血清终止反应我是用马血清;便宜;但是切忌不要用国产血清;污染几率很大..把消化用的培养皿放在冰上冷却;然后垫高一面使得液体集中在另外一面便于吹打..整个体系静止2-3分钟;然后仔细的去掉上层的液体;保留消化过的组织块8.实验成败最关键最关键的:吹打..笔者经验是;没有必要用巴斯德管..对于胎鼠和10天之内的新生鼠;一般的1ml蓝枪头就可以达到满意的效果..在一边被垫高的皿里面;加入1-1.5ml新鲜培养基和少量DNA酶;吹打10次..吹打时候要注意;切忌过快;要缓慢吹打..吸入时候要很缓慢;吹出的时候可以略快;但是也要缓慢..轻柔吹打是细胞成活关键要用进口枪头;国产的有时候会过细..我们采用的是最合理的分步吹打法..每个10次吹打过后;静止2分钟..这时候游离的单细胞在上面;而团块会沉到下面去..上面的那层就是你要的单个悬浮细胞;把它们挪到指定容器里一样要冷在冰上..下面沉淀下去的细胞团块不要丢弃;再加1-1.5ml新鲜培养液;重复刚才的步骤;轻柔吹打;再静止2分钟;转移上层的单细胞到刚才指定容器里;一共这样分步吹打3次..3次之后如果还有团块在下面;请果断丢弃..造成这个的原因就是刚才的血管膜没剥好..这个分步吹打的过程保证了每一个单细胞都避免了过度吹打;而分布吹打又保证了尽可能多的收获单细胞..重申一下;为了保证最大程度收获活细胞;每次吹打之前;可以加入少量DNA酶..这招可以更容易收获大量高质量细胞;尤其是实验材料不足时候..消化和吹打非常忌讳操作的细胞过多;这样反而会使大量细胞死亡..实验者如果是用的皮层这种原材料充足的话;切忌不要放入过多皮层..9可选可不选;但是我建议全选神经元进一步纯化.. 一般来说;收获的细胞悬液已经是很纯的神经细胞了..但是;由于实验者等个人或者客观原因;血管膜可能不会被完全剥离;杂细胞可能也被吹入了细胞悬液;操作者手法不熟练;造成死细胞过多;影响种板等等原因;还是会影响到培养的神经元的质量..为了克服这些;笔者查了一些文献;经过反复测试采用了nycoprep低转速密度-梯度离心..现在这个梯度溶液已经被公司升级为optiprep.稀释时候;NycoPrep 1.077 推荐稀释成 60%;就用刚才悬浮细胞的培养液来配置..注意:1.077被广泛应用于血液中分离血细胞;如果你拿到的nycoprepoptiprep不是1.077;那么请换算一下稀释度值得注意的是;皮层和海马可能需要的稀释度稍微有不同..一般来说;一个10ml梯度离心管中;2-4ml稀释过的梯度液体就够用;取决于你收获的细胞量多少..把你收获的细胞悬液小心的加到稀释好的梯度剂的上面一层;经过1500转不是15000请注意5-8分钟;神经元被高度压缩到梯度面和上面的培养液分界面那一层..而最下面的梯度剂下的沉淀则是细胞团块;血细胞;杂细胞;以及部分小胶质细胞..而最上层最浮头;则是细胞碎片..这样;你就得到了最大限度去除了杂细胞;和细胞碎片;并去掉了一定的胶质细胞的最干净最纯粹的神经元..如果实验者技术精湛;也可以不用这步;但是用这步会效果更好..注意:母液稀释到60%只是参考;具体实验室不同可能略微有差别;要摸条件而不是死记硬背..另外皮层和海马浓度也稍微有差别..有些文献曾经报道;在梯度一定的情况下;一些特定梯度可以分开神经元和少突和其他胶质细胞;但是笔者从来没成功过..10.种板..这一步没有那么非常重要;但是如果没认真弄;浓度的不均一也会使得整个实验失败..记住的是;神经元原代培养是细节决定成败;任何一个环节没处理好;整个实验都会失败..种板密度过低是非常有害的..首先;神经元互相接触的几率小;难以存活和成熟..其次;胶质细胞会大量分裂;导致神经元变得更少;从而得到失败的实验..密度过高也是很有害的..由于营养争抢;密度过高有可能导致局部细胞营养枯竭死亡..另外;高密度会导致细胞聚团;结果也是细胞不正常形态或者死亡;导致错误或失败的实验结果..笔者的经验是6孔板每一个孔要有7×100000 个正好如果是台酚蓝染色只记录活细胞;那么酌情减少密度..注意;注意由于神经元的成熟速度和接种密度有很大关系;所以细胞计数一定要非常严肃认真一定要所有格子都看如果发现计数板细胞不均匀;宁愿重新计数由于接触抑制现象;适当密度的神经元可以抑制胶质细胞生长;从而达到理想的实验由于neurbasal培养液相当昂贵;所以种板时候是不用它的;而第一次换液才用..一般种板时候我们用的是DMEM-HG或者DMEM-F12 +10%马血清..注意一定要进口的..国产杂质很多会导致莫名其妙的细胞死亡..马血清会抑制神经元凋亡并且抑制胶质细胞生长FBS也可以;但是太贵种板注意事项一:种板时候的溶液推荐使用含有谷氨酸的培养液..神经元的NMDA等谷氨酸毒受体一般3天后才会出现..所以种板时候有谷氨酸不会导致细胞毒性反应..而这时候的谷氨酸反而是有利条件;可以促使神经元贴壁;从而提高存活率..注意事项二严格注意种板后都是要摇晃板摇匀细胞;切记千万不能圈圈摇晃圈圈摇晃会导致神经元都集中到培养板的孔中间;导致浓度不均;生长不均匀;会直接导致实验失败..正确的摇法是左右平行翻转角度;然后前后平行翻转角度;千万不要让里面液体画圈注意事项三严格注意种板后过过一定时间要换成正常标准培养神经元的neurobasal+b27+谷氨酰胺的无血清培养液..很多paper是1小时就换液;理由是胶质细胞贴壁差;会除去..但是笔者的经验来看;这是极端错误的1小时换液;神经元还没有完全贴壁;这时候换液会吹下很多神经元;流到孔另外一侧再贴壁;直接照成一个孔内细胞分布不均匀;从而成熟的不均匀..严格来说这种事情直接实验失败..笔者的经验是种板4-6小时换液;但是千万不能超过12小时..原因是:超过12小时;混在细胞悬液中的大量细胞碎片也会牢牢贴壁;而细胞碎片直接影响神经元存活和正常代谢;另外12小时后;胶质细胞会开始分裂;这时候就很难抑制了..笔者推荐是4小时;但是别超过8小时..注意事项四:4小时后换液时;请轻轻摇晃板几下再换..原因:细胞碎片贴壁很慢而且不牢固;这时候的摇晃可以最大程度去掉细胞碎片..然后;注意:细胞请再用没有加血清的;种板时候用的培养液清洗一次;也是要轻微摇晃;来进一步去除细胞碎片;从而得到良好的细胞生长环境..有条件也可以洗两次;但是笔者觉得一次足够..洗了之后吸干液体;换成neurbasal+B27+谷氨酰胺的神经元专用无血清培养基..对于原代培养;是细节决定成败;一个地方不够好会满盘皆输;所以请严肃对待..德国队的fans于凌晨×××××××××ugly分界线××××××××××××下面是对版面一些犯错误的研究方法中最严重的错误之一的提醒;请大家注意;我把回帖编辑到这里了..enjoy.刚回答了一系列形形色色的问题;我把这个问题挑明了说..如果您不相信 neurobasal最好的而还是坚持用DMEM之类的培养液;麻烦检查一下你们的DMEM或者MEM或者DMEM-F12的invitrogen产品序列号;到网站上检查下相应的组成公式..很多这种培养液都是含有谷氨酸;而且是mmol级的..成熟的神经元10um的谷氨酸即可照成细胞凋亡;而谷氨酸受体4-6天就开始表达;如果胎鼠神经元经历了mmol浓度的培养液中的谷氨酸还不被毒死;那就是怪事了..新生鼠不受这个限制;新生鼠原代培养没有有功能的 NMDA受体;×××××××××××ugly 分界线××××××××最雷到我的;是板上有人用材料非常混乱;一会用胎鼠;一会用新生鼠;一会成年鼠的你们这叫对自己不负责任 we do all cases seriously 而不是凭借肉眼观察和猜测..做原代培养的;首先就是收集大量背景文献;看看你的相关要求的国际公认的做法是用哪种..胎鼠和新生鼠虽然形状一样;但是神经元的功能一点都不一样;表达受体都不一样.. 大部分实验都是用胎鼠;而新生鼠往往用来培养测一些LTD之类的not quite sure;文献太少;所以有人问我新生鼠的问题;我都是说你先看下背景文献是不是你搞错了..而成年鼠是另外的培养方法;要求更小心仔细和特别的处理..硬套胎鼠的原代培养必然导致失败..另外;在一些药物保护实验中;不要以为NGF就一定对神经有很大作用;笔者的经验是;想要细胞存活;FGF2是最佳选择但是;FGF2会导致细胞变化很大;所以还是别用在原代培养中;不过成年鼠培养一定要用FGF2..对于一些雷到我的;我只能再次说;请收集足够的背景文章在去选择合适材料;please do it carefully and seriously祝大家都能培养出健康漂亮的神经元;欢迎大家来问问题如果是美女的话;给个电话我会很开心声明;除了最后一幅图是来自别的文献;前面所有的经验都是我的原创;从无数失败总结出的我认为最完美的神经原代培养..最后提醒大家注意:nycoprepoptiprep可能有很多浓度;但是NycoPrep 1.077 以前是最常用的;笔者默认的就是用这个溶液配置的密度梯度..如果您买到的不是这个密度;请换算一下..笔者当时只有NycoPrep 1.077在中国能买到;现在可能会有更多浓度供选择;请大家注意换算..。
原代神经细胞培养

原代神经细胞培养新生鼠神经元细胞的培养准备工作:1、解剖器械一套(实验室有专门用于细胞间取材用的成套器械),需提前一天灭菌,过夜烤干。
2、试剂、溶液:Neuralbasal培养基(2mM glutamine);D-PBS缓冲液;0.25%trypsin/0.02%EDTA消化液;0.05%poly-lysin。
3、包被玻片:干烤灭菌12 x 12mm2玻片,12孔板。
包被过程:0.05%poly-lysin滴于置培养板中的盖玻片上(注意勿溢出玻片),37℃放置12hr后纯水洗3遍,晾干。
4、操作中所用枪头均用剪刀剪去枪头尖,然后在酒精灯上迅速过一下抛光。
取材:1、从-200C取出三个冰袋,预冷若干皿D-PBS,一大皿用于冷却剥出的脑子,另外的35mm的皿用于冷却分离出的脑组织,如:海马,皮层,下丘脑等,分离几个部位就预冷几小皿D-PBS,小皿盖子上做好标记,第三个冰袋上放一皿盖,上面放灭过菌的滤纸,用于剥离脑子的操作。
2、取1-3天鼠,75%酒精浸泡片刻后,用大剪子断头处死。
3、弯头眼科剪剪开颅骨,取出完整脑至盛有预冷D-PBS的培养皿中,在体式镜下分离相应部位的组织,分别放在相应的皿中。
4、将组织块用弯头眼科剪剪碎,每小块约2mm3左右,用枪移入5mlEP中,稍为沉淀1分钟,吸弃上清,加入0.25%trypsin/0.02%EDTA,37℃消化10-15分钟左右,期间每3分钟颠倒几下。
注:每四个海马用胰酶1ml,每个皮层用胰酶2ml。
5、消化完毕,每毫升胰酶加入100ul血清以终止胰酶,颠倒混匀;1000rpm,4分钟,离心沉淀。
6、吸弃上清,注意不要将组织吸出。
7、加入37℃预热的1-2ml DMEM/10%FBS,用枪头吹打20下左右,此时液体浑浊,组织块明显变小。
8、放置沉淀3分钟左右,可见组织块沉底,吸取上清,其中包含所要的细胞。
9、计数,将细胞密度调整至2-4 x 10 5 /ml后接种于预先用poly-lysine包被过的盖玻片或皿上,100ul每玻片(12 x 12mm玻片)。
神经元原代培养

神经元原代培养一、准备1. 试剂1)胎牛血清灭活:血清室温自然融解,摇匀。
选用与血清瓶同规格的对照瓶一个,并加入与血清等体积的水。
对照瓶内插入1~2支温度计(保证测试温度的准确性)。
将血清瓶与对照瓶同时放入恒温水浴箱,待温度计所示温度上升至56℃时,定时30分钟。
灭活后分装,10ml/瓶,一瓶放在培养箱做无菌实验,其余-20~-70℃保存。
2)D-Hank’s液(g/L):NaCl: 8, KCl: 0.4, KH2PO4: 0.06, Na2HPO42H2O: 0.06, NaHCO3: 0.35; 酚红:0.02,超纯水溶解,调节PH至7.2,高压灭菌(购自南方医院临床试验中心)。
3)多聚赖氨酸:避光,用超纯水配制好后过滤,200μl或400μl/tube分装, -20度储存。
母液浓度为5mg/ml,工作液浓度为0.1mg/ml(购自Sigma,P2636,100mg)。
配置方法:20ml 超纯水溶解,分装为1ml/管。
用时加入49ml超纯水稀释为0.1mg/ml的工作液。
4)50mM谷氨酸:超纯水溶解,滤过除菌,分装50μl/tube,-20℃保存。
使用时需将终浓度调整为25μM(购自Sigma,G8415,100g,分子量:147.13)。
配制方法:电子天平称量谷氨酸1.4713g,即0.01mol=10mmol,溶于200ml超纯水中,配制为50mM的谷氨酸母液。
例:500ml的Neurobasal Medium中加入Glutamate母液(50mM)250μl,此时终浓度约为25μM。
5)胰蛋白酶:0.25%(购自Hyclone,SH30042.01,100ml)。
6)阿糖胞苷(AraC)母液:10mM,(阿糖胞苷,1:1000稀释用)(购自Sigma,C1768,100mg,分子量:243.22);配制方法:100mg/243.22≈0.41mM,加入41ml超纯水溶解,配制成10mM的AraC母液。
神经元原代培养 丁香园

(丁香园protocol 2008)一、常规的试剂配制:1、培养基:选用高糖型DMEM/F12 (1:1)培养基干粉(含15 mmol Hepes),每升培养液中加入碳酸氢钠1.8 g,调节pH值7.0,0.22 μm的微孔滤膜过滤除菌后,加入谷氨酰胺、无菌的青霉素和链霉素液,使其终浓度分别为0.5 mmol/L 、100 U/ml和100 μg /ml,分装后置于4℃保存,临用前加入2%的B27。
神经细胞代谢旺盛,选用高糖型培养基;谷胱甘肽可保持培养基还原状态,营养成分稳定;B27是公认的刺激神经元生长的营养因子,不过价格挺贵;PH值很关键,Hepes是优秀的缓冲系统,pH值调好后能保持很长时间;2、多聚赖氨酸溶液的配制:称取1.5 mg L-多聚赖氨酸溶于100 ml PBS,0.22 μm的微孔滤膜过滤除菌,密封后置于4 ℃保存,可长期使用;3、磷酸盐缓冲液(PBS)的配制:称取8.0 g NaCl、0.2 g KCl、2.85 g Na2HPO4•12H2O 和0.2 g KH2PO4充分溶于900 ml dd H2O,调节pH值为7.2,补dd H2O至1000 ml并分装,高压灭菌(121~126 ℃),4 ℃备用;4、D-Hanks液的配制:称取KCl 0.4 g,KH2PO4 0.06 g,NaCl 8.0 g,NaHCO3 0.35 g,Na2HPO4•12H2O 0.08 g,溶于dd H2O中,调节PH值为7.2,定容至1000 ml,高压蒸汽灭菌(121~126 ℃),4℃备用;5、0.25%胰蛋白酶的配制:称取0.25 g胰蛋白酶粉末,少许D-Hanks溶液调成糊状,用D-Hanks定容至100 ml,混匀,冰箱内放置过夜,滤纸过滤后,0.22 m微孔滤膜过滤,分装,-20 ℃保存。
二、试验操作过程:1、包被:培养前晚上将培养瓶(板)用多聚赖氨酸包被10 min,吸除,无菌操作台上15min 自然晾干,置于培养箱中备用;2、取脑:选取新生24 h的SD大鼠数只,雌雄不限,75%酒精全身消毒,断头处死,无菌条件下分层剪开头皮、颅骨,用弯镊拉开脑区视野,小心取出全脑,D-hanks液洗数次;(预先准备至少4把眼科剪刀,分别用于断头、剪头皮和剪颅骨及第3步的剪组织这4个操作)3、分离:以脑中线为起点,小心拨开大脑颞叶皮层,暴露出新月状海马回,小心夹出海马组织,置于冰浴的D-hanks液中,仔细剔除微血管,用充分剪碎组织;4、消化:加入同体积0.125%的胰蛋白酶,瓶口用锡箔纸盖上,37 ℃培养箱内消化20 min 左右,期间轻摇数次;过滤:加入数滴胎牛血清终止消化,用尖头吸管轻轻吹打细胞数分钟,至液体成米糊状即停止吹打,动作要轻柔,用150目网筛过滤,收集细胞悬液;5、接种:以1100 rpm离心5 min,倾去上清,用DMEM/F12培养液重悬细胞,轻轻吹打,台盼蓝染色快速计数,根据计数结果,调整细胞终浓度为1 00000个/ml,加入终浓度为10%胎牛血清后接种于培养瓶(板)中,置于37 ℃、含5% CO2培养箱中培养;12小时内禁止晃动6、换液:接种后24 h将培养液全量换成无血清DMEM/F12培养液,第48 h时加入终浓度为10 μmol/L的阿糖胞苷,以抑制非神经元细胞的过度生长,随后每3 d半量换液。
原代神经元培养的概念

原代神经元培养的概念一、神经元的获取原代神经元培养的第一步是从动物脑组织中获取神经元。
通常,新生动物(如新生小鼠)的脑组织被用于这一目的,因为新生动物的神经元具有较强的增殖能力。
获取的神经元可以通过酶消化或物理分散法进行处理,使组织分离成单个细胞。
二、培养基的选择培养基是原代神经元生长的必要条件,它提供了神经元所需的营养物质、激素和生长因子。
为了维持神经元的生长和存活,需要选择适当的培养基,以满足神经元的特殊营养需求。
常用的培养基有DMEM、MEM、F-12等。
三、培养条件控制原代神经元培养需要在一定的条件下进行,包括温度、湿度、气体(如O2和CO2)浓度等。
这些条件对神经元的生长和分化有重要影响,因此需要精确控制。
温度的稳定性对于维持细胞活性至关重要,而气体浓度则影响细胞的代谢和pH值。
四、神经元的生长与分化在适宜的培养条件下,获取的神经元开始生长并增殖。
在生长过程中,神经元会通过突起与其他神经元建立连接,形成复杂的网络结构。
随着时间的推移,这些神经元还会分化成不同的类型,执行特定的功能。
五、神经元网络的建立在原代神经元培养过程中,神经元会形成复杂的网络结构。
这些网络通过突触连接,传递电化学信号,模拟生物体内的神经网络。
通过观察神经元网络的建立,可以深入了解神经元的生长和功能机制。
六、培养过程中的监测与观察在原代神经元培养过程中,需要定期对细胞进行监测和观察,以了解细胞的生长状态、形态变化以及是否存在异常。
可以使用显微镜、电生理技术等方法对神经元进行监测。
通过这些手段,可以及时发现并解决培养过程中出现的问题。
七、培养物的应用与价值原代神经元培养在科学研究、药物筛选和疾病模型构建等方面具有广泛的应用价值。
通过原代神经元培养,可以深入了解神经系统的生理和病理机制,为药物研发提供有效的工具。
此外,原代神经元培养还为神经系统疾病的动物模型提供了替代方案,有助于研究疾病的发病机制和治疗方法。
总的来说,原代神经元培养是一种重要的实验手段,有助于推动神经系统科学的发展和进步。
原代DRG神经元培养

原代DRG神经元培养主要试剂和溶液的配置Dulbecco缓冲液(1L): 0.2g KCl,0.2g KH2PO4,8.0 g NaCl,Na2HPO4•12H2O 2.89g溶于900 ml ddH2O中,溶解完全后调pH值至7.2-7.4,定容至1 L。
0.3%胰酶(250 ml)(神经元用):称取0.75 g胰酶(Sigma)溶于200 ml Dulbecco缓冲液中,定容至250 ml,0.22 μm微孔小滤器过滤除菌。
0.5%胶原酶IA (250 ml):称取1. 25 g 胶原酶(Sigma)溶于200 ml Dulbeaco缓冲液中,定容至250 ml,0.22 μm微孔小滤器过滤除菌。
DMEM培养液:称取DMEM固体培养基(Gibco-BRL, USA) 13.48 g溶于800 ml ddH2O中,用3.7 g NaHCO3调pH值至7.2-7.4,定容至1 L。
0.22 μm微孔滤膜过滤除菌,4℃保存。
临用前配制成含10% FBS (Hyclone, USA)的完全培养基。
Neurobasal (Gibco): 4℃保存,临用前按50:1的比例加入B27无血清培养液添加剂(Gibco)。
Poly-D-lysine (Sigma): 1 mg/ml,可回收利用。
0.25%胰酶(250 ml)(细胞系用):NaCl 2 g,KCl 0.05 g,KH2PO40.05 g,Na2HPO4·12H2O 0.721 g,胰酶(Sigma) 0.3125 g,EDTA 0.05 g,苯酚红0.001 g,碳酸氢钠0.145 g。
先将胰酶和EDTA 入瓶内溶1-2 h(4 o C),然后加入KH2PO4,Na2HPO4·12H2O及苯酚红的混合液混匀,溶解2 h (4 o C),在混合后的最后10 min加入0.145 g碳酸氢钠,调pH值至7.0~7.5,立即过滤以防碳酸挥发。
世界上最完善最详细的神经元原代培养完全黄金版讲解

世界上最完善最详细的神经元原代培养完全黄金版[精华]序言:国内外关于原代培养有很多文献,不幸的是,没有一篇是详细的,没有一篇解答过why. 为了让大家少走弯路,根据我杀过3000只老鼠的经验,我把我这几年摸索的经验和大家分享,请求多投几票得几个叮当。
相信你follow我的这篇文章一定会做的很好。
我的标题说的很像吹牛,但是我是严肃认真的说的,I earn it.note:这篇文章全是我个人的经验,99。
9%原创,经过无数失败的到的最完美的原代神经元培养法,除了最后那一副图是来自下面第2行那篇文献,所以我才说是99.9%原创。
(注:附录的图是常用的消化酶对实验的存活率影响,来自下面链接里的文献)另外,成年鼠的原代培养我没有摸索过,推荐一篇文献在我以前的帖子(无需叮当)(/bbs/post/view?bid=156&id=17651341&sty=3)本篇专门讨论胎鼠和新生鼠神经元的原代培养。
1.材料选择。
一定要严格按照国际上,NCBI数据库,其他文献的材料一致。
胎鼠就要胎鼠,新生鼠就要新生鼠,不能混淆。
因为新生鼠有一些受体,在培养的工作中失去功能,会不能再生,比如NMDA受体。
和文献不同会导致错误结论,甚至困惑。
很多人写信问我新生鼠的培养问题,我回答用新生鼠的实验很少,八成是你自己搞错了实验对象,请确认国际上的相关研究文献所用老鼠,再来问我下面的问题。
2,培养基选择(neurbasal/neurobasal-a,invitrogen Co.ltd)。
我强烈不建议用血清培养。
原因是:首先,血清刺激胶质细胞和杂细胞分裂,最后非常影响神经元产量;其次,为了抑制胶质生长,往往要加入阿糖孢苷。
严重的毒性会影响许多灵敏实验;再次,最重要的,血清培养的细胞状态很不均一,从正常到凋亡都有,严重影响试验准确,而无血清专用培养基所有的正常细胞都处于一样的状态,要么都好,要么都差,高度一致。
Invitrogen/GIBCO 无血清培养基neurobasal(neurbasal-A)被认为是最标准,最好的培养基。
皮层神经元的原代培养

皮层神经元的原代培养
一、背景
大脑皮层是调节躯体运动的高级中枢。
它由初级感觉区、初级运动区和联合区三部分构成。
人类大脑皮层的神经细胞约有140亿个,面积约2200平方厘米,主要含有锥体形细胞、梭形细胞和星形细胞(颗粒细胞)及神经纤维。
体外原代培养神经元作为神经科学研究的有力手段,越来越受到广大科研工作者的重视。
由于神经元是一种高度分化的细胞,动物出生后很少分裂,相对于其它细胞而言,神经元在体外更难以存活和生长。
因此,体外培养神经元要求的培养方法和营养条件均较为特殊。
二、实验步骤
1. 无菌条件下,从怀孕18天的SD 大鼠腹中取出胚胎放入预冷的无钙、镁平衡盐溶液中;
2.在解剖显微镜下,分离取出胚胎大脑皮层,除去脑膜,剪碎组织(1mm3),加入0.125%胰酶-EDTA 消化液后放入 37℃孵箱内消化10 分钟,期间摇晃一次;
3.去掉上清,加入终止液终止消化,吹打10-15次,不能有气泡,收集上清;剩余沉淀加入终止液继续吹打5-10次;
4.所收集的上清过滤后,将细胞悬液于4℃1000rpm离心10min ,弃上清,管内加入新鲜培养液重悬;
5.台盼蓝染色计数,以 3 x 104cells/孔种入包被的24孔板,37℃,5%CO2,95%饱和湿度的培养箱中培养;
6.每周换液两次,每次半量换液。
世界上最完善最详细的神经元原代培养完全黄金版讲解

世界上最完善最详细的神经元原代培养完全黄金版[精华]序言:国内外关于原代培养有很多文献,不幸的是,没有一篇是详细的,没有一篇解答过why. 为了让大家少走弯路,根据我杀过3000只老鼠的经验,我把我这几年摸索的经验和大家分享,请求多投几票得几个叮当。
相信你follow我的这篇文章一定会做的很好。
我的标题说的很像吹牛,但是我是严肃认真的说的,I earn it.note:这篇文章全是我个人的经验,99。
9%原创,经过无数失败的到的最完美的原代神经元培养法,除了最后那一副图是来自下面第2行那篇文献,所以我才说是99.9%原创。
(注:附录的图是常用的消化酶对实验的存活率影响,来自下面链接里的文献)另外,成年鼠的原代培养我没有摸索过,推荐一篇文献在我以前的帖子(无需叮当)(/bbs/post/view?bid=156&id=17651341&sty=3)本篇专门讨论胎鼠和新生鼠神经元的原代培养。
1.材料选择。
一定要严格按照国际上,NCBI数据库,其他文献的材料一致。
胎鼠就要胎鼠,新生鼠就要新生鼠,不能混淆。
因为新生鼠有一些受体,在培养的工作中失去功能,会不能再生,比如NMDA受体。
和文献不同会导致错误结论,甚至困惑。
很多人写信问我新生鼠的培养问题,我回答用新生鼠的实验很少,八成是你自己搞错了实验对象,请确认国际上的相关研究文献所用老鼠,再来问我下面的问题。
2,培养基选择(neurbasal/neurobasal-a,invitrogen Co.ltd)。
我强烈不建议用血清培养。
原因是:首先,血清刺激胶质细胞和杂细胞分裂,最后非常影响神经元产量;其次,为了抑制胶质生长,往往要加入阿糖孢苷。
严重的毒性会影响许多灵敏实验;再次,最重要的,血清培养的细胞状态很不均一,从正常到凋亡都有,严重影响试验准确,而无血清专用培养基所有的正常细胞都处于一样的状态,要么都好,要么都差,高度一致。
Invitrogen/GIBCO 无血清培养基neurobasal(neurbasal-A)被认为是最标准,最好的培养基。
神经元的生长发育和死亡41页PPT

15、机会是不守纪律的。——雨果
56、书不仅是生活,而且是现在、过 去和未 来文化 生活的 源泉。 ——库 法耶夫 57、生命不可能有两次,但许多人连一 次也不 善于度 过。— —吕凯 特 58、问渠哪得清如许,为有源头活水来 。—— 朱熹 59、我的努力求学没有得到别的好处, 只不过 是愈来 愈发觉 自己的 无知。 ——笛 卡儿
神经元的生长发育和死亡
11、战争满足了,或曾经满足过人的 好斗的 本能, 但它同 时还满 足了人 对掠夺 ,破坏 以及残 酷的纪 律和专 制力的 欲望。 ——查·埃利奥 特 12、不应把纪律仅仅看成教育的手段 。纪律 是教育 过程的 结果, 首先是 学生集 体表现 在一切 生活领 域—— 生产、 日常生 活、学 校、文 化等领 域中努 力的结 果。— —马卡 连柯(名 言网)
拉
60、生活的道路一旦选定,就要勇敢地 走到底 ,决不 回头。 ——左
原代神经细胞培养方法

神经细胞培养体外神经细胞的培养已成为神经生物学研究中十分有用的技术手段。
神经细胞培养的主要优点是:(1)分散培养的神经细胞在体外生长成熟后,能保持结构和功能上的某些特点, 而且长期培养能形成髓鞘和建立突触联系,这就提供了体内生长过程在体外重现的机会。
(2)能在较长时间内直接观察活细胞的生长、分化、形态和功能变化,便于使用各种不同的技术方法如相差显微镜、荧光显微镜、电子显微镜、激光共聚焦显微镜、同位素标记、原位杂交、免疫组化和电生理等手段进行研究。
(3)易于施行物理(如缺血、缺氧)、化学和生物因子(如神经营养因子)等实验条件, 观察条件变更对神经细胞的直接或间接作用。
(4)便于从细胞和分子水平探讨某些神经疾病的发病机制,药物或各种因素对胚胎或新生动物神经细胞在生长、发育和分化等各方面的影响。
我们实验室从80年代始开展了神经细胞的体外培养工作,取得了一些经验,现将培养细胞分类及方法简要介绍如下:一.鸡胚背根神经节组织块培养主要用于神经生长因子(NGF)等神经营养因子的生物活性测定。
在差倒置显微镜下观察以神经突起的生长长度和密度为指标半定量评估NGF的活性。
1.材料和方法(1)选正常受精的鸡蛋,置于37℃生化培养箱内孵化,每日翻动鸡蛋一次。
(2)取孵化8-12 d 的鸡蛋, 用70% 酒精消毒蛋壳,从气室端敲开蛋壳,用消毒镊剥除气室部蛋壳。
(3)用弯镊钩住鸡胚颈部,无菌条件下取出鸡胚置小平皿内,除去头部后,腹侧向上置灭菌毛玻璃片上,用眼科弯镊子打开胸腹腔,除去内脏器官。
(4)在解剖显微镜下,小心除去腹膜,暴露脊柱及其两侧,在椎间孔旁可见到沿脊柱两侧排列的背根节(图1),用一对5号微解剖镊小心取出。
(5)置背根节于解剖溶液内,用微解剖镊去除附带组织,接种于涂有鼠尾胶的玻璃或塑料培养瓶中,在DMEM无血清培养液中培养。
2.结果鸡胚背根神经节在含神经生长因子(NGF, 2.5S,20ng/ml)的无血清培养液中培养24 h,神经节长出密集的神经突起。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
世界上最完善最详细的神经元原代培养完全黄金版[精华]序言:国内外关于原代培养有很多文献,不幸的是,没有一篇是详细的,没有一篇解答过why. 为了让大家少走弯路,根据我杀过3000只老鼠的经验,我把我这几年摸索的经验和大家分享,请求多投几票得几个叮当。
相信你follow我的这篇文章一定会做的很好。
我的标题说的很像吹牛,但是我是严肃认真的说的,I earn it.note:这篇文章全是我个人的经验,99。
9%原创,经过无数失败的到的最完美的原代神经元培养法,除了最后那一副图是来自下面第2行那篇文献,所以我才说是99.9%原创。
(注:附录的图是常用的消化酶对实验的存活率影响,来自下面链接里的文献)另外,成年鼠的原代培养我没有摸索过,推荐一篇文献在我以前的帖子(无需叮当)(/bbs/post/view?bid=156&id=17651341&sty=3)本篇专门讨论胎鼠和新生鼠神经元的原代培养。
1.材料选择。
一定要严格按照国际上,NCBI数据库,其他文献的材料一致。
胎鼠就要胎鼠,新生鼠就要新生鼠,不能混淆。
因为新生鼠有一些受体,在培养的工作中失去功能,会不能再生,比如NMDA受体。
和文献不同会导致错误结论,甚至困惑。
很多人写信问我新生鼠的培养问题,我回答用新生鼠的实验很少,八成是你自己搞错了实验对象,请确认国际上的相关研究文献所用老鼠,再来问我下面的问题。
2,培养基选择(neurbasal/neurobasal-a,invitrogen Co.ltd)。
我强烈不建议用血清培养。
原因是:首先,血清刺激胶质细胞和杂细胞分裂,最后非常影响神经元产量;其次,为了抑制胶质生长,往往要加入阿糖孢苷。
严重的毒性会影响许多灵敏实验;再次,最重要的,血清培养的细胞状态很不均一,从正常到凋亡都有,严重影响试验准确,而无血清专用培养基所有的正常细胞都处于一样的状态,要么都好,要么都差,高度一致。
Invitrogen/GIBCO 无血清培养基neurobasal(neurbasal-A)被认为是最标准,最好的培养基。
需要的添加剂为B27和谷氨酰胺。
培养出的细胞状态均一,胶质细胞几乎不分裂。
其中,neurobasal 是胎鼠培养基,而-A为新生鼠培养基。
不过根据笔者实验,没什么太大区别,都能很轻松培养成功,但是,切忌中途换培养基,要从一而终。
很多实验室是不加抗生素的,也有的实验室加。
由于很多初学者操作不熟练,不注意,很多人会污染,所以我还是建议加双抗。
注意!由于无血清培养基添加剂不是血清,而是人工合成的B27(也有用N2的,但是推荐B27,只差10美金),血清有复杂的解毒作用而B27没有,双抗对细胞的干扰,无血清培养基要大的多。
所以,如果你在neurobasal里添加抗生素,记得用量只要标准量的一半。
另一个添加成分谷氨酰胺溶液不稳定,所以每次用时候要现配现加。
大概是0.5-2mmol/l大部分人用0.5mmol/l.虽然笔者试过,没加这个也正常生长,但是几乎所有文献都添加,we just simply follow.3.解剖过程一定要全程在冰上预冷。
这就意味着,从你处死母鼠后,胎鼠的大脑的温度要最快的降低到0度。
另外,在解剖过程中,胎鼠直到皮层或海马被剪碎的过程,有时候可能需要2小时,如果样品多。
一定要注意多准备冰袋子。
确保你的任何过程都在冰浴的培养液中进行(消化步骤除外)。
这样可以大大提高存活率。
在解剖大脑时候,有人用hank's,在其中解剖。
根据我的经验,即使是0度,神经元还是在进行着很大程度代谢。
所以,hanks的无糖环境很不利。
我个人建议,用DMEM-高糖或DMEM-F12+马血清来浸泡解剖过程中的大脑,来供给大脑的代谢,血清可以抑制神经凋亡。
这其中还有一个问题,就是DMEM培养液会在空气中变碱性。
国外有的实验室用L-15培养液来浸泡解剖过程中的脑,因为L-15不会再空气中变碱性。
没有L-15的(国内几乎没有),可以全程用DMEM-HG或者DMEM-F12+血清浸泡解剖过程中的脑。
注意注意:一切操作都在冰浴的液体中,所以要多准备冰袋。
(中途冰袋没了的是猪头)4.关于培养皿的问题。
一般来说,6孔板是最佳选择。
神经元让人苦恼的问题是,神经元发育的情况和密度相关。
大于6孔板(比如6CM DISH)会使得中间很难和周围的细胞密度一样,造成板中间和边缘成熟度不一样,而这会给实验带来很大干扰。
而太小的话(96孔或128孔)细胞会倾向于向中间集中也会照成发育不均匀。
所以笔者经验是6孔板是最佳选择。
神经原代培养一定是要包被,用POLY-LYS或者胶原。
关于POLY-LYS包被,我们是包被30分钟,吸干晾过夜,然后洗两次,或者只洗一次但是要30分钟接着晾干。
由于neurobasal培养法相当成熟,所以没必要用难固定的胶原。
5.处死母鼠并把胎鼠解剖时候,一定要注意母鼠状态,是否有死胎,胎儿胎盘是不是正常,有多少胎,母鼠是多少天的(一般是E16-E18)这些信息也要严格记录。
处死孕鼠后,要立即取出子宫放入冰冷培养液中。
注意孕鼠处死后要短暂的浸泡酒精消毒,防止污染。
一般来说,冰袋笔者选择一面蓝一面白的。
蓝的朝上,可以很清晰看到海马结构。
而去除血管膜的时候,白的那面朝上,这样可以很清楚看到血管膜。
从处死老鼠到大脑被剥离出这段是人就会做,我就不多说了。
不过有一点要注意,不管你是用皮层还是海马,不要取一个分离一个,而是要快速把所有胎鼠大脑都迅速取出放到冰预冷的培养液中。
我建议胎鼠取出来的时候胎鼠就放在冰冷的缓冲液中。
就是说,处死后要迅速把大脑的温度降到0.6:最影响原代培养的成败因素之一:这小段请大家一定注意看。
无论皮层还是海马,上面都是有一层血管膜。
注意:一定要尽可能的去掉所有的血管和血管膜。
可以用解剖显微镜。
胎鼠很好剥离,而新生鼠则比较难。
这里千万不能粗心大意!!血管膜没剥离干净的后果是:1 吹打时候很难吹打下来神经元,被迫多次吹打,造成神经元大量死亡;2 血管膜一部分细胞会混入原代培养中,这些细胞往往会分裂,导致你的培养成果根本不能用。
可以说,剥离的不好不干净,实验就是失败的,不可能得到高质量的神经元。
注意的事情二:如果是要的是皮层的神经元原代培养,而皮层的细胞很多,切忌不要放太多皮层消化吹打!过多的细胞反而会导致神经元大量死亡。
另外,请仔细注意文献中要的是皮层哪一部位。
没有详细要求的话一般是取顶端。
从大脑到单个海马的剥离我不细说了。
我现在以海马神经元为例。
在所有海马都成功剥离后,请仔细的去除血管膜,原因上面说过。
最后,请再仔细检查一下是不是都去掉了。
确认后,海马片段被移入一个小烧杯里,加少量培养液,用剪刀剪成0.5-1立方毫米的小块,放置冰上准备消化,7:实验成败的关键之二:消化。
一般来说,我看很多人都用0.125%的胰酶消化。
实话说,胰酶消化非常的难以掌握速度,而且消化效果真的是--很烂!稍微不注意,就会消化过头,细胞都消化光了。
而胰酶消化的快,还会出现细胞快速破裂释放出DNA,和其他蛋白缠结成絮状包裹在组织块外面,使得块里面的不到应有的消化。
所以我个人不推荐胰酶。
想做一个完美成功的消化,我强烈推荐木瓜酶+DNA酶。
木瓜酶是消化过后,存活率最高的酶(见附图)。
笔者是配成2mg/ml的木瓜酶。
另外要用的是DNA酶。
DNA酶的作用是,消化掉破裂细胞的DNA,避免了释放的DNA和蛋白缠结在组织块表面而阻碍进一步消化。
DNA酶由于各个公司的活性不一样,很难有标准浓度,需要摸条件,不过不是很严格,只要能防止DNA缠结就好。
木瓜酶的特点是不会消化过头,非常温和。
缺点是一定要现配(请直接用无血清的培养液配来保证神经元的营养代谢,而且一定要现用现配。
消化的技巧:消化时候,很多人有不良习惯,喜欢用个50ml试管装着所有东西。
结果是组织都沉底,下面消化不足,上面消化的没细胞。
笔者的技巧是,用一个6或10CM 培养皿来消化。
这样消化液在培养皿里形成浅而广阔的一层,均匀分布着组织块,从而彻底解决了上述问题。
消化酶是2mg/ml木瓜蛋白酶+适量DNA酶。
木瓜蛋白酶很便宜请放心。
消化过程是20-30分钟。
由于木瓜蛋白酶温和,因此不会消化过头,使得你的实验消化的很稳定。
消化时候请在37度温箱里,每5分钟稍微摇动一下。
注意:1)木瓜酶不要用巯基化合物激活,激活后消化好很多但是成活率坏很多;2)尽量用不含血清的培养液来配置木瓜酶而不是用hanks,使得神经元保持营养;3)木瓜酶一定要现用现配。
再次重申:除了消化过程中要37度,其余任何时刻都是要浸泡在0度(冰浴)的液体中。
消化过后可以用1ml血清终止反应(我是用马血清,便宜,但是切忌不要用国产血清,污染几率很大)。
把消化用的培养皿放在冰上冷却,然后垫高一面使得液体集中在另外一面便于吹打。
整个体系静止2-3分钟,然后仔细的去掉上层的液体,保留消化过的组织块8.实验成败最关键最关键的:吹打。
笔者经验是,没有必要用巴斯德管。
对于胎鼠和10天之内的新生鼠,一般的1ml蓝枪头就可以达到满意的效果。
在一边被垫高的皿里面,加入1-1.5ml新鲜培养基和少量DNA酶,吹打10次。
吹打时候要注意,切忌过快,要缓慢吹打。
吸入时候要很缓慢,吹出的时候可以略快,但是也要缓慢。
轻柔吹打是细胞成活关键!要用进口枪头,国产的有时候会过细。
我们采用的是最合理的分步吹打法。
每个10次吹打过后,静止2分钟。
这时候游离的单细胞在上面,而团块会沉到下面去。
上面的那层就是你要的单个悬浮细胞,把它们挪到指定容器里(一样要冷在冰上)。
下面沉淀下去的细胞团块不要丢弃,再加1-1.5ml新鲜培养液,重复刚才的步骤,轻柔吹打,再静止2分钟,转移上层的单细胞到刚才指定容器里,一共这样分步吹打3次。
3次之后如果还有团块在下面,请果断丢弃。
造成这个的原因就是刚才的血管膜没剥好。
这个分步吹打的过程保证了每一个单细胞都避免了过度吹打,而分布吹打又保证了尽可能多的收获单细胞。
重申一下,为了保证最大程度收获活细胞,每次吹打之前,可以加入少量DNA酶。
这招可以更容易收获大量高质量细胞,尤其是实验材料不足时候。
消化和吹打非常忌讳操作的细胞过多,这样反而会使大量细胞死亡。
实验者如果是用的皮层这种原材料充足的话,切忌不要放入过多皮层。
9(可选可不选,但是我建议全选)神经元进一步纯化。
一般来说,收获的细胞悬液已经是很纯的神经细胞了。
但是,由于实验者等个人或者客观原因,血管膜可能不会被完全剥离;杂细胞可能也被吹入了细胞悬液;操作者手法不熟练,造成死细胞过多,影响种板等等原因,还是会影响到培养的神经元的质量。
为了克服这些,笔者查了一些文献,经过反复测试采用了nycoprep低转速密度-梯度离心。
现在这个梯度溶液已经被公司升级为optiprep.稀释时候,NycoPrep™ 1.077 推荐稀释成60%,就用刚才悬浮细胞的培养液来配置。