巯基乙酸钠含量的测定
化妆品中巯基乙酸的检测方法

附件4:化妆品中巯基乙酸的检测方法1.适用范围本方法规定了采用液相色谱法测定化妆品中巯基乙酸(CAS:68-11-1)的方法。
本方法适用于头发烫卷剂或烫直剂、脱毛膏类化妆品中巯基乙酸的测定。
2.方法提要样品在经过提取后,经高效液相色谱仪分离,紫外检测器检测,根据保留时间定性,峰面积定量,以标准曲线法计算含量。
本方法对巯基乙酸的检出限为0.004μg,定量下限为0.015μg。
若取0.25g样品,本方法对巯基乙酸的检出浓度为35.6μg /g,最低定量浓度为118.7μg/g。
3.试剂和材料除另有规定外,所用试剂均为分析纯,水为一级实验用水。
3.1巯基乙酸,纯度≥99%。
3.2乙腈,色谱纯。
3.3 乙腈水溶液,乙腈+水(10+90)。
3.4 磷酸二氢钾(KH2PO4),色谱纯。
3.5 磷酸,优级纯。
3.6 巯基乙酸使用前需进行标定,标定方法参见2007年版《化妆品卫生规范》。
3.7 巯基乙酸标准储备液:取巯基乙酸0.05g,精确到0.0001g,置50 mL棕色容量瓶中,用乙腈水溶液(3.3)溶解并定容,摇匀,配成质量浓度为1 g/L的标准储备溶液。
常温保存,3日内稳定。
3.8 巯基乙酸标准工作溶液:配制浓度分别为5μg/mL、20μg/mL、50μg/mL、80μg/mL、110μg/mL和150μg/mL的巯基乙酸标准工作溶液。
4.仪器4.1高效液相色谱仪,配紫外检测器。
4.2涡旋振荡器。
4.3超声波清洗器。
4.4分析天平:感量0.0001g 。
5.测定步骤5.1 样品前处理称取样品0.25g ,精确至0.001g ,置于25mL 具塞刻度管中,加入20 mL 乙腈水溶液(3.3),涡旋使样品分散,超声(功率:400W )提取15 min ,取出,冷却至室温后用乙腈水溶液(3.3)定容,混匀,取上层液经0.45 μm 有机系滤膜过滤后用乙腈水溶液(3.3)稀释10倍,稀释液作为待测样液,备用。
硫酸铜中铜含量测定实验报告

硫酸铜中铜含量测定实验报告实验报告:硫酸铜中铜含量测定一、实验目的1.学习和掌握硫酸铜中铜含量的测定方法;2.通过实验操作掌握分光光度法的原理和操作技巧;3.培养实验操作的仔细、严密和精确。
二、实验原理硫酸铜溶液中的铜离子可以与巯基乙酸钠(又称为巯基乙酸钠盐)生成红色络合物,络合物的紫外吸收峰值为780nm。
按比例测量络合物溶液的吸光度,就可以计算出溶液中铜离子的浓度。
三、实验仪器和药品仪器:分光光度计、天平、移液器、烧杯、比色皿等;药品:硫酸铜、巯基乙酸钠、一定浓度的硫酸溶液。
四、实验步骤1.准备工作(1)将分光光度计预热10分钟;(2)准备一系列不同浓度的铜标准溶液,用硫酸铜和硫酸溶液配制;(3)按比例配制不同浓度的巯基乙酸钠溶液;(4)用硫酸溶液清洗烧杯、比色皿等仪器。
2.测定实验样品(1)取一定体积的硫酸铜溶液,转移到干净的烧杯中;(2)加入适量的硫酸溶液稀释;(3)加入适量的巯基乙酸钠溶液并充分搅拌;(4)加入去离子水稀释至标定容量;(5)取标准比色皿,清洗干净并标定容量。
3.浓度测定(1)设置分光光度计波长为780nm,进行零吸光度调零;(2)取一定体积的标准铜溶液,转移到标准比色皿中;(3)将标准比色皿放入分光光度计中,测量吸光度;(4)取实验样品溶液,按照上述操作进行测量吸光度。
五、结果与分析根据实验测量得到各浓度标准铜溶液的吸光度值,绘制吸光度与浓度之间的标准曲线。
根据实验样品测量的吸光度值,在标准曲线上找到相应的浓度,即为实验样品中铜的浓度。
六、误差分析1.实验仪器的误差:分光光度计的波长设置和调零操作的准确性会影响实验结果的准确性;2.实验药品的误差:标准铜溶液和巯基乙酸钠溶液的配制和稀释过程中的误差会影响实验结果的准确性;3.实验操作的误差:取样体积、加入试剂量、搅拌均匀程度等操作操作不准确都会影响实验结果的准确性。
七、实验结论通过本实验,我们成功地测定了硫酸铜中铜的含量。
海波法合成铜钼矿抑制剂巯基乙酸钠

Synthesis of the coppermolybdenum ore inhibitor sodium thioglycolate by sodium thiosulphate
CHEN Guobao,YANG Hongying,LING Xue
( School of Materials & Metallurgy,Northeastern University,Shenyang 110819 ,China) Abstract: The separation of copper and molybdenum from copper molybdenum ore is a challenge in mineral processing. Sodium thioglycolate is a preferable chalcopyrite inhibitor in molybdenite dressing due to its high efficiency, low toxicity and environmental friendly. The synthesis of sodium thioglycolate is studied by sodium thiosulphate in this study. The optimal synthesis conditions are as follows: 7. 5 of pH, 70℃ of temperature of condensation reaction and 20℃ of thiolation reaction temperature. The yield of sodium thioglycolate can reach 96. 56% . The chemical structure of obtained products is confirmed by infrared spectrum. The flotation of coppermolybdenum ore are conducted using the obtained products as inhibitor. The results show that, when the dosage of sodium thioglycolate is 0. 5 kg / t, the concentration of molybdenum increases from 0. 62% to 3. 21% and the recovery rate of molybdenum reached 80. 71% . When the dosage of sodium thioglycolate is 2. 5 kg / t, the recovery rate of molybdenum and concentration of molybdenum respectively. It suggests that the homemade sodium thioglycolate possesses high inhibiting is 91. 92% and 5. 56% , ability towards copper molybdenum ore. Key words: sodium thioglycolate; synthetic; separation process of copper and molybdenum; inhibitor
巯基乙酸-中国食品药品检定研究院

附件1化妆品中巯基乙酸的检测方法(征求意见稿)1 范围本方法规定了离子色谱法测定化妆品中巯基乙酸的含量。
本方法适用于化妆品中巯基乙酸及其盐类和酯类含量的测定。
2 方法提要样品中的巯基乙酸经水溶解提取后,用离子色谱仪分离巯基乙酸根与无机离子,电导检测器检测,以保留时间定性,峰面积定量。
本方法巯基乙酸的检出限5.8ng,定量下限20ng。
取样量为0.5g时,检出浓度为46μg/g,最低定量浓度0.15mg/g。
3 试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T6682规定的一级水。
3.1 巯基乙酸,优级纯。
3.2 甲醇,优级纯。
3.3 三氯甲烷,分析纯。
3.4 氢氧化钠,分析纯。
3.5 硫酸溶液[φ(H2SO4)=10%]:取硫酸(ρ20=1.84g/ml)10mL,缓慢加入到90mL水中,混匀。
3.6 盐酸溶液[φ(HCl)=10%]:取盐酸(ρ20=1.19g/ml)10mL,加入90mL水中,混匀。
3.7 淀粉溶液(10g/L):称可溶性淀粉1g,加水5mL调成溶液,再加入沸水95mL,煮沸,并加水杨酸0.1g或氯化锌0.4g防腐。
3.8 氢氧化钠溶液(500g/L):称取氢氧化钠50g,加水适量使溶解并至100mL。
再量取一定量用经超声脱气的水稀释到淋洗液浓度。
3.9 重铬酸钾标准溶液[c(1/6K2Cr2O7)=0.1000mol/L]:准确称取已于120℃±2℃电烘箱中干燥至恒重的重铬酸钾基准物质4.9031g ,溶于水并转移至1000mL 量瓶中,定容至刻度,摇匀。
3.10 硫代硫酸钠标准溶液(0.1mol/L):称取硫代硫酸钠(Na 2S 2O 3▪5H 2O )26g (或无水硫代硫酸钠16g )溶于1000mL 新煮沸放冷的水中,加入氢氧化钠0.4g 或无水碳酸钠0.2g ,摇匀,贮存于棕色瓶内,放置两周后过滤,用重铬酸钾标准溶液标定其浓度,标定方法如下:准确吸取重铬酸钾标准溶液(3.9)25.00mL 于500mL 碘量瓶中,加碘化钾2.0g 和硫酸溶液(10%)20mL ,立即密塞,摇匀,于暗处放置10min 。
固相测定法测定巯基含量

固相测定法测定巯基含量
固相测定法是一种常用的测定巯基含量的方法。
巯基是一种含有硫原子的官能团,常见于半胱氨酸和囊氨酸等氨基酸中。
测定巯基含量在生物化学和药物化学中具有重要意义,因为巯基参与了许多生物活性分子的结构和功能。
固相测定法通常包括以下步骤:
1. 样品制备,首先需要将待测样品进行适当的制备处理,通常包括提取、纯化和去除干扰物质等步骤。
2. 衍生化反应,巯基常常需要进行衍生化反应,以便形成可以在固相柱上进行分离和测定的化合物。
常用的衍生化试剂包括二硫代硫酸酯和氯乙酰氯等。
3. 固相柱层析,经过衍生化反应后的样品通常需要通过固相柱层析来分离目标化合物和其他杂质。
固相柱层析是利用固定在柱内的填料对化合物进行分离的方法。
4. 检测方法,固相柱层析后,巯基衍生物可以通过吸收检测器
或荧光检测器等进行定量分析。
这些检测方法可以测定巯基含量,并且通常具有较高的灵敏度和选择性。
在实际操作中,需要根据样品的性质和巯基含量的预期范围选择合适的固相柱、衍生化试剂和检测方法。
此外,还需要注意样品制备和操作过程中的干扰物质的去除和控制,以确保测定结果的准确性和可靠性。
总的来说,固相测定法是一种可靠且常用的测定巯基含量的方法,通过合理的样品制备、衍生化反应、固相柱层析和检测方法选择,可以准确地测定样品中巯基的含量。
巯基乙酸钠分光光度法测量水中的铁含量

巯基乙酸钠分光光度法测量水中的铁含量
徐洁鸿;吴丹虹;应琪;任旭滨;王羽
【期刊名称】《化工设计通讯》
【年(卷),期】2022(48)6
【摘要】重点研究了使用巯基乙酸钠作为显色剂,使用分光光度法测定水中铁的含量。
在室温条件下氨水调节pH为9.4~10.8,同时采用酒石酸来屏蔽干扰元素,铁离子与巯基乙酸钠产生紫红色的络合物,在UV测试中530nm处有最大的吸光度。
为此创建了铁离子浓度为(0.5~10.0)×10^(-6)的曲线,该曲线符合朗博比尔定律且有较好的线性。
【总页数】3页(P112-114)
【作者】徐洁鸿;吴丹虹;应琪;任旭滨;王羽
【作者单位】宁波市中普检测技术服务有限公司
【正文语种】中文
【中图分类】O657
【相关文献】
1.普鲁士蓝分光光度法测定饮用水中总铁的含量
2.巯基棉富集—火焰原子吸收分光光度法测定水中的微量铁
3.硫氰酸盐分光光度法测定煤层气排采水中r铁含量的不确定度评定
4.分光光度法测定水中总铁含量的探讨
5.关于二氮杂菲分光光度法检测生活饮用水和水源水中的铁含量
因版权原因,仅展示原文概要,查看原文内容请购买。
化学冷烫液中巯基乙酸含量的测定

化学冷烫液中巯基乙酸含量的测定(碘量法)√一、实验原理HI I COOH HSCH COOH SCH COOHSCH 22|2222+→+二、仪器及试剂1、仪器:容量瓶、碘量瓶、量筒、移液管、电炉、碱式滴定管、烧杯 2、试剂:30%、0.5%淀粉指示剂、0.05mol/L 碘液、0.1mol/L 标液、、3mol/L 、KI 固体、试样:化学冷烫液。
三、实验步骤1、0.1mol/L 硫代硫酸钠标液的配制与标定:配制:称取 12.5g 硫代硫酸钠和0.1g 碳酸钠溶于500ml 水中。
标定:准确称取3份重铬酸钾,每份约0.13~0.15g ,分别置于500ml 碘量瓶中,加蒸馏水25ml ,溶解后,取其中一个碘量瓶加3mol/L 硫酸15ml 和KI 固体2g ,混匀后盖上瓶塞,瓶口封少量蒸馏水,在暗处静置5min,然后加150ml 蒸馏水,立即用0.1mol/L 硫代硫酸钠标液滴定至浅黄绿色,加入3ml0.5%淀粉指示剂,继续滴定至溶液由黄色变为亮绿色,即为终点。
计算公式如下:3227227223226O S Na O Cr k O Cr k O S Na V M m c ⨯=式中—722O Cr k M =294.19g/mol 。
2、1/2I 2溶液的配制与标定:标定:量取35.00~40.00ml 配置好的碘溶液置于碘量瓶中,加150ml 蒸馏水,用硫代硫酸钠标液滴定,近终点时加2ml 淀粉指示剂,继续滴定至蓝色消失。
计算公式:23223222I O S Na O S Na I V V c c =3、样品测定:取冷烫液样品2.00ml 于锥形瓶中,加入2滴甲基红,用1:1盐酸调节PH 至溶液呈红色,加入2滴淀粉指示剂,用碘标液滴至溶液便为蓝褐色及为终点。
计算公式如下:00.2mg/ml 22巯基乙酸)巯基乙酸(M V c I I式中:巯基乙酸M=92.11,g/mol 。
四、实验记录及数据处理1、硫代硫酸钠标液的标定:相对平均极差为:0.045%。
化妆品中巯基乙酸等8种原料的检验方法2023年
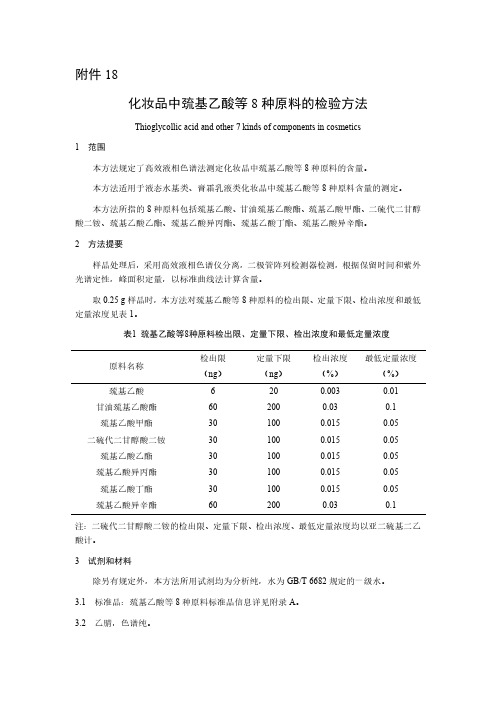
附件18化妆品中巯基乙酸等8种原料的检验方法Thioglycollic acid and other7kinds of components in cosmetics1范围本方法规定了高效液相色谱法测定化妆品中巯基乙酸等8种原料的含量。
本方法适用于液态水基类、膏霜乳液类化妆品中巯基乙酸等8种原料含量的测定。
本方法所指的8种原料包括巯基乙酸、甘油巯基乙酸酯、巯基乙酸甲酯、二硫代二甘醇酸二铵、巯基乙酸乙酯、巯基乙酸异丙酯、巯基乙酸丁酯、巯基乙酸异辛酯。
2方法提要样品处理后,采用高效液相色谱仪分离,二极管阵列检测器检测,根据保留时间和紫外光谱定性,峰面积定量,以标准曲线法计算含量。
取0.25g样品时,本方法对巯基乙酸等8种原料的检出限、定量下限、检出浓度和最低定量浓度见表1。
表1巯基乙酸等8种原料检出限、定量下限、检出浓度和最低定量浓度原料名称检出限(ng)定量下限(ng)检出浓度(%)最低定量浓度(%)巯基乙酸6200.0030.01甘油巯基乙酸酯602000.030.1巯基乙酸甲酯301000.0150.05二硫代二甘醇酸二铵301000.0150.05巯基乙酸乙酯301000.0150.05巯基乙酸异丙酯301000.0150.05巯基乙酸丁酯301000.0150.05巯基乙酸异辛酯602000.030.1注:二硫代二甘醇酸二铵的检出限、定量下限、检出浓度、最低定量浓度均以亚二硫基二乙酸计。
3试剂和材料除另有规定外,本方法所用试剂均为分析纯,水为GB/T6682规定的一级水。
3.1标准品:巯基乙酸等8种原料标准品信息详见附录A。
3.2乙腈,色谱纯。
3.3磷酸,优级纯。
3.4磷酸溶液(0.1%):取磷酸(3.3)1mL,加水至1000mL,混匀。
3.5磷酸溶液(0.05%):取磷酸(3.3)0.5mL,加水至1000mL,混匀。
3.6含0.05%磷酸的乙腈溶液:取磷酸(3.3)0.5mL,加乙腈(3.2)至1000mL,混匀。
巯基乙酸异辛酯纯度分析方法比较

巯基乙酸异辛酯纯度分析方法比较王爱红;李建丰;李继文;陈雍;龙守奎【摘要】通过比较化学分析法(一)、化学分析法(二)、气相色谱分析法分析巯基乙酸异辛酯纯度的检测结果表明,化学分析方法(一)与以上两种方法检测结果绝对误差在0.20%~0.35%,误差较大,容易造成检验误判;化学分析方法(二)与气相色谱检测结果绝对误差0.02%以内,误差小,分析方法(二)与气相色谱可以作为巯基乙酸异辛酯进厂检测方法,供业内相关厂家参考使用.【期刊名称】《塑料助剂》【年(卷),期】2019(000)001【总页数】3页(P39-41)【关键词】巯基乙酸异辛酯;纯度;分析;方法比较【作者】王爱红;李建丰;李继文;陈雍;龙守奎【作者单位】湖北南星化工总厂,宜昌,443200;湖北南星化工总厂,宜昌,443200;湖北南星化工总厂,宜昌,443200;湖北南星化工总厂,宜昌,443200;湖北南星化工总厂,宜昌,443200【正文语种】中文聚氯乙烯热稳定剂硫醇甲基锡主要原材料巯基乙酸异辛酯没有行业和国家标准,巯基乙酸异辛酯的纯度分析方法可以采用化学分析法(一):其原理是利用直接碘量法测定巯基乙酸中总巯基,同时利用碘量法测定其中游离的巯基乙酸,然后经过计算得到巯基乙酸异辛酯的纯度。
也可以采用化学分析方法(二):其原理是利用直接碘量法测定巯基乙酸中总巯基,同时利用酸碱测定法测定其中游离的巯基乙酸,然后经过计算得到巯基乙酸异辛酯的纯度。
也可以采用气相色谱法进行分析检测,检测器为氢火焰离子化检测器,通过色谱数据处理工作站,直观得到巯基乙酸异辛酯纯度。
本文采用三种检测方法对巯基乙酸异辛酯的纯度进行分析,分析结果表明,化学分析方法(一)与气相色谱方法的绝对误差范围在0.20%~0.35%,误差较大。
容易造成误判;化学方法(二)与气相色谱方法分析的结果误差较小,绝对误差范围在0.00%~0.02%以内,这两种方法均可作为巯基乙酸异辛酯进厂检测方法。
巯基乙酸钠含量的测定

巯基乙酸钠含量的测定1、主题内容与适用范围本主题的内容是用碘酸钾与碘化钾氧化法测定巯基乙酸含量的方法,适用于分析生产过程中含巯基乙酸(或其盐类)的各种物料,如尾液、酸化液、除臭液、萃取尾液以及各种含巯基乙酸的产品,分析结果均以巯基乙酸百分含量表示。
2、原理碘酸钾在弱酸性溶液中与碘化钾作用,析出的碘氧化巯基乙酸成二硫撑酯酸,反应式如下:KIO3 + 5KI+ 3H2SO4 →3I2 + 3K2SO4 + 3H2O2HSCH2COOH + I2 →HOOCCH2SSCH2COOH + 2HI3、试剂及标准溶液3.1、硫酸溶液,1+1将100 ml 浓硫酸慢慢地加入100 ml 水中,冷却后使用。
3.2、硫酸溶液,20%量取128 ml 浓硫酸,慢慢地加入约700 ml 水中,冷却稀释到1000 ml。
3.3、碘化钾3.4、淀粉指示剂,5 g/L称取0.5g淀粉,加入5ml 水使成糊状。
在搅拌下将糊状物加到90ml沸腾的水中,煮沸1-2分钟,冷却稀释到100 ml。
3.5、硫代硫酸钠标准溶液,C(NaS2O3)-0.1mol/L3.5.1 配制:称取26g硫代硫酸钠,溶于1000ml水中,缓缓煮沸10分钟。
冷却放置两周后过滤备用。
3.5.2 标定:称取0.15g于120℃烘至恒重的基准试剂重铬酸钾,称准至0.001g,置于500ml碘量瓶中,溶于25ml水中,加2g 碘化钾及20 ml 硫酸溶液(20%),摇匀,于暗处放置10分钟加150ml水,用配好的硫代硫酸钠溶液滴定,近终点时加3ml淀粉指示剂,继续滴定至溶液由蓝色变为亮绿色,同时作空白试验。
3.5.3 计算硫代硫酸钠标准溶液浓度按下式计算:C(NaS2O3)= m/(V1-V2)*0.04903式中: V1---硫代硫酸钠溶液之用量,mlV2---空白试验硫代硫酸钠溶液之用量,mlC(NaS2O3)---硫代硫酸钠标准溶液之物之的量浓度,mol/Lm---称取重铬酸钾的量,g0.04903---重铬酸钾,1/6 重铬酸钾之毫摩尔质量,g/m mol3.5.4 标定允差,平行测定不得少于四次,四次平行测定值的极差不大于0.0001 mol/L时取平均值。
盐溶液Ph值对巯基乙酸钠生产品位的影响

3 原 因 分 析
将 氯乙酸和碳酸钠通过 中和反应制成 盐溶液 , 分别控制其 P h值 为 6 ~7 、7 ~7 . 5和 8 ~9 ,其它条件不变 ,进行实验 ,并测 定每个 实验 中产生 的硫单质 的含量。实验结果分别见表 l 、表 2和表 3 。
4 .1 9 4 .0 3 3 .6 8 4 .2 1 4 .1 1
表 3在 P h值为 8 ~9 时巯基 乙酸钠和硫单质百分含量
从 实验结果表 3可以明显看 出:盐溶液 P b值增大 ,巯基 乙酸钠 的平均生产 品位 下降幅度增大,而且品位波动较大 ,硫单质含 量增 大 ,产 品质量下 降,生产成本上升 。 通过盐溶液 P h 值变化对整个反应 过程影 响的理 论分析 发现: ( 1 )如果制盐 时 P h值过 小,则会发 生如下副反应 :
品
位( %)
7. 2 3 6. 7 7 6. 91
硫 单质含 量 ( %)
3 . 9 4 4 .3 7 4 .1 4
编 号
l 1 l 2 l 3
品 位
( % )
7 .7 4 7 .4 3 7 .0 1
硫单 质含 量
( % )
科 技 论坛
盐溶液 P h 值对巯基 乙酸钠生产陕西华光实业有 限责任公 司,陕西 华县 7 1 4 1 0 2)
【 摘 要】 盐溶 液 P h 值 对巯基 乙酸钠 的生产品位影响较大。 通
编 号
1 2 3
过实验研究发现 , 如 果控 制盐溶液的 P h值 为 7 ~7 . 5 ,不仅能提 高巯 基 乙酸钠的生产品位 ,而且 能大量减少产品 中的硫 单质 ,进一步提 高产品的质量。
巯基乙酸生产工艺研究

巯基乙酸生产工艺研究刘新胜【摘要】以氯乙酸与硫脲为原料,采用硫脲法合成了巯基乙酸,讨论了物料配比、反应温度、反应时间、水解反应中NaOH和BaCl2的用量、水解时间等因素对收率的影响,确定了最佳反应条件.结果表明氯乙酸与硫脲物料配比为1:1,反应温度为80℃,反应时间为1 h,水解反应中分别加入理论计量的NaOH和BaCl2,反应时间为45 min时,能够获得较好的收率.【期刊名称】《广州化工》【年(卷),期】2016(044)024【总页数】3页(P68-70)【关键词】巯基乙酸;合成工艺;硫脲【作者】刘新胜【作者单位】宁夏医科大学基础医学院,宁夏银川 750004【正文语种】中文【中图分类】O657.7+1;O657.63巯基乙酸(SHCH2COOH)又名硫代乙醇酸、氢硫基乙酸(简称TGA),具有广泛的用途。
由巯基乙酸制得的巯基乙酸单甘油酯或单乙醇胺酯可作为有效的头发卷发剂[1]。
巯基乙酸酯作为聚氯乙烯树脂加工时的稳定剂[2-4],具有低毒或无毒性,能使产品的透明性和热稳定性得到提高,广泛用于聚氯乙烯上水管道、水泵等产品的加工、食品包装、食品加工厂的设备管道等。
巯基乙酸可与多种金属离子形成络合物[5-6],显示出特殊颜色,常被用作分光光度分析试剂。
此外,巯基乙酸还可用作聚合反应催化剂[7]、金属精密加工中的防锈剂、聚丙烯加工成型时的结晶成核剂、涂料及纤维的改性剂、石油勘探缓蚀剂、润滑油添加剂等[8]。
随着工业需求的扩大,为了提高巯基乙酸的收率,探讨巯基乙酸的合成方法及生产工艺条件具有重要的意义。
本文采用硫脲法合成了巯基乙酸,对实验最佳条件进行了探讨,并对实验结果进行了分析与讨论。
1.1 实验所用主要仪器、药品及试剂1.1.1 实验仪器723型可见分光光度计(1 cm玻璃比色皿),上海精密科学仪器有限公司;电热鼓风干燥箱,上海福玛实验设备有限公司;数显恒温水浴锅,常州国华电器有限公司;电子精密天平(0.001 g),梅特勒—托利多仪器(上海)有限公司。
浅淡巯基乙酸钠的生产及在选矿中的应用

内蒙某铜钼 矿铜钼分 离进行抑制 剂试验 , 用我 公司生产 的新型 HB铜 抑制剂对 该矿进行 铜钼分 离试验 , 并 且进行 了 实验 室 闭路 试 验 , 验 证 抑制 剂效 果 ,开路 试验 流 程 如 图 1 , 试验结 果见表 1 。
i n hi bi t or of s od i um c ya ni d e p oi s on,H y pe r i o n me t hod f or t he pr od uc t i o n o f s odi um t hi og l y c o l a t e ma i nl y s a l t pr oc e s s , c onde ns a t i o n pr oc e s s ,c he mi c a 1 or de r s e ve r a l s t e ps s uc h a s t hi o l ,a dd i ng ne w e x t r a ct i on a nd i m pur i t y r e m ova l a nd c onc e nt r a t i on pr oc e s s ,e ns u r e t he q ua l i t y of t he p r od uc t .Af te r a l a r g e numbe r of l a bo r a t o r y t e s t s a nd i n d us t r i a l p r od uc t i o n t e s t , c a n a c hi e v e be t t e r be n e ic f i a t i on r e s u l t s .
ห้องสมุดไป่ตู้
Ab s t r a c t : T h e u s e o f s o d i u m t h i o g l y c o l a t e i s c h a r a c t e r i s t i c o f D O p o l l u t i o n , h i g h e ic f i e n c y , a n d g r a d u a l l y r e p l a c e d t h e
巯基乙酸

附件1化妆品中巯基乙酸的检测方法(征求意见稿)1 范围本方法规定了离子色谱法测定化妆品中巯基乙酸的含量。
本方法适用于化妆品中巯基乙酸及其盐类和酯类含量的测定。
2 方法提要样品中的巯基乙酸经水溶解提取后,用离子色谱仪分离巯基乙酸根与无机离子,电导检测器检测,以保留时间定性,峰面积定量。
本方法巯基乙酸的检出限5.8ng,定量下限20ng。
取样量为0.5g时,检出浓度为46μg/g,最低定量浓度0.15mg/g。
3 试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T6682规定的一级水。
3.1 巯基乙酸,优级纯。
3.2 甲醇,优级纯。
3.3 三氯甲烷,分析纯。
3.4 氢氧化钠,分析纯。
3.5 硫酸溶液[φ(H2SO4)=10%]:取硫酸(ρ20=1.84g/ml)10mL,缓慢加入到90mL水中,混匀。
3.6 盐酸溶液[φ(HCl)=10%]:取盐酸(ρ20=1.19g/ml)10mL,加入90mL水中,混匀。
3.7 淀粉溶液(10g/L):称可溶性淀粉1g,加水5mL调成溶液,再加入沸水95mL,煮沸,并加水杨酸0.1g或氯化锌0.4g防腐。
3.8 氢氧化钠溶液(500g/L):称取氢氧化钠50g,加水适量使溶解并至100mL。
再量取一定量用经超声脱气的水稀释到淋洗液浓度。
3.9 重铬酸钾标准溶液[c(1/6K2Cr2O7)=0.1000mol/L]:准确称取已于120℃±2℃电烘箱中干燥至恒重的重铬酸钾基准物质4.9031g ,溶于水并转移至1000mL 量瓶中,定容至刻度,摇匀。
3.10 硫代硫酸钠标准溶液(0.1mol/L):称取硫代硫酸钠(Na 2S 2O 3▪5H 2O )26g (或无水硫代硫酸钠16g )溶于1000mL 新煮沸放冷的水中,加入氢氧化钠0.4g 或无水碳酸钠0.2g ,摇匀,贮存于棕色瓶内,放置两周后过滤,用重铬酸钾标准溶液标定其浓度,标定方法如下:准确吸取重铬酸钾标准溶液(3.9)25.00mL 于500mL 碘量瓶中,加碘化钾2.0g 和硫酸溶液(10%)20mL ,立即密塞,摇匀,于暗处放置10min 。
巯基乙酸钠

巯基乙酸钠
巯基乙酸钠(Sodium Thioglycolate),也叫做硫代乙醇酸钠。
是一种有机金属盐。
在铜钼矿浮选中,用作铜矿和镁铁矿的抑制剂,也用作分析试剂或化学冷烫液的配制。
性能
白色粉末状结晶,有时略显浅粉色,非易燃、易爆品,有潮解性,易溶于水,微溶于醇,避光密闭保存。
用途
它在铜钼矿浮选中,用作铜矿物和硫铁矿的抑制剂。
采用先进的技术以及优良的工艺精制而成,对铜,硫以及其他物质有明显的抑制作用,从而有效地提高了钼精矿的品位。
作为一种新型的硫化矿的有效抑制剂在选钼生产中已经成功应用多年,可完全替代剧毒抑制剂氰化钠。
主要的是在选钼过程中本产品不仅抑制了铅、锌、铁、铜等金属杂质,而且还对硅、硫等非金属物质的降低也起到了很好的作用。
该药剂使用剂量少,用法简单方便,能更好的节约成本,增加经济效益。
不仅提高了产品的质量,而且无污染、无毒害、对生产区域的环境保护起到了积极的作用。
是国家环保部门积极推荐的环保型无污染产品。
巯基乙酸钠106691_SDS_CN_ZH

登陆来获取目录中产品的安全数据单。
页码 2 的 8
化学品安全技术说明书 根据欧盟(EC) No. 1907/2006的法规
物品编号 物品名称
106691 巯基乙酸钠 for microbiology
燃烧性材料 起火时可能引发产生危害性气体或蒸气. 着火可能引起生成出: 硫氧化物, 硫化氢
图形符号
Xn
有害的
R - 类 警示句 S-类 警示句 EC-编号
22-43 24-37 206-696-4
误吞对人体有害。 接触皮肤可引起过敏。 避免沾及皮肤。 戴好适当的手套。
2.3 其它危险 未见报道。
部分 三 成分/组成资料 分子式 化学文摘编号(CAS No.) EC-编号 分子量
C₂H₃NaO₂S (Hill) 367-51-1 206-696-4 114.1 g/mol
大约200 kg/m³
部分 十 稳定性和反应活性 10.1 反应性 一般而言, 以下应用及准备时, 易燃有机性物质与粉尘混合, 可能被认定有爆炸的潜在危险.
10.2 化学稳定性 对光敏感 对空气敏感。
10.3 危险反应的可能性 可能与之发生剧烈反应: 强氧化剂, 强酸
登陆来获取目录中产品的安全数据单。
物品编号 microbiology
个人的防护措施 需依照工作环境的情况与危险物质的浓度与数量选择适当的防护衣物. 防护衣物对于化学物质的抗化测试表可向其供货商索取.
卫生措施 立即更换受污染衣物. 使用皮肤保护乳液. 使用此物质后须洗手及洗脸. 在通风橱下操作. 避免吸入该物质.
对紧急情况处理人员的建议: 防护装备见第8部分。
6.2 环境预防措施 禁止直接排入下水道。
巯基乙酸钠的合成

巯基乙酸钠的合成
一、引言
巯基乙酸钠是一种重要的有机化合物,具有良好的螯合性能和生物活性,广泛应用于医药、农业、环境保护等领域。
本文将介绍巯基乙酸钠的合成方法及其反应机理。
二、巯基乙酸钠的合成方法
1. 巯基乙醇与碳酸氢钠反应法
将巯基乙醇与碳酸氢钠在水中反应,得到巯基乙酸钠:
C2H5SH + NaHCO3 → C2H5SNaO2 + CO2 + H2O
该方法简单易行,但产率较低。
2. 巯代硫酸钠与氢氧化钠反应法
将巯代硫酸钠与氢氧化钠在水中反应,得到巯基乙酸钠:
NaOSO3Na + NaOH → C2H5SNaO2 + Na2SO4
该方法产率较高,但需要使用较多的试剂和设备。
3. 巴比妥与硫代乙醇反应法
将巴比妥和硫代乙醇在碱性条件下反应,得到差不多纯度的巯基乙酸钠:
C4H6N2O3 + C2H5SH → C2H5SNaO2 + C4H5N2O3
该方法产率较高,但需要使用较多的试剂和设备。
三、巯基乙酸钠的反应机理
巯基乙酸钠是一种螯合剂,其主要反应机理为配位作用。
巯基乙酸钠中的硫原子可以与金属离子形成配合物,从而改变金属离子的性质和活性。
四、结论
巯基乙酸钠是一种重要的有机化合物,具有广泛的应用前景。
其合成方法主要包括巯基乙醇与碳酸氢钠反应法、巯代硫酸钠与氢氧化钠反
应法和巴比妥与硫代乙醇反应法。
其中,后两种方法产率较高,但需要使用较多的试剂和设备。
其主要反应机理为配位作用。
巯基乙酸甲酯含量的测定

巯基乙酸甲酯含量的测定刘 燕(贵州省化工研究院,贵阳,550002) 摘 要 本文介绍了巯基乙酸甲酯测定的原理及方法,并对其精密度及准确度进行了实验。
关键词 巯基乙酸甲酯 巯基 碘 中图分类号 TQ651121 引 言 巯基乙酸甲酯是一种合成医药、烟用香料和食品香料的重要中间体原料。
贵州省化工研究院易生精细化工有限公司已成功合成了该产品,并用其进一步合成香料原料:3—羰基—2—甲氢噻吩甲酸甲酯和4—羰基—2—四氢噻吩甲酸甲酯。
该公司研究的工艺路线是国内外唯一的,由于其工艺的特殊性,要求对产品每一步的巯基乙酸甲酯含量进行准确的测定,因此笔者对巯基乙酸甲酯含量的测定作了大量的实验,确定了其含量测定的分析方法,对工艺路线的确定起了重要的作用。
2 原理及分析方法211 原理 巯基乙酸甲酯为无色透明液体,不溶于水,易溶于醇、醚等有机溶剂中。
由于硫比碳容易被氧化得多,而且硫氢键又比氧氢键容易断裂,因此,巯基远比羰基容易被氧化,它极易被氧化成二硫化合物,并且无论有无催化剂存在,于低温下用空气即可把它氧化成二硫化合物。
我们利用碘易把它氧化成二硫化合物的特性来对其进行测定。
HSCH2COOCH3+I2CH3OH・H2OCH3OOCCH2S-SCH2COOCH3+2HI该反应是先生成CH3OOCCH2S-游离基,再结合成二硫化合物。
由于反应的完全性,可根据碘的消耗量,测出其巯基乙酸甲酯的含量。
212 分析方法21211 C(12I2)=011mol/L碘标准溶液配制及标定2121111 配制:称取13g碘及35g碘化钾,溶于100ml水中,稀释至1000ml,摇匀,保存于棕色具塞瓶中。
2121112 标定:称取0115g预先在硫酸干燥器至恒重的基准三氧化二砷,称准至010002g,置于碘量瓶中,加4ml1M氢氧化钠溶液[C(NaOH)=1mol/L],加50ml水,加2滴1%酚酞指示液,用1M硫酸[C(12H2SO4)=1mol/L]中和,加3g碳酸氢钠及3ml015%淀粉指示液,用0.1M碘溶液[C(12I2)=0.1mol/L]滴定至溶液呈浅蓝色。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
巯基乙酸钠含量的测定
1、主题内容与适用范围
本主题的内容是用碘酸钾与碘化钾氧化法测定巯基乙酸含量的方法,适用于分析生产过程中含巯基乙酸(或其盐类)的各种物料,如尾液、酸化液、除臭液、萃取尾液以及各种含巯基乙酸的产品,分析结果均以巯基乙酸百分含量表示。
2、原理
碘酸钾在弱酸性溶液中与碘化钾作用,析出的碘氧化巯基乙酸成二硫撑酯酸,反应式如下:
KIO3 + 5KI+ 3H2SO4 →3I2 + 3K2SO4 + 3H2O
2HSCH2COOH + I2 →HOOCCH2SSCH2COOH + 2HI
3、试剂及标准溶液
3.1、硫酸溶液,1+1
将100 ml 浓硫酸慢慢地加入100 ml 水中,冷却后使用。
3.2、硫酸溶液,20%
量取128 ml 浓硫酸,慢慢地加入约700 ml 水中,冷却稀释到1000 ml。
3.3、碘化钾
3.4、淀粉指示剂,5 g/L
称取0.5g淀粉,加入5ml 水使成糊状。
在搅拌下将糊状物加到90ml沸腾的水中,煮沸1-2分钟,冷却稀释到100 ml。
3.5、硫代硫酸钠标准溶液,C(NaS2O3)-0.1mol/L
3.5.1 配制:
称取26g硫代硫酸钠,溶于1000ml水中,缓缓煮沸10分钟。
冷却放置两周后过滤备用。
3.5.2 标定:
称取0.15g于120℃烘至恒重的基准试剂重铬酸钾,称准至0.001g,置于500ml碘量瓶中,溶于25ml水中,加2g 碘化钾及20 ml 硫酸溶液(20%),摇匀,于暗处放置10分钟加150ml水,用配好的硫代硫酸钠溶液滴定,近终点时加3ml淀粉指示剂,继续滴定至溶液由蓝色变为亮绿色,同时作空白试验。
3.5.3 计算
硫代硫酸钠标准溶液浓度按下式计算:
C(NaS
2O
3
)= m/(V1-V2)*0.04903
式中: V1---硫代硫酸钠溶液之用量,ml
V2---空白试验硫代硫酸钠溶液之用量,ml
C(NaS
2O
3
)---硫代硫酸钠标准溶液之物之的量浓度,mol/L
m---称取重铬酸钾的量,g
0.04903---重铬酸钾,1/6 重铬酸钾之毫摩尔质量,g/m mol
3.5.4 标定允差,平行测定不得少于四次,四次平行测定值的极差不大于0.0001 mol/L时取平均值。
注:也可以准确称取5g烘至恒重的重铬酸钾溶于1000ml容量瓶中,在此准确量取30ml重铬酸钾标准溶液用以标定硫代硫酸钠溶液。
3.6 碘酸钾标准溶液,C(1/6KIO
)=0.1mol/L
3
3.6.1配制
称取3.5669g碘酸钾,溶于100ml水中,并用水稀释至1000ml,混匀。
3.6.2 标定
准确量取30ml已配好的碘酸钾标准溶液三份,分别放入300 ml烧杯中,各加水50毫升,碘化钾2克,加1:1盐酸5毫升,摇匀,于暗处放置5分钟,以硫代硫酸钠标准溶液滴定至淡黄色,然后加0.5%淀粉溶液5毫升,继续滴定至蓝色消失,即为终点,同时作空白试验。
3.6.3 计算
碘酸钾标准溶液的浓度按下式计算:
)=(V1-V2)*C1/V
C(1/6KIO
3
式中:C(1/6KIO
)--- 碘酸钾标准溶液之物质量浓度
3
C1---硫代硫酸钠标准溶液之物质量浓度;
V1---硫代硫酸钠标准溶液之用量,ml;
V2---空白试验硫代硫酸钠标准溶液之用量,ml;
V----量取的碘酸钾溶液体积,ml。
3.6.4 标定允差
两次平行结果相差不大于0.5时,取平均值报告测定结果。
)=0.02mol/L
3.7碘酸钾标准溶液,C(1/6KIO
3
准确量取0.1mol/L的碘酸钾标准溶液200.00ml,置于1000ml容量瓶中,用煮沸又冷却的水稀释至刻度,摇匀。
4、操作手续
于300 ml烧杯中加100 ml水,用注射器称取试样0.3g,准确至0.0001g,加入烧杯中使溶于水,加2 ml硫酸溶液(1+1),及1g碘化钾,溶解后加1ml 淀粉指示剂,用碘酸钾标准溶液(除分析萃取尾液用0.02mol/L溶液外,分析其它物料均使用0.1mol/L溶液)滴定至蓝色为终点。
5、计算
用巯基乙酸表示的百分含量X按下式计算:
X=C×V×92.1×100/W×1000
式中: C---碘酸钾标准溶液之物质的量浓度,mol/L;
V---碘酸钾标准溶液之用量,ml;
W---称取试样的量,g;
92.1---巯基乙酸之摩尔质量,g/mol.。