ICH关于遗传毒性体外、体内试验的建议--ICHS2(R1)人用药物遗传毒性试验和结果分析指导原则介绍(二)
遗传毒性试验

遗传毒性试验能检出DNA损伤及其损伤的固定。
根据检测的遗传学终点分为4种类型:1检测基因突变(比如:Ames试验);2检测染色体畸变(比如:微核试验);3检测染色体组畸变(比如:体外CHL细胞染色体畸变、精原细胞染色体畸变试验);4检测DNA原始损伤(比如:单细胞凝胶电泳分析(singlecellgeleletrophoresis,SCGE))。
以上检测结果为呈阳性(除外假阳性)的化合物,为潜在人类致癌剂和/或致突变的物质。
FDA于2006年制定了遗传毒性试验结果的综合分析法指导原则(Guidanceforindustyandreviewstaff:Recommendedapproachestointegrationofgenetict oxicologystudyresults)对遗传毒性试验的阳性结果评价和处理:ICHS2(R1)中的遗传毒性结果评价和追加试验策略。
目前已建立的遗传毒性短期检测法已超过200种。
1 现行组合试验方案,用一组试验配套进行试验。
200多种检测方法中,真正经过验证有合适灵敏度和特异度的大概不到10种。
目前多数国家规定,如体内诱变试验显示1个或以上试验呈阳性结果,则需要进行生殖细胞遗传毒性测试。
2 各类遗传毒性试验方法的研究进展2.1 检测基因突变2.1.1 Ames试验Ames试验是检测化学物质基因突变的常用方法。
常规的Ames试验选用四个测试菌株(TA97、TA98、TA100、TA102),最近有人提出增加TA1535测试菌株,该菌株特别适用于检测混合物的致突变性。
目前出现的新生菌株具有更高的敏感性和特异性,如YG7014、TG7108,缺乏编码O6-甲基鸟嘌呤DNA甲基化转移酶的ogtST基因,专用于对烷化剂引起的DNA损伤检测;引入乙酰转移酶基因的YG1024、YG1029菌株,对硝基芳烃和芳香胺的敏感性比原菌株高100倍以上[4]。
测试代谢活化系统一般采用由Aro-clor1254(PCBs)诱导大鼠肝微粒体酶的S9;国外也有用人肝S9的报道,试验证明其代谢活性明显高于鼠S9[5,6]。
CH药物遗传毒性研究指导原则的最新进展介绍

CH药物遗传毒性研究指导原则的最新进展介绍黄芳华王庆利审评二部黄芳华审评四部王庆利ICH的指导原则在药品的研究与开发中有较好的参考意义。
从2006年9月开始,ICH提出对遗传毒性指导原则进行修订改版,ICH专家组对遗传毒性试验一系列问题进行了讨论修订,但目前尚未达成一致意见,预期需2年的时间完成该指导原则的修订。
鉴于目前我们起草的指导原则参考的是已有的ICH指导原则的基本原则,及时跟踪国际上,包括ICH遗传毒性研究指导原则在内的相关领域的进展情况,对我们进行药物的研发和评价有借鉴作用。
因此,本文拟对ICH关于遗传毒性研究指导原则的相关进展进行简介。
针对遗传毒性研究,ICH分别于1995年和1997年发布了两个指导原则,即ICH S2A:Guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals(药物遗传毒性试验的特殊性指导原则)和ICH S2B:Genotoxicity:a Standard Battery for Genotoxicity Testing of Pharmaceuticals(遗传毒性:药物遗传毒性试验标准组合)。
由于遗传毒性试验大部分是短期试验,新技术发展迅速,且对于涉及基因突变过程的不同类型遗传损伤和不同作用方式的性质和相关性的科学认识也在不断发展,使得对遗传毒性试验有了新的认识。
在这种情况下,ICH于2006年启动了遗传毒性指导原则的修订工作,并于2006年9月和2007年5月的ICH会议上进行了讨论,最近的会议在2007年10月底至11月初召开。
ICH修订指导原则的起因主要源于两方面:其一是现有遗传毒性试验的进展与原指导原则推荐的试验方法存在着一些问题。
这些年来,一系列的体内和体外遗传毒性试验有了新发展并积累了大量的数据使得具有加入到原指导原则中的价值,如体外微核试验、体内彗星试验、转基因模型等。
29个ICH三级指导原则转化实施建议
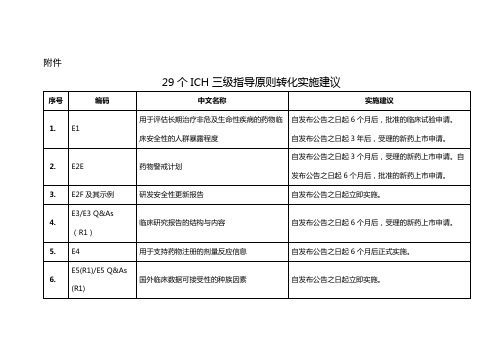
E2F及其示例
研发安全性更新报告
自发布公告之日起立即实施。
4.
E3/E3 Q&As(R1)
临床研究报告的结构与内容
自发布公告之日起6个月后,受理的新药上市申请。
5.
E4
用于支持药物注册的剂量反应信息
自发布公告之日起6个月后正式实施。
6.
E5(R1)/E5 Q&As (R1)
国外临床数据可接受性的种族因素
自发布公告之日起6个月后正式实施。
14.
E16
与药物或生物制品研发相关的生物标志物:资质提交材料的背景、结构以及格式
自发布公告之日起6个月后正式实施。
15.
E17
多区域临床试验计划与设计的总体原则
自发布公告之日起立即实施。
16.
S1A
药物致癌性试验必要性指导原则
申请人需在现行技术要求基础上尽早按照ICH指导原则开展研究;自发布公告之日起,6个月后开始的非临床研究按照ICH指导原则执行。
附件
29个ICH三级指导原则转化实施建议
序号
编码
中文名称
实施建议
1.
E1
用于评估长期治疗非危及生命性疾病的药物临床安全性的人群暴露程度
自发布公告之日起6个月后,批准的临床试验申请。
自发布公告之日起3年后,受理的新药上市申请。
2.
E2E
药物警戒计划
自发布公告之日起3个月后,受理的新药上市申请。自发布公告之日起6个月后,批准的新药上市申请。
S10
药物光安全评价
29.
M7(R1)
评估和控制药物中的DNA活性(致突变)杂质以限制潜在的致癌风险
申请人需在现行技术要求基础上尽早按照ICH指导原则开展研究;自发布公告之日起,6个月后开始的研究,以试验记录时间点为准,按照ICH指导原则执行。
ICHQSEM指导原则有哪些?马上给您列出来

ICHQSEM指导原则有哪些?马上给您列出来一、总目录类别主要内容ICH指导原则数量Quality Guidelines 质量指导原则化工、医药、质量保证相关指导原则44Safety Guidelines 安全性指导原则实验室动物实验等临床前研究相关指导原则16Efficacy Guidelines有效性指导原则人类临床研究相关指导原则30Multidisciplinary Guidelines多学科指导原则内容交叉涉及以上三个分类,不可单独划入任何一类的指导原则59总数149二、分目录2.1质量(Quality Guidelines)编号英文题目中文译文发布时间Q1 Stability/稳定性Q1A(R2): Stability Testing ofNew Drug Substances andProductsQ1A(R2):新型原料药和药品的稳定性测试2003.2.6Q1B: Stability Testing:Photostability Testing of NewDrug Substances and ProductsQ1B: 稳定性测试: 新型原料药和药品的光稳定性测试1996.11.6Q1C: Stability Testing for NewDosage FormsQ1C:新剂型的稳定性1996.11.6测试Q1D: Bracketing and MatrixingDesigns for Stability Testingof New Drug Substances andProductsQ1D :新型原料药和药品稳定性测试的交叉和矩阵设计 2002.2.7Q1E: Evaluation for StabilityDataQ1E :稳定性数据的评价2003.2.6Q1F: Stabilitiy Guidelines_WHO Q1F :WHO 稳定性指导原则2009Q2 Analytical Validation/分析方法验证 Q2(R1): Validation of Analytical Procedures Text and Methodology Q2(R1): 分析过程和方法的确证2005.11 Q3A - Q3DImpurities/杂质Q3A(R2): Impurities in New Drug Substances Q3A(R2): 新型原料药中的杂质问题 2006.10.25Q3B(R2): Impurities in New DrugProductsQ3B(R2): 新型药品中的杂质问题2006.6.2Q3C(R6): Impurities Guideline for Residual Solvents Q3C(R6):杂质:残留溶剂的指导原则2016.10.20Q3D: Guideline for Elemental Impurities Q3D :元素杂质的指导原则2014.12.16Q4 - Q4BPharmacopoeias/药典 Q4B: Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions Q4B :ICH 区域所用药典文本的评价和建议2007.11.1Q4B Frequently Asked Questions Q4B :常见问题与解答2012.4.26Q4B Annex 1 (R1): Residue onIgnition/Sulphated Ash General ChapterQ4B 附录1(R1): 关于灼烧残渣/灰分 常规篇2010.9.27Q4B Annex 2 (R1): Test for Extractable Volume of Parenteral Preparations General Chapter Q4B 附录2(R1): 关于注射剂可提取容量测试2010.9.27常规篇Q4B Annex 3 (R1): Test for Particulate Contamination: Sub-Visible Particles General Chapter Q4B 附录3(R1): 关于颗粒污染物测试:不溶性微粒 常规篇2010.9.27Q4B Annex 4A (R1):Microbiological Examination ofNon-SterileProducts: MicrobialEnumeration Tests GeneralChapterQ4B 附录4A(R1):非无菌药品的微生物检查:微生物计数试验 常规篇2010.9.27Q4B Annex 4B (R1):Microbiological Examination ofNon-Sterile Products Tests for Specified Micro-OrganismsGeneral ChapterQ4B 附录4B(R1): 非无菌产品的微生物检查—特定微生物 常规篇 2010.9.27Q4B Annex 4C (R1):Microbiological Examination ofNon-SterileProducts: AcceptanceCriteria for PharmaceuticalPreparations and Substancesfor Pharmaceutical UseGeneral ChapterQ4B 附录4C(R1): 非无菌产品的微生物检查:药物制备以及药物使用物质的接受标准 常规篇2010.9.27Q4B Annex 5 (R1):Disintegration Test General ChapterQ4B 附录5(R1):崩解试验 常规篇2010.9.27Q4B Annex 6 Uniformity of Dosage Units General Chapter Q4B 附录6: 统一剂量单位常规篇2013.11.13Q4B Annex 7 (R2): Dissolution Test General Chapter Q4B 附录7(R2): 溶出试验 常规篇2010.11.11Q4B Annex 8 (R1): Sterility Test General Chapter Q4B 附录8(R1): 无菌2010.9.27试验 常规篇Q4B Annex 9 (R1): Tablet Friability General Chapter Q4B 附录9(R1): 片剂易碎性 常规篇2010.9.27Q4B Annex 10 (R1):Polyacrylamide GelElectrophoresis GeneralChapterQ4B 附录10(R1): 聚丙烯酰胺凝胶电泳 常规篇2010.9.27Q4B Annex 11: Capillary Electrophoresis General Chapter Q4B 附录11:毛细管电泳 常规篇2010.6.9 Q4B Annex 12: AnalyticalSieving General ChapterQ4B 附录12:分析筛选 常规篇 2010.6.9Q4B Annex 13: Bulk Density andTapped Density of Powders General ChapterQ4B 附录13:粉末的堆密度和振实密度 2012.6.7Q4B Annex 14: BacterialEndotoxins Test GeneralChapterQ4B 附录14:细菌内毒素试验 常规篇2012.10.18Q5A - Q5E Quality of Biotechnological Products/生物技术产品质量 Q5A(R1): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin Q5A(R1):人或者动物细胞系来源的生物技术产品的病毒安全性评估 1999.9.23Q5B: Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products Q5B: 关于重组DNA 来源的蛋白质产品生产所用的细胞中表达构建的分析1995.11.30 Q5C: Stability Testing ofBiotechnological/BiologicalProductsQ5C: 生物技术/生物产品的稳定性试验1995.11.3Q5D:Derivationand Q 5D: 用于生1997.7.16Characterisation of Cell Substrates Used for Production of BiotechnologicalBiological Products 产生物技术/生物产品的细胞底物的起源和特征描述Q5E: Comparability of BiotechnologicalBiological Products Subject to Changes in their Manufacturing Process Q5E: 基于不同生产工艺的生物技术产品/生物产品的可比较性2004.11.18 Q6A- Q6BSpecifications/规格 Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances Q6A: 质量规格:新原料药和药品的检验程序和可接收标准:化学物质 1999.10.6 Q6B: Specifications: TestProcedures and AcceptanceCriteria for Biotechnological/Biological ProductsQ6B: 质量规格:生物技术/生物产品的检验程序和可接收标准1999.3.10Q7 GoodManufacturingPractice/GMP Q7: Good ManufacturingPractice Guide for Active Pharmaceutical Ingredients Q7: 原料药GMP 指南 2000.11.1Q7 Questions and Answers Q7 问答部分2015.6.10Q8 Pharmaceutica l Development/药物研发 Q8(R2): Pharmaceutical DevelopmentQ8(R2): 药物研发 2009.8Q8, Q9 and Q10 Questions & Answers (R4) Q8/Q9/Q10问答部分(R4)2010.11.11Q9 Quality RiskManagement/质量风险管理Q9: Quality Risk ManagementQ9: 质量风险管理 2005.11.09Q10 Pharmaceutical Quality System/药物质量体系Q10: Pharmaceutical Quality System Q10: 药物质量体系2008.6.4Q11 Development and Manufacture of Drug Substances/化Q11: Development and Manufacture of Drug Substances (Chemical Entities and Q11:化学药品的研发与生产(化2012.5.1学药品的研发与生产 Biotechnological/BiologicalEntities)学实体以及生物科技/生物制品)Q11:Questions and AnswersQ11:问答部分 2016.10.132.2安全性(Safety Guidelines)编号英文题目 中文译文 发布时间S1A - S1C Carcinogenicity Studies/致癌性研究S1A: Need for Carcinogenicity Studies of Pharmaceuticals S1A: 药物致癌性的研究需求 1995.11.29 S1B: Testing forCarcinogenicity of PharmaceuticalsS1B: 药物致癌性测试1997.7.16S1C(R2): Dose Selectionfor Carcinogenicity Studies of PharmaceuticalsS1C(R2): 药物致癌性研究的剂量选择2008.3.11S2 GenotoxicityStudies/基因毒性研究S2(R1): Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use S2(R1): 关于人用药基因毒性试验和数据解读的指导原则2011.11.9S3A - S3BToxicokinetics andPharmacokinetics/毒代动力学和药代动力学S3A: Note for Guidance onToxicokinetics: TheAssessment of Systemic Exposure inToxicity StudiesS3A :毒理动力学指导原则说明:毒性研究中系统性暴露的评价1994.10.27S3A Implementation Working Group Questions and Answers S3A 实施工作组问答:毒代动力学指导原则说明:毒性研究中的全身暴露量评价集中于微量采样(中文版:征求意见稿)2016.1.19 S3B: Pharmacokinetics Guidance for Repeated Dose S3B :关于重复剂量组织1994.10.27TissueDistribution Studies分布研究的药代动力学指导原则 S4 ToxicityTesting/毒性试验S4: Duration of Chronic Toxicity Testing in Animals (Rodent and Non Rodent Toxicity Testing) S4:动物慢性毒性试验的持续时间(啮齿动物和非啮齿动物毒性试验)1998.9.2S5 Reproductive Toxicology/生殖毒性 S5(R2):Detection ofToxicity to Reproductionfor Medicinal Products &Toxicity to Male FertilityS5(R2): 检测药品的生殖毒性以及对雄性生殖能力的毒性 2000.11S6 Biotechnological Products/生物技术产品 S6(R1): Preclinical SafetyEvaluation ofBiotechnology-Derived PharmaceuticalsS6(R1): 生物科技来源药品的临床前安全性评价2011.6.12S7A - S7B Pharmacology Studies/药理学研究 S7A: SAFETY PHARMACOLOGY STUDIES FOR HUMAN PHARMACEUTICALS S7A :人用药的安全性药理学研究 2000.11.8 S7B: The Non-ClinicalEvaluation of thePotential forDelayed Ventricular Repolarization (QTInterval Prolongation) byHuman PharmaceuticalsS7B :人用药延迟心室复极化(QT 间期延长)潜力的非临床评价2005.5.12S8 Immunotoxicology Studies 免疫毒理学研究 S8: Immunotoxicity Studiesfor Human PharmaceuticalsS8:人用药免疫毒性研究2005.9.15S9 NonclinicalEvaluation forAnticancerPharmaceuticals/抗癌药物的非临床评价S9: Nonclinical Evaluation for Anticancer Pharmaceuticals S9:抗癌药物的非临床评价2009.10.29S9 Implementation Working Group Questions and Answers S9 实施工作组问答部分2016.6.8S10 Photosafety Evaluation/光安全性评价 S10: Photosafety Evaluation of Pharmaceuticals S10:药物的光安全性评价2013.11.132.3有效性(Efficacy Guidelines)编号英文题目中文译文发布时间E1 Clinical Safety for Drugs used in Long-Term Treatment/长期使用的药物的临床安全性 E1: The extent of PopulationExposure to Assess ClinicalSafety for Drugs Intended for Long-term Treatment of Non-life-threateningConditionsE1: 用于评估长期治疗非危及生命性疾病的药物临床安全性的人群暴露程度1994.10.27E2A - E2FPharmacovigilance/药物警戒性E2A: Clinical Safety DataManagement: Definitions and Standards for Expedited ReportingE2A: 临床安全性数据管理:快速报告的定义和标准(中文版:征求意见稿) 1994.10.27E2B(R3):ImplementationGuide for ElectronicTransmission ofIndividual Case SafetyReports (ICSRs) E2B(R3)Data Elements and MessageSpecificationE2B(R3):个例安全报告(ICSR )电子传输执行指导原则 E2B (R3)数据元素和信息规范元素(中文版:征求意见稿) 2016.11.10 E2B(R3) QA document_v2_1 E2B(R3) 问答文件(中文版:征求意见稿) 2017.6.1E2C(R2): Periodic Benefit-Risk Evaluation Report E2C(R2):定期获益—风险2012.12.17间评估报告E2C(R2) Implementation Working Group Questions & Answers E2C(R2)实施工作组问答部分2014.3.31E2D: Post-Approval SafetyData Management: Definitions and Standards for Expedited ReportingE2D: 上市后安全性数据的管理:快速报告的定义和标准(中文版:征求意见稿) 2003.11.12E2E: Pharmacovigilance Planning E2E:药物警戒规性划2004.11.18E2F: Development Safety Update Report E2F: 研发安全性更新报告 2010.8.17E3 Clinical StudyReports/临床研究报告 E3: Structure and Content of Clinical Study Reports E3: 临床研究报告的结构与内容 1995.11.30E3 Questions & Answers(R1) : Structure and Content of Clinical Study ReportsE3 实施工作组 问答部分2012.7.6E4 Dose-Response Studies/剂量反应研究 E4: Dose-Response Information to Support Drug Registration E4: 用于支持药物注册的剂量反应信息 1994.3.10E5 Ethnic Factors/种族因素E5(R1): Ethnic Factors in the Acceptability of Foreign Clinical Data E5(R1):国外临床数据可接受性的种族因素1998.2.5E5 Implementation WorkingGroup Questions & Answers(R1)E5 实施工作组 问答部分(R1)2006.6.2间E6 GCP/药物临床试验管理规范 E6(R1): Guideline for Good Clinical Practice E6(R1):药物临床试验管理规范指导原则 1996.6.10E6(R2):Integrated Addendum to Good Clinical Practice (GCP) E6(R2):药物临床试验管理规范综合附录2016.11.9E7 Clinical Trials inGeriatric Population/老人中开展的临床试验E7: Studies in Support of Special Populations: GeriatricsE7: 特殊人群的支持性研究:老人病学 1993.6.24E7 Questions & AnswersE7 问答部分 2010.7.6E8 GeneralConsiderations for Clinical Trials/临床试验的一般性考虑 E8: General Considerations for Clinical Trials E8: 临床试验的一般性考虑1997.7.17E9 StatisticalPrinciples for Clinical Trials/临床试验的统计原则E9: Statistical Principles for Clinical Trials E9: 临床试验的统计原则1998.2.5E10 Choice of Control Group in Clinical Trials/试验中对照组的选择 E10: Choice of Control Group and Related Issues in Clinical Trials E10: 临床试验中对照组的选择以及相关问题2000.7.20E11 Clinical Trials inPediatric Population/儿童人群临床研究 E11: Clinical Investigation of Medicinal Products in the Pediatric Population E11:儿科人群药物临床试验 2000.7.20E11(R1): Addendum: Clinical Investigation of Medicinal Products in the Pediatric Population E11(R1): 儿科人群药物临床试验补充2017.8.18 E12 Clinical Evaluation by Therapeutic Category/根据治疗类别进行临床评价 E12: Principles for Clinical Evaluation of New Antihypertensive Drugs E12: 新型抗高血压药物的临床评价原则2000.3.2间E14 Clinical Evaluationof QT/QT 临床评价E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs E14:非抗心律失常药物QT/QTc 间期延长及致心律失常潜力的临床评价2005.5.12E14 Implementation Working Group Questions & Answers (R3) E14 实施工作组 问答部分(R3)2015.12.10 E15 Definitions in Pharmacogenetics/Pharmac ogenomics/药物基因组学以及遗传药理学相关定义 E15: Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmac ogenetics, Genomic Data and Sample Coding Categories E15: 基因组生物标志物、药物基因组学、遗传药理学、基因组数据以及样本编码分类的定义2007.11.1 E16 Qualification of Genomic Biomarkers/基因组生物标志物的合格条件 E16: Biomarkers Related to Drug or Biotechnology Product Development: Context, Structure and Format of Qualification Submissions E16:与药物或生物制品研发相关的生物标志物:资质提交材料的背景、结构以及格式2010.8.20 E17 Multi-Regional Clinical Trials/多地区临床试验 E17: General principle on planning and Designing Multi-Regional Clinical Trials E17:计划和设计多地区临床试验的一般性原则2016.5.6 E18 Genomic Sampling/基因组取样 E18: Genomic Sampling and Management of Genomic Data E18:基因组采样和基因组数据管理指导原则(中2015.12.10间文版:征求意见稿)2.4多学科(MultidisciplinaryGuidelines)编号 英文题目中文译文 发布时间M1 MedDRA Terminology 监管活动医学词典 MedDRA Data Retrieval and Presentation: Points to Consider MedDRA 数据检索与呈现: 考虑要点(中文版:征求意见稿) 2017.9.1 MedDRA Term Selection: Points to Consider MedDRA 术语选择: 考虑要点(中文版:征求意见稿)2017.9.1 M2 Electronic Standards 电子标准 ICH M2 EWG Work Plan M2专家工作组工作计划2017.3.27 M2: ElectronicStandards for the Transfer of Regulatory Information Final Concept PaperM2监管信息转移的电子标准终版概念文件1994.10.27 ElectronicTransmission of Individual Case Safety Reports Message Specification个例病例安全性报告的电子传输信息规范 2000.11.9 ICH M2 EWG The eCTDBackbone File Specification for Study Tagging FileseCTD 研究标签文件主文件规范2008.6.3 Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) General Recommendation -Procedure监管信息转移的电子标准一般性建议—程序 2015.6.11 Electronic Standards for the Transfer of Regulatory Information (ESTRI) General Recommendation – ESTRI 监管信息转移的电子标准一般性建议—ESTRI 网关 2015.6.11Gateway Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) File Format Recommendation – PDF监管信息转移的电子标准文件格式建议—PDF2011.4.5Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) File Format Recommendation – XML监管信息转移的电子标准文件格式建议—XML2005.11.10 Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) File Format Recommendation – PDF/A监管信息转移的电子标准文件格式建议—PDF/A2014.6.2Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) File Format Recommendation – DOCX监管信息转移的电子标准文件格式建议—DOCX2015.6.11Electronic Standardsfor the Transfer ofRegulatory Information (ESTRI) Controlled Vocabularies Recommendation -Genericode监管信息转移的电子标准控制词汇建议—代码2015.6.11Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) Information Transfer Recommendation – EDIINT AS1/AS2监管信息转移的电子标准信息转移建议—EDIINT AS1/AS22010.6.10Electronic Standardsfor the Transfer of Regulatory Information (ESTRI) File Integrity – MD5监管信息转移的电子标准文件完整性—MD52010.6.10Electronic Standards for the Transfer of 监管信息转移的电子标准文件完2015.6.11Regulatory Informaation (ESTRI) File IntegrityRecommendation - SHA-256整性建议—SHA-256M2 Glossary of Terms and Abbreviations M2术语和简写词汇表2015.6.11 ICH M2 File Format Criteria M2文件格式标准 2014.11.10Use of OIDs & UUIDs in ICH Messages OIDs & UUIDs 在ICH 信息中的使用2015.6.11 M3 Nonclinical Safety Studies 非临床研究 M3(R2) Questions and Answers (R2)M3(R2)问答 (R2) 2012.3.5 M3(R2): Guidance onNonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for PharmaceuticalsM3(R2):关于实施药物人体临床试验以及上市批准非临床安全性研究的指导原则2009.6.11M4 : The Common Technical Document 通用技术文件 M4 (R4): Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human UseM4(R4):人用药物注册通用技术文档的组织(中文版:征求意见稿) 2016.6.15 M4 Implementation Working Group Questions & Answers (R3) M4执行工作组问答(R3)(中文版:征求意见稿) 2004.6.10 The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality – M4Q(R1)M4Q (R1):人用药物注册通用技术文档:药学部分(中文版:征求意见稿) 2002.9.12 M4Q Implementation Working Group Questions & Answers (R1) M4Q 执行工作组问答(R1)(中文版:征求意见稿)2003.7.17 The Common Technical Document for the M4S (R2):人用药物注册通用技2002.12.2Registration of Pharmaceuticals for Human Use: Safety – M4S(R2)术文档:安全性部分(中文版:征求意见稿)M4S Implementation Working Group Questions & Answers (R4) M4S 执行工作组问答 (R4)(中文版:征求意见稿)2003.11.11 Efficacy- M4E(R2) M4E (R2):人用药物注册通用技术文档:有效性部分(中文版:征求意见稿)2016.6.15M4E Implementation Working Group Questions & Answers (R4) M4E 执行工作组问答(R4)(中文版:征求意见稿)2004.6.10 M5 Data Elements and Standards for Drug Dictionaries 药物词典的数据要素和标准 The Re-development ofthe Standard forE2B(R3) and the Development of Standards for the Identification of Medicinal Products(IDMP)(ICH M5)ICH M5:E2B(R3)标准的再制定及医药产品鉴定标准的制定2010.11.1 ICH E2B(R3)Implementation Working Group ICH E2B(R3) Guideline: Electroni c Transmission of Individual Case Safety Reports (ICSRs) E2B(R3)实施工作组个例病例安全报告的电子传输问答部分 2016.11.10 Appendix I (B) to theImplementation Guide for Electronic Transmission of Individual Case SafetyReports (ICSRs)个例病例安全报告的电子传输实施指南附录 I (B) 2016.11.10 Appendix I (G) to theImplementation Guide for Electronic Transmission of Individual Case Safety个例病例安全报告的电子传输实施指南附录 I (G)2016.11.10Reports (ICSRs) Implementation Guidefor Electronic Transmission of Individual Case Safety Reports(ICSRs)个例病例安全报告的电子传输实施指南 2016.11.10 M6 Gene Therapy 基因治疗 Final Concept Paper M6: Guideline on Virus and Gene Therapy Vector Shedding and TransmissionM6: 病毒和基因治疗载体的脱落和传播终版概念文件 2009.8.26 General Principles to Address Virus and Vector Shedding 解决病毒和基因治疗载体脱落的基本原则 2009.6An inventory of shedding data from clinical gene therapy trials临床基因疗法试验脱落数据目录2007.7.30 Final Business Plan M6: Guideline on Virus and Gene Therapy Vector Shedding and TransmissionM6: 病毒和基因治疗载体的脱落和传播终版业务计划 2009.8.27 M7 Genotoxic Impurities 遗传毒性杂质 M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk M7:评估和控制药物中的DNA 活性(致突变)杂质以限制潜在的致癌风险 2014.6.23 M7(R1): Addendum to M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic RiskM7(R1): 评估和控制药物中 DNA 反应性(致突变)杂质以限制潜在的致癌风险(中文版:征求意见稿) 2017.3.31 M8 Electronic Common Technical Document (eCTD) 电子通用技术文件 Electronic Common Technical Document Specification V3.2.2电子通用技术文件规范 V3.2.2 2008.7.16 M8 : Electronic Common M 8: 电子通用2015.12.9Technical Document Concept Paper 技术文件概念文件ICH M8 EWG/IWG Work Plan M8: 电子通用技术文件工作计划2017.3.13 Support Documentation for M8: eCTD EWG eCTD v4.0 Implementation Package v1.2 M8:eCTD 专家工作组eCTD v4.0实施包 v1.2 支持性证明文件2016.11 Orientation Material forM8: eCTD EWG eCTD v4.0 Implementation Package v1.2 M8:eCTD 专家工作组eCTD v4.0实施包 v1.2 培训材料2016.11 ICH Electronic CommonTechnical Document (eCTD) v4.0 Implementation Guidev1.2ICH eCTD v4.0 实施指南 v1.2 2016.11.10 eCTD v4.0 Implementation Package v1.2eCTD v4.0 实施包 v1.2 USFDA eCTD v4.0 Implementation Package History v1.1 美国FDA eCTDv4.0 实施包历史 v1.1USFDA Module 1Electronic Common Technical Document (eCTD) v4.0 ImplementationGuide v1.1美国FDA 模块1 eCTD v4.0 实施指南 v1.1 2017.2.20 ICH eCTD v4.0 Requirements ICH eCTD v4.0 要求ICH M8 Expert WorkingGroup Specification for Submission Formats for eCTDeCTD 提交格式规范 2016.11.1Change Control Process for the eCTD eCTD 变更控制过程2017.4 Request for change 请求变更表M9 Biopharmaceutic s Classification System-based M9: Biopharmaceutics Classification System-based Biowaivers Final M9:基于生物药剂学分类系统的生物豁免业务计2016.10.7Biowaivers 基于生物药剂学分类系统的生物豁免 endorsed Business Plan划ICH M9 EWG Work Plan M9 专家工作组工作计划2017.2.9 M9: Biopharmaceutics Classification System-based Biowaivers Final endorsed Concept PaperM9:基于生物药剂学分类系统的生物豁免概念文件 2016.10.7 M10 Bioanalytical Method Validation 生物样品分析的方法验证 M10: Bioanalytical Method Validation Final endorsed Business Plan M10: 生物样品分析的方法验证业务计划 2016.10.7ICH M10 EWG Work Plan M10: 专家工作组工作计划 2017.3.10 M10: Bioanalytical Method Validation Final endorsed Concept Paper M10: 生物样品分析的方法验证概念文件2016.10.7文章来源:龙腾整理。
总局关于发布药物遗传毒性研究技术指导原则

总局关于发布药物遗传毒性研究技术指导原则附件:药物遗传毒性研究技术指导原则食品药品监管总局2018年3月12日药物遗传毒性研究技术指导原则一、概述遗传毒性研究(GenotoxicityStudy)是药物非临床安全性评价的重要内容,与其他研究尤其是致癌性、生殖毒性等研究有着密切的联系,是药物进入临床试验及上市的重要环节。
拟用于人体的药物,应根据受试物拟用适应症和作用特点等因素考虑进行遗传毒性试验。
遗传毒性试验是指用于检测通过不同机制直接或间接诱导遗传学损伤的受试物的体外和体内试验,这些试验能检测出DNA损伤及其损伤的固定。
以基因突变、较大范围染色体损伤或重组形式出现的DNA损伤的固定,通常被认为是可遗传效应的基础,并且是恶性肿瘤多阶段发展过程的重要因素(恶性肿瘤发展变化是一个复杂的过程,遗传学改变可能仅在其中起部分作用)。
染色体数目的改变也与肿瘤发生有关,并可提示生殖细胞出现非整倍体的可能性。
在遗传毒性试验中呈阳性的化合物为潜在的人类致癌剂和/或致突变剂。
由于在人体中已建立了某些致突变/遗传毒性化合物的暴露与致癌性之间的相关性,而对于遗传性疾病尚难以证明有类似的相关性,因此遗传毒性试验主要用于致癌性预测。
但是,因为生殖细胞突变与人类疾病具有明确的相关性,所以也应同样重视化合物引起潜在可遗传性效应的风险。
此外,遗传毒性试验结果可能对致癌性试验的结果分析有重要作用。
因此,在药物开发的过程中,遗传毒性试验的目的是通过一系列试验来预测受试物是否有遗传毒性,在降低临床试验受试者和药品上市后使用人群的用药风险方面发挥重要作用。
本指导原则重点阐述遗传毒性试验的基本原则,介绍标准试验组合方案,阐述体内外试验的基本原则,以及对试验结果的分析评价与追加研究策略。
本指导原则适用于中药、天然药物和化学药物。
二、基本原则(一)实验管理药物遗传毒性试验必须执行《药物非临床研究质量管理规范》(GLP)。
(二)具体问题具体分析遗传毒性试验的设计,应该在对受试物认知的基础上,遵循“具体问题具体分析”的原则。
体内微核试验的自动化分析20110804

您现在的位置:电子刊物 >> 电子刊物列表 >> 电子刊物详细发布日期20110804栏目化药药物评价>>非临床安全性和有效性评价标题体内微核试验的自动化分析作者黄芳华部门药理毒理学部正文内容遗传毒性评价是新药非临床安全评价的重要组成部分,采用标准试验组合的方式。
由于体内试验具有考虑到了与人体应用相关的吸收、分布、排泄、代谢的优点,体内试验在遗传毒性评价策略中意义重大。
啮齿类动物体内微核试验由于其检测终点明确、重复性好且易于开展,是目前广泛应用的体内遗传毒性检测方法,是ICH指导原则中规定的药物遗传毒性标准试验组合中唯一一项体内试验,也是我国药物遗传毒性研究指导原则所规定的一项试验。
目前,体内微核试验采用人工显微镜阅片的方法。
该方法虽然程序简单,但存在一些明显的缺陷。
首先,人工阅片时间长,费时费力,工作效率较为低下。
其次,人工阅片的准确性依赖于检测人员的水平,而在目前需大量进行该试验的情况下,检测人员水平不一,将导致检测结果具有很大的主观性。
最为重要的是,微核的发生率低,在一组动物上能否检出小的微核升高率的能力受到计数误差以及动物个体之间差异性的限制,而且,在单个动物上能否检测出小的微核升高率的灵敏度受到低微核数的计数误差以及在检测过程中个体动物高的自发变异性的限制。
基于这种实际,ICH的国际遗传毒性试验工作组(theInternational Workshop on GenotoxicityTesting,IWGT)建议,应尽可能计数足够数量的细胞,以使计数误差低于动物间微核率的变异性[1]。
也就是说,为了达到充分的检测灵敏度,只有检测大量细胞才能保证检出微核。
虽然2007年颁布的《药物遗传毒性研究指导原则》已将检测细胞数由原来的1000个嗜多染红细胞提高为至少2000个,但是仍难以解决灵敏度的问题。
因此,目前需大量检测化合物与相对较慢速度的微核人工检测方法之间、待检任务和检测能力之间的矛盾变得比较突出,需要以新技术、新方法来提高体内微核试验的检测能力。
ICH毒代动力学指导原则:毒性研究中全身暴露量的评价

该指导原则为ICH (人用药品注册技术要求的国际协调会议)三方(欧盟、日本和美国)协调的指导原则。
根据ICH程序,该指导原则由ICH专家工作组(安全性)起草,并提交管理部门讨论协商。
1994年10月27日,在ICH程序的第4阶段会议上,该指导原则被ICH筹备委员会推荐给欧盟、日本和美国的行政管理部门采纳。
1995年3月,该指导原则发布在美国FDA的Federal Register± (60 FR 11264),适用于化学药物和生物制品〔医学整理发布〕。
1前言本指导原则所涉及的毒物代谢动力学(毒代动力学)仅与拟开发作为人用的药品有尖。
毒代动力学是药代动力学在全身暴露评价中的延伸,为非临床毒性研究的一个组成部分,或为某一特殊设计的支持研究。
研究结果可用于阐明毒理学发现及其与临床安全性的矢系(文中其它术语的定义见注释1)。
制定该指导原则是为了使人们理解毒代动力学的意义和应用,指导毒代动力学的试验设计。
本指导原则强调毒性试验需与毒代动力学相结合,这将有助于解释毒理学发现和制定合理的试验设计。
毒代动力学测定通常是结合于毒性研究中,故又被称为“伴随毒代动力学”。
毒性试验的试验程序有助于获得受试动物多剂量的毒代动力学数据。
如果在毒性试验中测定了合适的指标或参数,毒代动力学研究可避免重复的毒性试验。
有时,模拟毒性试验的支持研究也可获得相应的毒代动力学数据。
获取数据的优化设计可以减少试验动物数。
非临床药代动力学和代谢过程的研究,对解释毒理学的发现可能有价值,但毒代动力学数据侧重于新药毒性研究中的动力学。
因此,毒代动力学是非临床试验设计的组成部分,在理解毒性试验结果和临床人体用药风险性、安全性时可提高毒理学资料的价值。
毒代动力学已成为毒性试验的组成部分,成为非临床和临床试验间的桥梁,其研究重点是解释毒性试验结果,而不是为描述受试物的基本药代动力学参数特征。
由于药品开发是在非临床和临床间反馈的动态过程,因此毒代动力学研究无严格的、详细的试验程序,也无必要在全部研究中获取毒代动力学数据,应该科学地判断什么情况下需要进行。
ICH遗传毒性指导原S2(R1)修订要点及相关背景介绍

[ Ke y wo r d s ] g e n o t o x i c i t y a s s a y ;I C H s 2( R 1 ) ; m j a o r r e v i s i o n p o i n t s
背景 情况 。
[ 关键词 ] 遗传 毒性试 验 ; I C H s 2( R1 ) ; 修订 要 点 [ 中图分类 号 ]R 9 6 8 [ 文献 标 志码 ]A [ 文章编 号 ] 1 0 0 3— 3 7 3 4 ( 2 0 1 3 ) 0 2— 0 1 4 6— 0 5
[ A b s t r a c t ] I C H g u i d a n c e o n g e n o t o x i c i t y t e s t s 2( R 1 )w a s i s s u e d i n N o v e m b e r 2 0 1 1 , w h i c h i s a r e f e r e n c e
ZHANG Mi n g 。
,
HU A N G F a n g - h u a , Z HO U C h a n g - h u i , C H A N G Y a n , WA N G Q i n g — l i
( 1 C h i n a S t a t e I n s t i t u t e o f P h a r m a c e u t i c a l I n d u s t r y N a t i o n a l S h a n g h a i C e n t e r f o r N e w D r u g S a f e t y E v a l u a t i o n& R e s e a r c h , S h a n g h a i 2 0 1 2 0 3 ,C h i n a ; 2 C e n t e r f o r D r u g E v a l u a t i o n , S t a t e F o o d a n d D r u g A d mi n i s t r a t i o n , B e i j i n g 1 0 0 0 3 8 , C h i n a )
FDA《推荐的遗传毒性试验结果综合分析法指导原则》

FDA《推荐的遗传毒性试验结果综合分析法指导原则》介绍审评四部王庆利审评二部黄芳华审评五部彭健摘要:FDA于2006年1月正式发布了《推荐的遗传毒性试验结果综合分析法指导原则》,该指导原则介绍了目前对遗传毒性结果的综合分析方法,尤其是在遗传毒性试验结果出现阳性结果时提出了一些推荐性的建议和意见。
现在原草案翻译稿的基础上,重新翻译整理了该指导原则,以期为药物研究者提供一些参考性信息。
关键词:遗传毒性;致癌性一、简介本指导原则的目的是为了向企业和CDER的审评人员说明,CDER如何看待药物开发过程中出现的遗传毒性试验阳性结果。
当遗传毒性试验结果提示药物具有潜在的致癌性或遗传危害时,本指导原则为如何继续进行临床试验并保证受试者的安全性提供建议。
本指导原则中讨论了单次给药和多次给药临床试验相关的管理决策。
本指导原则适用于经口、静脉、局部和其他途径给药的药物。
FDA的指导原则,包括本指导原则在内,并不具有法律上的强制性,而是描述了当前FDA 对某一问题的想法,应该被认为仅是一种建议,除非是援引了特殊的管理性的或法令性的要求。
FDA指导原则中的“应该”一词是指建议的或推荐某些东西,而不是要求某些东西。
二、背景遗传毒性试验的时间安排和如何进行在ICH指导原则M3、S2A和S2B中已有描述。
我们建议参考这些指导原则,而本指导原则可认为是仅作为附件性指导原则。
通常是在啮齿类动物试验中评估致癌性风险,包括周期为2年的试验或采用替代模型而周期较短的试验。
通过ICH程序,企业和管理者接受了遗传毒性核心组合试验方案。
这些试验是用于确定化合物的遗传毒性,包括:■ 一项细菌基因突变试验■ 一项采用哺乳动物细胞进行的体外染色体损伤评估试验,或体外小鼠淋巴瘤tk+/- 试验■ 一项采用啮齿类动物造血细胞进行的体内染色体损伤试验。
以下讨论是根据当前的指导原则文件进行的。
我们建议在进行I期临床试验前完成体外遗传毒性试验。
三、遗传毒性试验结果的综合分析当遗传毒性结果为阳性结果时,对进入临床试验是否安全,FDA会考虑所有的安全性资料。
ICH指导原则目录

Q4 Pharmacopoeias(药典)
Q4
Pharmacopoeias
Q4A
Pharmacopoeial Harmonisation
Q4B
Step 5
2007-11-1
Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions
关于ICH区域内药典附录的评 价及建议-不溶性微粒检查法
Q4B
Annex 4A (R1)
Step 5
2010-9-27
Microbiological Examination of Non-Sterile Products: Microbial Enumeration Tests General Chapter
关于ICH区域内药典附录的评 价及建议-非无菌产品的微生物 检查:原料药及其制剂的判定 标准
Q4B Annex 5 (R1)
Step 5
2010-9-27
Disintegration Test General Chapter
关于ICH区域内药典附录的评 价及建议-崩解时限检查法
Q4B Annex 6
来源于人或动物细胞系的生物 技术产品的病毒安全性评价
Q5B
Step 5
1995-11-30
Quality of Biotechnological Products: Analysis of the Expression Construct in
Cells Used for Production of r-DNA Derived Protein Products
纳米材料遗传毒性试验选择指南 草案

附件1纳米材料遗传毒性试验选择指南(草案)纳米材料独特的物理化学性质及其与生物体相互作用特性为其在食品加工、抗菌产品生产、药物载体开发、肿瘤诊疗、体内示踪等领域的应用提供了广泛的前景。
新型纳米材料的诞生为人类带来机遇的同时也带来了挑战。
大量纳米材料的涌现,极大程度上增加了人体与纳米材料接触的机会,纳米材料的安全性评价成为关注的热点。
如何合理评价纳米材料对人体的潜在毒性,特别是慢性或者长期毒性,预测其潜在毒性风险是目前全科学界及各相关监管部门亟需解决的问题。
国际标准(ISO / TS 80004-2:2015)[1]对纳米粒子(nanoparticle)的定义为:微小的纳米物体(长度范围约1 nm至100 nm),所有外部尺寸在纳米级,其中纳米物体最长和最短轴的长度没有显著差异。
如果尺寸差异显著(通常超过三倍),则为纳米纤维或纳米板。
纳米粒子的尺寸效应和表面活性高等特点使它极易透过细胞膜,可在表面活性和蓄积性的共同作用下,对细胞遗传物质产生直接或间接的相互作用。
尤其是携带金属离子的纳米粒子进入体内后有可能通过氧化应激等作用机制诱发染色体或DNA断裂[2-5]。
此外,纳米材料进入人体后可能在脏器蓄积并长期存在。
纳米材料的遗传毒性,现已发展成一个专门的亚分支研究领域—纳米遗传毒理学[6-7]。
对纳米材料的潜在致癌和致畸作用评估,是对纳米材料对人体安全性评价中极为重要的一环。
纳米材料与生物体作用方面存在一定特殊性,与大多数化学品和环境诱变剂有所差异,导致现行常规使用的遗传毒性标准化评价组合可能无法有效而可靠地进行评价。
美国FDA医疗器械和辐射健康中心(Center for Devices and Radiological Health, FDA)在2010年9月指出,有必要对标准遗传毒性试验方法进行评价,并分析传统方法是否适用于纳米材料的毒性评价[8]。
经合组织(The Organization for Economic Cooperation and Development,OECD)纳米材料产品工作组(Working Party on Manufactured Nanomaterials,WPMN)于2013年11月18-19日在加拿大渥太华召开纳米材料产品遗传毒性专题研讨会,就是否需要在现有OECD遗传毒性试验指导原则范畴内对纳米材料遗传毒性测试方法进行特别调整,以及/或需要制定新的试验指导原则或指导性文件进行讨论。
制药企业药品上市前安全风险管理

制药企业药品上市前安全风险管理摘要:药物警戒越来越宽阔的视野,也将使其包含更多超出现有药理学和药动学研究的、与很多行为相关的、有可能导致安全问题的因素,从而最终决定药品的安全使用,包括优化患者和护理者使用该药品的行为以及处方者选择和监测这种药品的信息和行为。
因此,患者和医生对该药品风险管理的了解,有助于权衡这种治疗方法的风险和受益,从而可更有效地监控药品不良反应。
关键词:制药企业;药品;上市前;安全风险;管理引言本文旨在集中讨论上市前新化学实体的药品警戒及安全风险管理的重要性及管理工作。
无论企业申办方在研发过程的哪个阶段开始,这些原则均可以且应该被广泛使用。
而且这些原则也应该应用于生物制品、复方制剂、医疗器械以及其他医疗产品的任何研究阶段。
1制药企业药品上市前安全风险管理的重要性1.1药物警戒的定义世界卫生组织对药物警戒的定义是“监测、评估、理解并预防不良反应或者其他药品相关问题的科学和活动。
药物警戒的目的是提高与药品使用相关的医疗质量并加强其安全性。
同时提供可靠的、均衡的信息来进行药品风险一收益的有效评估,以便为公众健康提供更好的服务”。
从实际情况看,这个定义亦适用于其他医疗产品,如疫苗、生物制品和各种医疗设备。
1.2上市前安全风险管理的重要性良好的药物警戒是最理想的药品风险管理方式,它可以最大程度的了解某种药品的所有信息,并且发现它潜在的安全问题,甚至有可能发现其预料之外的好处。
鉴于这个理念,新化学实体 (newchemicalentity) 在研发初期就必须进行动物和人体安全性测试,以便更好地促进并保护公众健康。
中国古话所说的“是药三分毒”,说明用来治疗疾病的人造或者天然药物都同时具有风险性和收益性。
近代以来,医学观察变得更加理性,当药品或其他医学产品进入市场后并引发不良反应甚至悲剧时,可以很快对其做出反应。
如在欧洲和美国曾发生著名的”反应停”事件。
20 世纪 5O ~ 60 年代,沙利度胺 ( 反应停 ) 因可有效阻止女性妊娠早期孕吐而在 2O 多个国家被广泛使用。
ICH 指导原则文件目录(中英文)

人用药品注册技术要求国际协调会(ICH)文件目录ICH的论题主要分为四类,因此ICH根据论题的类别不同而进行相应的编码分类:1. “Q”类论题:Q代表QUALITY,指那些与化工和医药,质量保证方面的相关的论题。
Q1/Q2...Q10都属于这类。
2. “S”类论题:S代表SAFETY,指那些与实验室和动物实验,临床前研究方面的相关的论题。
3. “E”类论题:E代表EFFICACY,指那些与人类临床研究相关的课题。
4. “M”类论题:M代表MULTIDISCIPLINARY, 指那些不可单独划入以上三个分类的交叉涉及的论题。
同时M又细分为5个小类:M1: 常用医学名词(Med DRA)M2: 药政信息传递之电子标准M3: 与临床试验相关的临床前研究时间的安排M4: 常规技术文件(CTD)M5: 药物词典的数据要素和标准一、ICH. 质量部分(Quality)稳定性1.Quality质量2.Q1: Stability稳定性3.Q1A(R2): Stability Testing of New Drug Substances and Products 新原料药和制剂的稳定性试验4.Q1B: Photostability Testing of New Drug Substances and Products 新原料药和制剂的光稳定性试验5.Q1C: Stability Testing for New Dosage Forms 新剂型的稳定性试验6.Q1D: Bracketing and Matrixing Designs for Stability Testing of Drug Substances and Drug Products原料药和制剂稳定性试验的交叉和矩阵设计 Q1E: Evaluation of Stability Data 稳定性数据的评估7.Q1F: Stability Data Package for Registration Applications in Climatic Zones III andIV在气候带III和IV,药物注册申请所提供的稳定性数据8.Q2: Analytical Validation分析验证9.Q2(R1): Validation of Analytical Procedures: Text and Methodology分析程序的验证:正文及方法论10.Q3: Impurities 杂质11.Q3A(R2): Impurities in New Drug Substances 新原料药中的杂质12.Q3B(R2): Impurities in New Drug Products (Revised Guideline) 新制剂中的杂质13.Q3C(R3): Impurities: Guideline for Residual Solvents 杂质:残留溶剂指南Impurities: Guideline for Residual Solvents (Maintenance) 杂质:残留溶剂指南(保留)PDE for Tetrahydrofuran (in Q3C(R3)) 四氢呋喃的日允许接触剂量PDE for N-Methylpyrrolidone (in Q3C(R3)) N-甲基吡咯烷酮的日允许接触剂量14.Q4: Pharmacopoeias药典15.Q4A: Pharmacopoeial Harmonisation 药典的协调16.Q4B: Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions药典内容的评估及推荐为用于ICH地区17.Q4B Annex1 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Residue on Ignition/Sulphated Ash General Chapter附录1 药典内容的评估及推荐为用于ICH地区关于灼烧残渣/灰分常规篇18.Q4B Annex2 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Test for Extractable Volume of Parenteral Preparations General Chapter附录2 药典内容的评估及推荐为用于ICH地区关于注射剂可提取容量测试常规篇19.Q4B Annex3 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Test for Particulate Contamination: Sub-Visible Particles General Chapter附录3 药典内容的评估及推荐为用于ICH地区关于颗粒污染物测试:不溶性微粒常规篇20.Q5: Quality of Biotechnological Products 生物技术制品质量21.Q5A(R1): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin来源于人或者动物细胞系的生物技术产品的病毒安全性评估22.Q5B: Quality of Biotechnological Products: Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products生物技术产品的质量:源于重组DNA的蛋白质产品的生产中所用的细胞中的表达构建分析23.Q5C: Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products生物技术产品的质量:生物技术/生物产品的稳定性试验24.Q5D: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products用于生产生物技术/生物产品的细胞底物的起源和特征描述25.Q5E: Comparability of Biotechnological/Biological Products Subject to Changes inTheir Manufacturing Process基于不同生产工艺的生物技术产品/生物产品的可比较性26.Q6: Specifications规格27.Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (including decision trees) 质量规格:新原料药和新制剂的检验程序和可接收标准:化学物质(包括决定过程)28.Q6B: Specifications: Test Procedures and Acceptance Criteria for29.Biotechnological/Biological Products质量规格:生物技术/生物产品的检验程序和可接收标准30.Q7: Good Manufacturing Practices (GMP)31.Q7A: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients活性药物成份的GMP指南32.Q8: Pharmaceutical Development药物研发33.Annex to Q8Q8附录34.Q9: Quality Risk Management质量风险管理35.Q10: Pharmaceutical Quality System药物质量体系二、ICH.安全性部分(Safety) 致癌试验1.S1A Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals 药物致癌试验的必要性2.S1B Testing for Carcinogenicity of Pharmaceuticals 药物致癌试验3.S1C(R2) Dose Selection for Carcinogenicity Studies of Pharmaceuticals药物致癌试验的剂量选择4.S1C’药物致癌试验的剂量选择的附件:补充剂量限度和有关注释遗传毒性5.S2(R1) Guidance on Genotoxicity Testing and Data Interpretation forPharmaceuticals Intended for Human Use 人用药物的遗传毒性试验和数据分析指导原则6.S2A药物审评遗传毒性试验的特殊性指导原则7.S2B遗传毒性:药物遗传毒性试验标准组合药代8.S3A Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposurein Toxicity Studies 毒代动力学指导原则:毒性研究中全身暴露的评价9.S3B Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies药代动力学:重复给药的组织分布研究指导原则慢性毒性10.S4Duration of Chronic Toxicity Testing in Animals (Rodent and Non RodentToxicity Testing) 动物慢性毒性试验的周期(啮齿类和非啮齿类)生殖毒性11.S5(R2) Detection of Toxicity to Reproduction for Medicinal Products andToxicity to Male Fertility (the Addendum dated November 1995 has beenincorporated into the core guideline in November 2005 )12.S5A药品的生殖毒性检测13.S5B雄性生育力毒性其他14.S6Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals 生物技术药品的临床前安全性试验15.S7A Safety Pharmacology Studies for Human Pharmaceuticals 人用药物的安全性药理研究16.S7B The Non-clinical Evaluation of the Potential for Delayed VentricularRepolarization (QT Interval Prolongation) by Human Pharmaceuticals人用药延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则17.S8Immunotoxicity Studies for Human Pharmaceuticals人类药品的免疫毒性研究18.S9 Nonclinical Evaluation for Anticancer Pharmaceuticals抗癌药物的临床前评价19.S10 Photosafety Evaluation三、ICH.临床部分(Efficacy)1.E1The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions 评价临床长期给药方案的安全性2.E2A Definitions and Standards for Expedited Reporting 快速报告的定义和标准3.E2B(R3) Data Elements for Transmission of Individual Case Safety Reports个体病例安全性报告传递的数据要素4.E2C Periodic Benefit-Risk Evaluation Report 上市药品定期安全性更新报告5.E2D Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting批准后安全性数据管理:快速报告的定义和标准6.E2E Pharmacovigilance Planning药物警戒计划7. E2F Development Safety Update Report8.E3Structure and Content of Clinical Study Reports 临床研究报告的结构与内容9.E4Dose-Response Information to Support Drug Registration 新药注册所需量-效关系的资料10.E5(R1)Ethnic Factors in the Acceptability of Foreign Clinical Data 对国外临床研究资料的种族因素的可接受性11.E6(R1) Good Clinical Practice: Consolidated Guideline 药品临床研究规范(GCP)一致性指导原则12.E7Studies in Support of Special Populations: Geriatrics 老年人群的临床研究13.E8General Considerations for Clinical Trials 临床试验的一般考虑14.E9Statistical Principles for Clinical Trials 临床试验统计原则15.E10Choice of Control Group and Related Issues in Clinical Trials 对照组的选择16.E11Clinical Investigation of Medicinal Products in the Pediatric Population 儿童人群的临床研究17.E12按治疗分类的各类药物临床评价E12 Principles for Clinical Evaluation of New Antihypertensive Drugs18.E14The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs 非抗心律失常药物致QT/QTc间期延长及潜在心律失常作用的临床评价19.E15 Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories20.E16 Biomarkers Related to Drug or Biotechnology Product Development: Context, Structure and Format of Qualification Submissions四、ICH.综合部分 (Multidisciplinary)1.M1医学术语Med DRA2.M2Electronic Transmission of Individual Case Safety Reports MessageSpecification (ICH ICSR DTD Version 2.1) companion document to E2B(R3)注册资料传递所需的电子代码3.M3Guidance on Nonclinical Safety Studies for the Conduct of Human ClinicalTrials and Marketing Authorization for Pharmaceuticals与临床研究有关的临床前研究的时间安排4.M4 Organisation of the Common Technical Document for the Registration ofPharmaceuticals for Human Use (Edited with Numbering and Section Header Changes, September 2002). Including the Annex : the Granularity Document(Revised November 2003).CTD(common technical document)(包括CTD、CTD-Q、CTD-S、CTD-E和eCTD)药品词汇的数据要素和标准5.M4Q (R1) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Quality (Edited with Numbering and Section Header Changes, September 2002)6.M4S (R2) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Safety (Edited with Numbering and SectionHeader Changes, September 2002)7.M4E (R1) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Efficacy (Edited with Numbering and Section Header Changes, September 2002)8.M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities inPharmaceuticals to Limit Potential Carcinogenic Risk Reference:1. 《ICH 药品注册的国际要求》2. 3. /health/Health/yx/yao/2007-08-07/6326.html。
适用的13个ICH非临床指导原则

序号
ICH编号
中文名称
1
S1A
药物致癌性试验必要性指导原则
2
S1B
药物致癌性试验
3
S1C(R2)
药物致癌性试验的剂量选择
4
S2(R1)
人用药物遗传毒性试验和结果分析指导原则
5
S3A
毒代动力学指导原则说明:毒性研究中的全身暴露量评价
S3A问答
毒代动力学指导原则说明:毒性研究中的全身暴露量评价问答
S9问答
抗肿瘤药物非临床评价指导原则问答
13
S10
药物光安全评价
6
S3B
药代动力学:重复给药的组织分布研究指导原则
7
S4
动物慢性毒性试验的期限(啮齿类和非啮齿类)
8
S6(R1)
生物制品的临床前安全性评价
9
S7A
人用药品安全药理学试验指导原则
10
S7B
人用药品延迟心室复极化(QT间期XX)潜在作用的非临床评研究
12
S9
抗肿瘤药物非临床评价指导原则
ICH 指导原则文件目录(中英文)
人用药品注册技术要求国际协调会(ICH)文件目录ICH的论题主要分为四类,因此ICH根据论题的类别不同而进行相应的编码分类:1. “Q”类论题:Q代表QUALITY,指那些与化工和医药,质量保证方面的相关的论题。
Q1/Q2...Q10都属于这类。
2. “S”类论题:S代表SAFETY,指那些与实验室和动物实验,临床前研究方面的相关的论题。
3. “E”类论题:E代表EFFICACY,指那些与人类临床研究相关的课题。
4. “M”类论题:M代表MULTIDISCIPLINARY, 指那些不可单独划入以上三个分类的交叉涉及的论题。
同时M又细分为5个小类:M1: 常用医学名词(Med DRA)M2: 药政信息传递之电子标准M3: 与临床试验相关的临床前研究时间的安排M4: 常规技术文件(CTD)M5: 药物词典的数据要素和标准一、ICH. 质量部分(Quality)稳定性1.Quality质量2.Q1: Stability稳定性3.Q1A(R2): Stability Testing of New Drug Substances and Products 新原料药和制剂的稳定性试验4.Q1B: Photostability Testing of New Drug Substances and Products 新原料药和制剂的光稳定性试验5.Q1C: Stability Testing for New Dosage Forms 新剂型的稳定性试验6.Q1D: Bracketing and Matrixing Designs for Stability Testing of Drug Substances and Drug Products原料药和制剂稳定性试验的交叉和矩阵设计 Q1E: Evaluation of Stability Data 稳定性数据的评估7.Q1F: Stability Data Package for Registration Applications in Climatic Zones III andIV在气候带III和IV,药物注册申请所提供的稳定性数据8.Q2: Analytical Validation分析验证9.Q2(R1): Validation of Analytical Procedures: Text and Methodology分析程序的验证:正文及方法论10.Q3: Impurities 杂质11.Q3A(R2): Impurities in New Drug Substances 新原料药中的杂质12.Q3B(R2): Impurities in New Drug Products (Revised Guideline) 新制剂中的杂质13.Q3C(R3): Impurities: Guideline for Residual Solvents 杂质:残留溶剂指南Impurities: Guideline for Residual Solvents (Maintenance) 杂质:残留溶剂指南(保留)PDE for Tetrahydrofuran (in Q3C(R3)) 四氢呋喃的日允许接触剂量PDE for N-Methylpyrrolidone (in Q3C(R3)) N-甲基吡咯烷酮的日允许接触剂量14.Q4: Pharmacopoeias药典15.Q4A: Pharmacopoeial Harmonisation 药典的协调16.Q4B: Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions药典内容的评估及推荐为用于ICH地区17.Q4B Annex1 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Residue on Ignition/Sulphated Ash General Chapter附录1 药典内容的评估及推荐为用于ICH地区关于灼烧残渣/灰分常规篇18.Q4B Annex2 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Test for Extractable Volume of Parenteral Preparations General Chapter附录2 药典内容的评估及推荐为用于ICH地区关于注射剂可提取容量测试常规篇19.Q4B Annex3 Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regionson Test for Particulate Contamination: Sub-Visible Particles General Chapter附录3 药典内容的评估及推荐为用于ICH地区关于颗粒污染物测试:不溶性微粒常规篇20.Q5: Quality of Biotechnological Products 生物技术制品质量21.Q5A(R1): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin来源于人或者动物细胞系的生物技术产品的病毒安全性评估22.Q5B: Quality of Biotechnological Products: Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products生物技术产品的质量:源于重组DNA的蛋白质产品的生产中所用的细胞中的表达构建分析23.Q5C: Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products生物技术产品的质量:生物技术/生物产品的稳定性试验24.Q5D: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products用于生产生物技术/生物产品的细胞底物的起源和特征描述25.Q5E: Comparability of Biotechnological/Biological Products Subject to Changes inTheir Manufacturing Process基于不同生产工艺的生物技术产品/生物产品的可比较性26.Q6: Specifications规格27.Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (including decision trees) 质量规格:新原料药和新制剂的检验程序和可接收标准:化学物质(包括决定过程)28.Q6B: Specifications: Test Procedures and Acceptance Criteria for29.Biotechnological/Biological Products质量规格:生物技术/生物产品的检验程序和可接收标准30.Q7: Good Manufacturing Practices (GMP)31.Q7A: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients活性药物成份的GMP指南32.Q8: Pharmaceutical Development药物研发33.Annex to Q8Q8附录34.Q9: Quality Risk Management质量风险管理35.Q10: Pharmaceutical Quality System药物质量体系二、ICH.安全性部分(Safety) 致癌试验1.S1A Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals 药物致癌试验的必要性2.S1B Testing for Carcinogenicity of Pharmaceuticals 药物致癌试验3.S1C(R2) Dose Selection for Carcinogenicity Studies of Pharmaceuticals药物致癌试验的剂量选择4.S1C’药物致癌试验的剂量选择的附件:补充剂量限度和有关注释遗传毒性5.S2(R1) Guidance on Genotoxicity Testing and Data Interpretation forPharmaceuticals Intended for Human Use 人用药物的遗传毒性试验和数据分析指导原则6.S2A药物审评遗传毒性试验的特殊性指导原则7.S2B遗传毒性:药物遗传毒性试验标准组合药代8.S3A Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposurein Toxicity Studies 毒代动力学指导原则:毒性研究中全身暴露的评价9.S3B Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies药代动力学:重复给药的组织分布研究指导原则慢性毒性10.S4Duration of Chronic Toxicity Testing in Animals (Rodent and Non RodentToxicity Testing) 动物慢性毒性试验的周期(啮齿类和非啮齿类)生殖毒性11.S5(R2) Detection of Toxicity to Reproduction for Medicinal Products andToxicity to Male Fertility (the Addendum dated November 1995 has beenincorporated into the core guideline in November 2005 )12.S5A药品的生殖毒性检测13.S5B雄性生育力毒性其他14.S6Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals 生物技术药品的临床前安全性试验15.S7A Safety Pharmacology Studies for Human Pharmaceuticals 人用药物的安全性药理研究16.S7B The Non-clinical Evaluation of the Potential for Delayed VentricularRepolarization (QT Interval Prolongation) by Human Pharmaceuticals人用药延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则17.S8Immunotoxicity Studies for Human Pharmaceuticals人类药品的免疫毒性研究18.S9 Nonclinical Evaluation for Anticancer Pharmaceuticals抗癌药物的临床前评价19.S10 Photosafety Evaluation三、ICH.临床部分(Efficacy)1.E1The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions 评价临床长期给药方案的安全性2.E2A Definitions and Standards for Expedited Reporting 快速报告的定义和标准3.E2B(R3) Data Elements for Transmission of Individual Case Safety Reports个体病例安全性报告传递的数据要素4.E2C Periodic Benefit-Risk Evaluation Report 上市药品定期安全性更新报告5.E2D Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting批准后安全性数据管理:快速报告的定义和标准6.E2E Pharmacovigilance Planning药物警戒计划7. E2F Development Safety Update Report8.E3Structure and Content of Clinical Study Reports 临床研究报告的结构与内容9.E4Dose-Response Information to Support Drug Registration 新药注册所需量-效关系的资料10.E5(R1)Ethnic Factors in the Acceptability of Foreign Clinical Data 对国外临床研究资料的种族因素的可接受性11.E6(R1) Good Clinical Practice: Consolidated Guideline 药品临床研究规范(GCP)一致性指导原则12.E7Studies in Support of Special Populations: Geriatrics 老年人群的临床研究13.E8General Considerations for Clinical Trials 临床试验的一般考虑14.E9Statistical Principles for Clinical Trials 临床试验统计原则15.E10Choice of Control Group and Related Issues in Clinical Trials 对照组的选择16.E11Clinical Investigation of Medicinal Products in the Pediatric Population 儿童人群的临床研究17.E12按治疗分类的各类药物临床评价E12 Principles for Clinical Evaluation of New Antihypertensive Drugs18.E14The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs 非抗心律失常药物致QT/QTc间期延长及潜在心律失常作用的临床评价19.E15 Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories20.E16 Biomarkers Related to Drug or Biotechnology Product Development: Context, Structure and Format of Qualification Submissions四、ICH.综合部分 (Multidisciplinary)1.M1医学术语Med DRA2.M2Electronic Transmission of Individual Case Safety Reports MessageSpecification (ICH ICSR DTD Version 2.1) companion document to E2B(R3)注册资料传递所需的电子代码3.M3Guidance on Nonclinical Safety Studies for the Conduct of Human ClinicalTrials and Marketing Authorization for Pharmaceuticals与临床研究有关的临床前研究的时间安排4.M4 Organisation of the Common Technical Document for the Registration ofPharmaceuticals for Human Use (Edited with Numbering and Section Header Changes, September 2002). Including the Annex : the Granularity Document(Revised November 2003).CTD(common technical document)(包括CTD、CTD-Q、CTD-S、CTD-E和eCTD)药品词汇的数据要素和标准5.M4Q (R1) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Quality (Edited with Numbering and Section Header Changes, September 2002)6.M4S (R2) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Safety (Edited with Numbering and SectionHeader Changes, September 2002)7.M4E (R1) The Common Technical Document for the Registration ofPharmaceuticals for Human Use: Efficacy (Edited with Numbering and Section Header Changes, September 2002)8.M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities inPharmaceuticals to Limit Potential Carcinogenic Risk Reference:1. 《ICH 药品注册的国际要求》2. 3. /health/Health/yx/yao/2007-08-07/6326.html。
ICH安全性指导原则(S)

阶段5
1995.11.29
有
S1B: Testing for Carcinogenicity of Pharmaceuticals
S1B:药物致癌性试验
阶段5
1997.7.16
有
S1C(R2): Dose Selection for Carcinogenicity Studies of Pharmaceuticals
安全性(Safety Guidelines)
主要内容:实验室动物实验等临床前研究相关指导原则
序号
英文题目
中文译文
阶段
发布时间
是否有中文译稿
1
S1A - S1C Carcinogenicity Studies/致癌性研究
S1A: Need for Carcinogenicity Studies of Pharmaceuticals
S2(R1):人用药物遗传毒性试验和结果分析指导原则
阶段5
2011.11.9
有
3
S3A - S3B Toxicokinetics and Pharmacokinetics/毒代动力学和药代动力学
S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies
有
6
S6 Biotechnological Products/生物技术产品
S6(R1): Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals
S6(R1):生物制品的临床前安全性评价
ICH遗传毒性标准试验组合的最新要求--ICHS2(R1)人用药物遗传毒性试验和结果分析指导原则介绍(一)

发布日期20080729栏目化药药物评价>>综合评价ICH遗传毒性标准试验组合的最新要求--ICHS2(R1)人用药物遗传毒性试标题验和结果分析指导原则介绍(一)作者黄芳华部门正文内容审评二部黄芳华从2006年9月开始,ICH提出对遗传毒性指导原则进行修订改版,ICH 专家组对遗传毒性试验一系列问题进行了讨论修订,并起草了S2(R1):Guidance on Genotoxicity Testing and Data Interpretation forPharmaceuticals Intended for Human Use(人用药物遗传毒性试验和结果分析指导原则)。
该指导原则于2008年3月6日达到ICH进程的第二阶段。
根据协调进程,该指导原则由ICH委员会向ICH三方的管理部门(欧盟、日本和美国)以征询国内外的意见。
该指导原则将替代ICH原来的遗传毒性研究的两个指导原则(S2A和S2B),这将对药物遗传毒性研究产生重大的影响。
及时了解国际遗传毒性技术要求的进展,对于国内遗传毒性的研究也有重大意义。
该指导原则对遗传毒性试验标准组合、体内外试验的要求及结果分析和评价方面提出了新的要求。
以下是ICHS2(R1)关于遗传毒性标准试验组合的要求。
一、基本原理药品注册要求对其潜在性遗传毒性进行全面评价。
大量回顾研究表明许多被细菌回复突变(Ames)试验检出是致突变剂的化合物是啮齿类动物致癌剂。
加上体外哺乳动物细胞试验可提高灵敏度和加宽遗传学物质的检测谱,但是同时也降低了预测的特异性,即提高了阳性结果发生率(而该结果与此啮齿类动物致癌性不相关)。
然而,组合方法仍是合理的,因为没有任何一个单一试验能检测所有与肿瘤发生相关的遗传毒性机制。
标准试验组合应具备的基本特征如下:i.用细菌回复突变试验评价致突变性。
该试验能检出相关的遗传学改变和大部分啮齿类动物和人类的遗传毒性致癌剂。
ii.遗传毒性还应采用与哺乳动物细胞体外和/或体内试验进行评价。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
发布日期20080729栏目化药药物评价>>综合评价ICH关于遗传毒性体外、体内试验的建议--ICHS2(R1)人用药物遗传毒性标题试验和结果分析指导原则介绍(二)作者黄芳华部门正文内容审评二部黄芳华前文已介绍了ICH关于遗传毒性标准试验组合的要求,以下介绍ICHS2(R1)Guidance on Genotoxicity Testing and Data Interpretation forPharmaceuticals Intended for Human Use(人用药物遗传毒性试验和结果分析指导原则)中关于体外、体内试验的建议。
一、对体外试验的建议1、试验重复和分析实验结果的重现性是涉及新方法或意外发现的研究的基本组成部分。
但是,用标准的、已广泛应用的遗传毒性试验进行常规试验时往往不需要完全重复。
这些试验都经过很好的充分验证且有有效的内部控制,对明确的阳性或阴性结果试验通常不需要重复。
理想状态是可明确宣称试验结果是明确的阳性或明确的阴性。
但是,试验结果有时达不到阳性或阴性称谓的预先设定的标准,因此被定为“可疑”。
统计学方法的应用有助于数据分析;但是,充足的生物学分析是至关重要的。
可疑试验的重复可致(i) 一个明确的阳性结果,因此作为整体阳性结果;(ii) 一个阴性结果,所以结果不需要重复和总体结果为阴性;或(iii)另一个可疑的结果,最后结论仍维持可疑。
2、对细菌突变试验的推荐方案OECE指导原则(1997)和IWGT报告(Gatehouse et al, 1994) 给出了对方案的建议。
2.1 高剂量水平的选择最高剂量水平当不受溶解度或细胞毒性限制时,推荐最高浓度为5000µg/皿。
溶解度的限制对于细菌培养,如果沉淀不干扰评分应对沉淀量进行评分,毒性不限制,最高剂量不超过5000µg/皿。
有证据表明在用细菌遗传毒性试验检测某些受试物时,在不溶解的浓度范围内也能检测出剂量相关性的遗传毒性。
另一方面,明显的沉淀物可干扰对集落的计算或使受试物无法进入细胞并与DNA相互作用。
如果未观察到细胞毒性,应以产生沉淀的最低浓度作为最高浓度。
如果观察到剂量相关的细胞毒性或诱变性,则不管溶解度如何,则应按下面所述中的细胞毒性确定最高浓度。
细胞毒性限制在细菌回复突变试验中,最高剂量应表现出明显的毒性,但不超过最高浓度5000µg/皿。
毒性可通过回复突变数目的减少,和/或背菌菌苔的清除或减少来检测。
2.2 试验设计/试验方案推荐的菌株组合(OECD)包括可检测碱基置换和移码突变的菌株,如下:鼠伤寒沙门氏菌TA98;TA100;TA1535;TA1573或TA97或TA97a;TA102或大肠埃希杆菌WP2 uvrA或大肠埃希杆菌WP2 uvrA(pKM101)。
与OECE和IWGT报告建议的一个不同点是:基于检测药物的经验,当结果是明确的阳性或阴性,且是在完全充分的方案下进行(包括在有和无代谢活化条件下使用所有菌株,合适的剂量范围以满足高剂量选择的标准,合适的阳性和阴性对照)时,一个单独的细菌突变(Ames)试验足够,而且,对于检测药物,无论是平板掺入法或预培养法对于该单一试验都是合适的(注释7)。
可疑或弱阳性结果可能提示重复度试验的必要性,可能需调整方案,如合适的剂量水平间距。
3、对哺乳动物细胞试验的建议OECE指导原则(1997)和IWGT出版物(Kirsch-Volders et al 2003; Moore et al 2006) 给出了对方案的建议。
此处将说明对于检测药物与这些建议的不同点,特别是与最高浓度、细胞毒性和浓度相关的高浓度的选择(详见下文)。
3.1 高浓度的选择最高浓度当不受溶解度或细胞毒性限制时,推荐的最高浓度是1 mM或0.5 mg/ml (选择任一更低者)(注释8)。
溶解度的限制当溶解度受限时,若不受细胞毒性限制,最高浓度应是培养基中产生最少可见沉淀的最低浓度(若其不干扰评定)。
应通过诸如光镜、连续稀释沉淀、或培养过程中的外观等方法来评价沉淀(在给药结束时)。
细胞毒性在中期相染色体畸变或微核的体外细胞遗传学试验中细胞生长减少无需超过50%(注释9和10),或在小鼠淋巴瘤tk 突变试验中RTG(相对总生长率)减少无需超过80%(注释9)。
3.2试验设计/试验方案在体外中期相细胞进行染色体损伤的细胞遗传学评价,试验方案包括在有或无代谢活化、合适的阳性和阴性对照情况下进行试验。
受试物应接触作用3-6小时,取样时间大约在从给药开始后的1.5个正常细胞周期。
在有或无活化代谢状态下短期给药结果为阴性或可疑结果时,在无代谢活化情况下必须持续给药至取样的大约1.5个正常细胞周期,同样的原理也适用于体外微核细胞试验,除了取样时间为从给药处理开始后的1.5-2个正常细胞周期以让细胞完成有丝分裂和进入下一个分裂间期。
对于两种体外细胞遗传学试验,某些化合物在更长时间的给药处理、延长取样时间或恢复期后更易检测到,如某些核苷类似物或亚硝胺类。
在中期相畸变试验中,通过记录中期相多倍体(包括核内复制的)占中期相细胞的百分比率可获得倍数状态信息。
有丝分裂指数(MI)或多倍体细胞出现频率的升高可提示化合物可能具有诱导多倍体的潜力。
对于小鼠淋巴瘤tk试验,试验方案包括有合适的阳性和阴性对照、加和不加代谢活化的试验,试验方案包括在有或无代谢活化、合适的阳性和阴性对照,受试物应接触作用3-4小时的情况下进行试验。
在有或无活化代谢状态下短期给药均得到阴性或可疑结果时,在无活化代谢情况下必须持续给药约24小时。
合适的小鼠淋巴瘤tk试验包括(1)含有主要诱导小集落的阳性对照;(2)应对阳性对照、溶剂对照、至少一个阳性受试物浓度(必不可少),包括使突变体发生率最高的培养基的集落大小进行测定。
对体外哺乳动物细胞试验,可采用诸如上述所列出(如不同的给药周期、有和无代谢活化的试验)的内在的确证因素。
在这些试验后,试验结果为明确的阴性或阳性或时通常无需进一步的确证性试验。
对于可疑或弱阳性结果可能需要重复试验,可能需调整方案,比如合适的受试物浓度间距。
3.3 阳性对照同期阳性对照是重要的,但是遗传毒性的体外哺乳动物细胞试验已充分标准化,使得染色体畸变试验和MLA试验可仅在活化状态下采用阳性对照(若与非活化试验同期进行),以证明代谢活化系统的活性和试验系统的反应性。
二、对体内试验的建议1、检测染色体损伤的体内试验采用骨髓细胞分析染色体畸变或检测微核化的嗜多染红细胞的体内方法均可用于检测断裂剂。
大鼠和小鼠均适用于骨髓微核试验。
微核也可通过小鼠外周血中未成熟(如嗜多染)红细胞或大鼠血液新生网织红细胞测定(注释3)。
同样,来源于被认为对检测断裂剂/非整倍体诱导剂有足够灵敏度的其他种属的骨髓或外周血的未成熟红细胞也可使用(注释3)。
染色体畸变也可通过给药啮齿类动物的外周淋巴细胞培养来分析(注释11)。
注意:当没有进行体外哺乳动物细胞试验(选择2)时,建议进行体内微核试验,而不是中期相染色体畸变试验,以包含更多直接检测染色体丢失(可能导致非整倍体)的能力。
2、微核的自动化分析如果已被验证适合,可采用自动化分析系统(图像分析和流式细胞计量术)(OECD, 1997; Hayashi et al 2000; 2007)。
3、其他体内遗传毒性试验与标准组合(选择2)中的第二种试验同样的体内试验,可用作追加试验以在评价体外或体内试验结果时加大证明力度(注释4和11)。
虽然体外试验中可见的作用类型和任何代谢信息可有助于指导体内试验的选择,染色体畸变或内源基因的基因突变的研究在很多组织上采用标准方法是不可行的;当在转基因啮齿类动物上检测突变时,需延长给药时间(如28天)以使突变表达/固定,尤其是在有极少细胞分裂的组织上时。
因此,第二种体内试验经常评价替代(DNA损伤)终点。
已有大量对方案方面的公开经验和建议的试验包括:DNA链断裂试验如单细胞凝胶电泳(“彗星”)试验和碱洗脱试验,体内转基因小鼠突变试验和DNA共价结合试验,所有这些试验均可采用多种组织(注释4),除了肝脏程序外DNA合成(UDS)试验。
4、体内遗传毒性试验啮齿类动物雄性/雄性的使用若检测性别特异性药物,试验应在适宜的性别中进行。
短期的体内试验通常仅在一种性别中进行(注释12)。
只有在已有的毒性/代谢资料提示在所用种属上存在明显的性别差异时,短期试验才考虑采用两种性别。
否则,对于短期试验单用雄性即可。
当该遗传毒性试验结合在两种性别的重复给药毒性试验中时,应对两种性别进行采样,但是如果毒性/代谢方面没有明显性别差异,应对单一性别进行评分。
对性别评分的剂量水平需符合合适的剂量水平标准(4.7.2和4.7.3节)。
类似的原则可应用于其他已建立的体内遗传毒性试验。
5、体内遗传毒性试验中多次给药的使用及与毒理试验的结合5.1 采样时间当微核试验结合在多周给药试验中时,血或骨髓的采样应在末次给药的次日(见下面的附加采血样的建议)。
当在多周试验(如28天)血或骨髓用于微核测定时,明显的血液毒性可影响检测微核的能力,即在短期给药后可诱导可检测到的微核升高的剂量也可能毒性太大,使多次给药后无法分析。
在给药2~4天时附加采血样是有用的(Hamada et al, 2001);见4.7.3节。
如果需要为染色体断裂剂和潜在非整倍体检测提供证据可采用早期取样(除了注释13和17外)。
对于其他遗传毒性试验,采样时间根据测定终点进行合理选择。
对于染色体损伤/链断裂测定,通常在末次给药的几个小时(如2~6小时)内取样。
原则上,只要最高剂量/暴露是合适的,任何周期的试验均是适当的。
5.2 可分析的动物样本数可分析的动物样本数取决于微核试验(OECD)或其他遗传毒性试验的当前建议,通常不包括毒性试验中所有的给药动物。
(用于遗传毒性分析的动物应随机选择)。
6、给药途径通常给药途径应与临床拟用途径一致,如口服、静脉或皮下,但是若为获得全身暴露需要,也可进行调整,如对于局部给药的化合物(见2.3.4节)。
7、体内试验的剂量选择应采用有代表性的三个剂量水平(Hayashi et al, 2005)。
7.1 短期试验对于短期试验(通常是给药1~2次)方案,遗传毒性试验推荐的最高剂量是:极限剂量2000mg/kg(若可耐受),或最大耐受量,例如:微核试验(OECD 474)采用产生明显毒性的剂量,基于同样的剂量范围,稍高一些剂量即预期会产生死亡。
(同样的建议也用于彗星试验[Hartmann et al, 2003])和转基因突变试验[Heddle et al, 2000]。
剂量选择时也应考虑骨髓红细胞抑制的发生。
稍低剂量通常应在该剂量下的约2~3倍间距。