选择性c-Met抑制剂类抗癌药Tivantinib
2022年10—11月FDA批准新药概况

2022年10—11月FDA批准新药概况作者:来源:《上海医药》2023年第21期2022年10月,FDA批出2個新生物制剂,分别为治疗肝细胞癌药物Imjudo(曲美木单抗)和治疗多发性骨髓瘤药物Tecvayli(特立妥单抗)。
11月,FDA批出2个新生物制剂,分别为治疗卵巢癌药物Elahere(mirvetuximabsoravtansine-gynx)和延缓1型糖尿病发作药物Tzield(teplizumab-mzwv)。
1 Imjudo(曲美木单抗)Imjudo为注射液,被批准与度伐利尤单抗(durvalumab)联合用药,用于治疗患有不可切除的肝细胞癌(hepatocellular carcinoma, HCC)的成年患者,为患者提供一种由双重免疫检查点抑制剂构成的全新免疫组合疗法。
HCC约占成人原发性肝癌的75%,晚期HCC患者5年存活率仅为7%。
Imjudo是一款人源化的抗细胞毒性T淋巴细胞相关蛋白4(cytotoxic T-lymphocyteassociated protein 4, CTLA-4)抗体,可以阻断CTLA-4、促进T细胞活化,从而启动免疫系统对抗癌细胞,诱导其死亡。
度伐利尤单抗是一款抗程序性细胞死亡受体配体1(programmed cell death-ligand 1, PD-L1)单克隆抗体,通过阻断PD-L1与程序性细胞死亡受体-1(programmed cell death-1, PD-1)和CD80蛋白的结合,解除肿瘤细胞对免疫反应的抑制。
2 Tecvayli(特立妥单抗)Tecvayli为注射液,被加速批准用于治疗复发或难治性多发性骨髓瘤(multiple myeloma,MM)成年患者,这些患者已经接受至少4种前期疗法,包括蛋白酶体抑制剂、免疫调节剂、抗CD38单抗。
MM是血液系统的第二常见恶性肿瘤,约占血液系统恶性肿瘤的10%。
Tecvayli是一款首创的B细胞成熟抗原(B cell maturation antigen, BCMA)/CD3双特异性T 细胞结合抗体,也是首款获批治疗MM和靶向BCMA的双特异性抗体疗法。
TKI类药物

过去十年,许多酪氨酸激酶抑制剂(TKI)药物已经在肿瘤领域应用,根据一篇最近的综述,看看截止到2013年8月份,由FDA 和EMA批准的所有TKI药物都有那些。
1、阿西替尼(axitinib,Inlyta)在2012年1月27日获FDA批准治疗对其它药物没有应答的晚期肾癌(肾细胞癌),由辉瑞(Pfizer)公司开发。
阿西替尼是多靶点酪氨酸激酶抑制剂,具体用法为5mg 空腹口服 2/日。
2、克唑替尼(crizotinib,XALKORI)用于治疗ALK阳性的局部晚期或转移的非小细胞肺癌,推荐剂量和方案是250 mg口服每天2次。
3、达沙替尼(Dasatinib,施达赛Sprycel)治疗对包括甲磺酸伊马替尼在内的治疗方案耐药或不能耐受的慢性髓细胞样白血病。
FDA也经正常程序批准达沙替尼治疗对其他疗法耐药或不能耐受的费城染色体阳性的急性淋巴细胞性白血病成人患者。
4、厄洛替尼(Erlotinib,特罗凯Tarceva)既往接受过至少一个化疗方案失败后的局部晚期或转移的非小细胞肺癌。
厄洛替尼单药用于非小细胞肺癌的推荐剂量为150mg/日,至少在进食前1小时或进食后2小时服用。
5、吉非替尼(Gefitinib,易瑞沙Iressa)适用于治疗既往接受过化学治疗或不适于化疗的局部晚期或转移性非小细胞肺癌。
推荐剂量为250mg(1片)每日1次,空腹或与食物同服。
6、伊马替尼(Imatinib,格列卫)用于治疗慢性粒细胞性白血病(CML),胃肠道间质瘤(胃肠道间质瘤)和其他一些疾病。
到2011年,该药已被FDA批准用于治疗10个不同的癌症。
7、拉帕替尼(Lapatinib;泰立沙Tykerb)联合卡培他滨治疗ErbB-2过度表达的,既往接受过包括蒽环类,紫杉醇,曲妥珠单抗(赫赛汀)治疗的晚期或转移性乳腺癌。
推荐剂量为1250mg,每日1次,第1~21天服用,与卡培他宾2000mg/d,第1~14天分2次服联用。
8、尼洛替尼(Nilotinib,达希纳Tasigna)适应症为对既往治疗(包括伊马替尼)耐药或不耐受的费城染色体阳性的慢性髓性白血病(Ph+ CML)慢性期或加速期成人患者。
伏美替尼和贝福替尼作用机制

伏美替尼和贝福替尼作用机制
伏美替尼和贝福替尼是两种常见的药物,它们分别用于治疗不同类型的癌症。
首先,我们来看看伏美替尼,它是一种酪氨酸激酶抑制剂,主要用于治疗慢性髓细胞白血病(CML)和胃肠道间质瘤(GIST)。
伏美替尼通过抑制BCR-ABL酪氨酸激酶的活性来发挥作用,这是CML的致病基础。
此外,伏美替尼还可以抑制肿瘤细胞中的PDGF受体和c-KIT受体,从而阻断肿瘤细胞的生长和增殖。
而贝福替尼是一种表皮生长因子受体(EGFR)酪氨酸激酶抑制剂,主要用于治疗非小细胞肺癌和胰腺癌。
贝福替尼通过抑制EGFR 的活性来发挥作用,从而阻断肿瘤细胞的生长和扩散。
此外,贝福替尼还可以抑制血管内皮生长因子受体(VEGFR)和PDGF受体的活性,从而阻止肿瘤血管的形成和肿瘤细胞的营养供应。
总的来说,伏美替尼和贝福替尼都是靶向治疗药物,通过干扰肿瘤细胞的生长信号通路来抑制肿瘤的生长和扩散。
它们的作用机制虽然不同,但都在一定程度上改善了特定类型癌症患者的预后,并为临床治疗提供了新的选择。
需要注意的是,使用这些药物时应严格遵医嘱,因为它们可能会引起一系列不良反应和副作用。
mvac化疗方案1

mvac化疗方案1化疗是一种通过使用化学药物来治疗癌症的治疗方法。
是许多癌症患者的首选治疗方法之一。
其中一种被广泛使用的化疗方案是Mvac方案。
本文将介绍Mvac化疗方案1的详细信息,包括药物组合、治疗流程和可能的副作用。
一、药物组合Mvac方案是由四种不同类型的药物组成:甲氨蝶呤(Methotrexate)、去甲氧基顺铂(Vinblastine)、依维莫仑(Adriamycin)和卡铂(Cisplatin)。
这些药物通过不同的方式作用于癌细胞,以抑制其生长和扩散。
1. 甲氨蝶呤(Methotrexate):甲氨蝶呤是一种抗代谢药物,能够抑制细胞的DNA和RNA的合成,从而干扰癌细胞的生长和分裂。
2. 去甲氧基顺铂(Vinblastine):去甲氧基顺铂是一种生物碱类药物,它能够干扰癌细胞的有丝分裂过程,阻止癌细胞的进一步增殖。
3. 依维莫仑(Adriamycin):依维莫仑是一种抗生素类药物,可以通过结合DNA和干扰其复制,进而导致癌细胞死亡。
4. 卡铂(Cisplatin):卡铂是一种铂类化合物,它可以与DNA结合,引发DNA链的断裂,阻止癌细胞的生长和增殖。
二、治疗流程Mvac化疗方案1通常需要在医院进行,治疗流程根据个体情况可能会有所不同。
1. 药物给药:首先,医生会根据患者的身体状况和病情确定化疗方案,然后通过输液的方式将药物注射入患者的血液中。
药物计量和注射频率会根据具体情况进行调整。
2. 化疗周期:Mvac化疗方案1通常是按照周期进行的,每个周期包括了特定的用药和休息时间。
通常,一个周期的长度为21天左右。
每个周期内,患者会经历一段化疗药物的注射期和一段恢复期。
3. 治疗阶段:Mvac化疗方案1通常包括预处理阶段、手术阶段和辅助化疗阶段。
- 预处理阶段:在一些情况下,患者需要在手术之前进行化疗来减小肿瘤的体积。
预处理阶段的化疗有助于手术的顺利进行。
- 手术阶段:在预处理阶段之后,患者可能需要进行手术来切除肿瘤。
avutometinib结构式

Avutometinib 结构式Avutometinib,也被称为AC0010,是一种新型的小分子靶向药物,属于酪氨酸激酶抑制剂。
它具有独特的化学结构和药理特性,被广泛用于肿瘤治疗领域。
化学结构Avutometinib 的化学名称是N-[4-[[[2-(dimethylamino)ethyl]amino]methyl]phenyl]-2-[(4-methoxyphenyl)amino]-1H-pyrazolo[3,4-b]pyridine-3-carboxamide。
从化学结构上看,它由苯环、吡唑环和吡啶环组成。
这种结构使得它能够与特定的酪氨酸激酶相互作用,并抑制其活性。
药理特性Avutometinib 通过选择性地抑制肿瘤细胞中的酪氨酸激酶活性来发挥治疗作用。
该药物与EGFR(表皮生长因子受体)、HER2(人类表皮生长因子受体2)和HER4等相关蛋白发生相互作用,阻断其信号传导通路。
这些信号通路在肿瘤细胞中起着重要的生长和增殖调节作用,因此抑制它们的活性可以有效地抑制肿瘤的生长。
此外,Avutometinib 还具有抗血管生成和免疫调节作用。
它可以通过抑制血管内皮生长因子受体(VEGFR)和PD-1等相关蛋白的活性,阻止肿瘤血管生成,并增强免疫细胞对肿瘤的攻击能力。
临床应用Avutometinib 目前已被证实在多种肿瘤治疗中具有显著的抗肿瘤活性。
以下是一些临床应用的示例:肺癌Avutometinib 在非小细胞肺癌(NSCLC)治疗中显示出良好的效果。
它可以通过抑制EGFR活性来减少肿瘤细胞的增殖,并有效地延长患者的生存期。
此外,该药物还可与其他化疗药物联合使用,以增强治疗效果。
乳腺癌Avutometinib 在HER2阳性乳腺癌治疗中显示出潜力。
HER2是乳腺癌中常见的过表达蛋白,与肿瘤的恶性程度和预后密切相关。
Avutometinib 可以选择性地抑制HER2活性,并抑制肿瘤细胞的生长和扩散。
抗癌药物作用机理及作用靶点
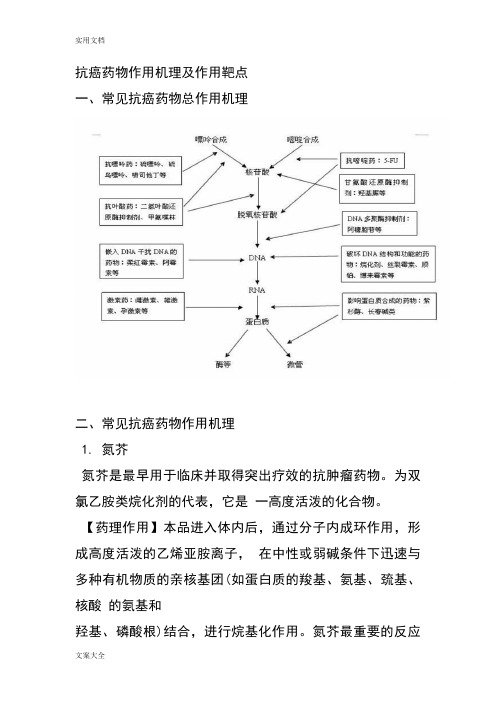
抗癌药物作用机理及作用靶点一、常见抗癌药物总作用机理二、常见抗癌药物作用机理1. 氮芥氮芥是最早用于临床并取得突出疗效的抗肿瘤药物。
为双氯乙胺类烷化剂的代表,它是一高度活泼的化合物。
【药理作用】本品进入体内后,通过分子内成环作用,形成高度活泼的乙烯亚胺离子,在中性或弱碱条件下迅速与多种有机物质的亲核基团(如蛋白质的羧基、氨基、巯基、核酸的氨基和羟基、磷酸根)结合,进行烷基化作用。
氮芥最重要的反应是与鸟嘌呤第7位氮共价结合,产生DNA 的双链内的交叉联结或DNA 的同链内不同碱基的交叉联结。
G1期及M 期细胞对氮芥的细胞毒作用最为敏感,由G1期进入S 期延迟。
【适应症】主要用于恶性淋巴瘤及癌性胸膜、心包及腹腔积液。
目前已很少用于其他肿瘤,对急性白血病无效。
与长春新碱(VCR)、甲基卡肼(PCZ)及泼尼松(PDN)合用治疗霍奇金病有较高的疗效,对卵巢癌、乳腺癌、绒癌、前列腺癌、精原细胞瘤、鼻咽癌(半身化疗法)等也有一定疗效;腔内注射用以控制癌性胸腹水有较好疗效;对由于恶性淋巴瘤等压迫呼吸道和上腔静脉压迫综合征引起的严重症状,可使之迅速缓解。
2.环磷酰胺环磷酰胺为氮芥与磷酰胺基结合而成的化合物,是临床常用的烷化剂类免疫剂。
【药理作用】该品在体外无抗肿瘤活性,进入体内后先在肝脏中经微粒体功能氧化酶转化成醛磷酰胺,而醛酰胺不稳定,在肿瘤细胞内分解成酰胺氮芥及丙烯醛,酰胺氮芥对肿瘤细胞有细胞毒作用。
环磷酰胺是双功能烷化剂及细胞周期非特异性药物,可干扰 DNA 及 RNA 功能,尤以对前者的影响更大,它与DNA 发生交叉联结,抑制DNA 合成,对S 期作用最明显。
【适应症】该品为最常用的烷化剂类抗肿瘤药,进入体内后,在肝微粒体酶催化下分解释出烷化作用很强的氯乙基磷酰胺(或称磷酰胺氮芥),而对肿瘤细胞产生细胞毒作用,此外本品还具有显著免疫作用。
临床用于恶性淋巴瘤,多发性骨髓瘤,白血病、乳腺癌、卵巢癌、宫颈癌、前列腺癌、结肠癌、支气管癌、肺癌等,有一定疗效。
培米替尼作用机制

培米替尼作用机制随着医学研究的不断深入,越来越多的药物被开发出来,以满足人类对于疾病的治疗需求。
其中,培米替尼作为一种新型的抗癌药物,其疗效已经被广泛认可。
本文将从培米替尼的作用机制方面进行介绍,以期更好地了解该药物的治疗原理。
一、培米替尼的基本信息培米替尼是一种小分子酪氨酸激酶抑制剂,属于口服治疗的靶向药物。
该药物能够特异性地抑制肿瘤细胞中的酪氨酸激酶,从而抑制肿瘤细胞的生长和扩散。
培米替尼的化学结构如下图所示:培米替尼的分子式为C23H21ClFN5O3,相对分子质量为473.9。
该药物主要通过口服的方式给药,其生物利用度高达90%以上,半衰期约为40小时。
二、培米替尼的作用机制培米替尼的作用机制主要与其抑制酪氨酸激酶有关。
酪氨酸激酶是一种重要的酶类蛋白,在细胞的信号转导中起着重要的作用。
该酶能够通过磷酸化作用,调节多种细胞信号通路的活性,从而影响细胞的生长、分化、凋亡等生物学过程。
而在肿瘤细胞中,酪氨酸激酶的活性往往会受到过度激活,从而导致肿瘤细胞的异常增殖和侵袭。
培米替尼能够特异性地抑制肿瘤细胞中的酪氨酸激酶,从而干扰肿瘤细胞的生物学过程,抑制肿瘤细胞的生长和扩散。
具体来说,培米替尼的作用机制主要包括以下几个方面:1. 抑制酪氨酸激酶的活性培米替尼能够结合酪氨酸激酶的ATP结合位点,从而抑制其活性。
这种结合方式是可逆的,因此培米替尼的作用具有一定的选择性和特异性。
在细胞内,培米替尼能够特异性地抑制多种酪氨酸激酶的活性,包括EGFR、HER2、ALK等。
这些激酶在多种肿瘤中都起着重要的作用,因此培米替尼具有广泛的抗肿瘤活性。
2. 干扰细胞信号通路酪氨酸激酶在细胞信号通路中起着重要的作用,包括EGFR信号通路、PI3K/Akt信号通路等。
培米替尼的抑制作用能够干扰这些信号通路的正常传导,从而抑制肿瘤细胞的生长和扩散。
此外,培米替尼还能够诱导肿瘤细胞凋亡,从而促进肿瘤的消退。
3. 抑制血管生成肿瘤的生长和扩散需要大量的血供,因此血管生成在肿瘤的发生和发展中起着重要的作用。
希美替尼结构式-概述说明以及解释

希美替尼结构式-概述说明以及解释1.引言1.1 概述希美替尼(Imatinib)是一种广泛应用于白血病和其他恶性肿瘤治疗的靶向治疗药物。
它是第一代酪氨酸激酶抑制剂,通过抑制异常的酪氨酸激酶活性,阻止了癌细胞的生长和扩散。
希美替尼已被证明在治疗慢性髓性白血病(CML)、急性淋巴性白血病(ALL)和一些消化道肿瘤等疾病中具有显著的疗效。
随着对希美替尼的深入研究,人们对其治疗机制和潜在的临床应用也有了更深入的了解。
本文将着重介绍希美替尼的化学结构、药理作用以及临床应用,以期为读者提供更全面的了解和认识。
1.2 文章结构文章结构部分主要是说明整篇文章的结构安排,帮助读者更好地理解文章的内容组织。
本文的结构分为引言、正文和结论三个部分。
引言部分主要包括概述、文章结构和目的三个方面。
在概述部分,会简要介绍希美替尼这种药物的背景和重要性。
文章结构部分即是本段,将会介绍整篇文章的结构安排。
目的部分则明确了本文撰写的目的和意义。
正文部分包括了希美替尼的化学结构、药理作用以及临床应用三个方面,分别介绍了这种药物的化学成分、作用机制以及临床使用的情况。
结论部分则对全文的内容进行总结,强调希美替尼的重要性和未来发展的展望。
同时,通过结论部分可以让读者更清晰地理解全文的核心意义和价值。
最后,结尾的结束语也是文章的收尾部分,可以对全文进行一个简短的总结或思考。
整个文章结构分明,逻辑清晰,能够很好地引导读者了解希美替尼这一药物的相关知识和重要性。
1.3 目的:本文的主要目的是详细介绍希美替尼这一药物的化学结构、药理作用以及临床应用,以便读者能够更全面地了解这一重要药物。
通过对希美替尼的研究和分析,可以帮助读者更好地认识并理解希美替尼在临床上的作用机制和应用领域,为医学研究和医疗实践提供参考和指导。
希美替尼作为一种重要的药物,对于肿瘤治疗以及其他相关疾病的治疗具有重要的意义,因此深入了解和研究希美替尼,可以为临床医生和研究人员提供更多选择和参考,有助于促进医疗领域的发展和进步。
2023仑伐替尼肝癌全病程应用中国专家指导意见(完整版)

2023仓健蕾尼肝癌金病程应用申国专家指导意见(完整版)摘要甲磺酸仓伐苔尼是一种针对血管内皮生长因子受体1~3、成纤维细胞生长因子受体1~4、血小板衍生生长因子受体a..干细胞生长因子受体以及转染重排墓因等靶点的口服醋氨酸激酶受体抑制剂。
该药于2018年9月4日经我国国家药晶监督管理局批准,用于治疗未接受过系统治疗的不可切除肝细胞癌患者。
截至2023年2月,仓伐苔尼已在我国上市4年余,积累了一系列临床研究证据。
为了临床上更加台理、高效使用仓伐苔尼,国内相关领域的多学科专家学者,采用德尔菲法,根据仓伐苔尼上市前后的临床实践,参考真他抗血筐生咸阳制剂的使用经验,经过多次共同讨论,反复修改,最终形成《仓伐苔尼肝癌全病程应用中国专家指导意见》,以供临床医师参考。
关键词肝肿瘤;仓伐苔尼;肝细胞癌;系统治疗;全程管理;合理应用;安全’性一、概述(-)背景肝细胞癌(以下简称肝癌)是常见的恶性肿瘤之一,我国每年肝癌新发病例和死亡病例均占全世界约50%[ 121H BV感染仍然是肝癌发生的主要危险因素,尤真是以我国为主的亚洲国家[3 1我国的肝癌与欧美国家及日本在发病特征、发病原因等多方面存在显著不同,真高高度异质性。
在我国,由于肝癌起病隐匿,症状不典型,大多数肝癌患者在初诊时已是中晚期,错过最佳手术切除机会旦进展迅速,导致整体预后差,长期生存率低。
药物治疗是中晚期肝癌治疗中不可或缺的手段,冥中靶向药物占据重要地位[41目前我国批准用于肝癌一线治疗方案包括:一线单药治疗(索拉非尼[5-61仓伐苔尼[7 ]和多纳非尼[8 ] i联合治疗(贝伐珠单克隆抗体联合阿苔利珠单克隆抗体[91信迪利单克隆抗体联合贝伐珠单克隆抗体类似饵10l阿帕苔尼联合卡瑞利珠单壳隆抗俐11]拟及FOLFOX4方案[12 1208年,墓于REFLECT的研究结果,仓伐苔尼在我国获批用于未接受过全身系统治疗的不可切除肝癌患者[71仓伐苔尼推荐剂量用法:体质量<60kg患者,推荐剂量为8mg(2粒,4mg/粒),口服,每日1次;体质量注60kg患者,推荐剂量为12mg( 3粒,4mg/粒),口服,每日1次。
EGFR-TKI 耐药机制及其个体化治疗策略

常见获得性耐药不常见获得性耐药表型改变EGFR突变患者在TKI治疗过程中获得性耐药通常是在苏氨酸看门氨基酸残基第790位发生第2位点突变,即T790M,发生率约50%。
T790M突变改变了ATP 的亲和性,导致EGFR-TKI不能有效阻断信号通路而产生耐药。
研究显示,L858R合并T790M突变对ATP的亲和力比单纯L858R强,而TKI是ATP竞争性激酶抑制剂,故导致TKI与激酶区结合率降低[PMID:15737014]。
p.C797S位点是EGFR第三代小分子抑制剂(如AZD9291)的诱导性耐药位点,该位点的突变会导致EGFR蛋白构象发生改变,阻止其与药物结合,从而产生耐药性[PMID:25948633;25939061]。
临床前研究显示,L858R和L747S突变对吉非替尼和厄洛替尼耐药[PMID:18309959]。
临床个案研究显示,L858R突变性肺腺癌患者在长期接受EGFR-TKI治疗后,会出现T854A诱导性耐药位点[PMID:19010870]。
体外实验研究显示,E762V/G突变对厄洛替尼产生耐药性[PMID18588508]。
体外实验研究显示,V766T/V突变对厄洛替尼产生耐药性[PMID18588508]。
体外实验研究显示,V769L突变对厄洛替尼产生耐药性[PMID18588508]。
体外实验研究显示,L777M突变对厄洛替尼产生耐药性[PMID18588508]。
体外实验研究显示,K852T/E突变对厄洛替尼产生耐药性[PMID18588508]。
体外实验研究显示,A859D突变对厄洛替尼产生耐药性[PMID18588508]。
有临床的个案报道,一例携带有EGFR p.A859T突变的非小细胞肺癌患者对吉非替尼(Gefitinib)的治疗耐受[PMID: 15710947]体外细胞学实验研究显示,L844V突变的NSCLC细胞株对WZ4002、CO-1686和阿法替尼产生耐药性,对AZD9291敏感[PMID:25948633;18588508]。
吡妥布替尼结构式-概述说明以及解释

吡妥布替尼结构式-概述说明以及解释1.引言1.1 概述概述:吡妥布替尼(英文名称为Imatinib)是一种靶向治疗白血病和其他恶性肿瘤的药物,是一种酪氨酸激酶抑制剂。
吡妥布替尼可以有效抑制癌细胞的生长和扩散,并且对特定基因突变导致的异常细胞增殖起到显著的抑制作用。
由于其独特的分子结构和药理作用,吡妥布替尼在临床上被广泛应用,并取得了显著的治疗效果。
本文将从吡妥布替尼的化学结构、药理作用和临床应用等方面进行详细介绍,旨在帮助读者更全面地了解和认识这一重要的抗癌药物。
1.2 文章结构文章结构部分将主要包括以下内容:1. 引言部分:对吡妥布替尼进行简要的介绍,引发读者对该化合物的兴趣和好奇心。
2. 正文部分:分为三个子部分,分别介绍吡妥布替尼的化学结构、药理作用和临床应用,以便深入了解吡妥布替尼在医药领域的重要性和价值。
3. 结论部分:总结吡妥布替尼在医学领域的重要性,并展望其未来的发展前景,最终得出结论。
通过这样的文章结构,读者可以系统地了解吡妥布替尼的相关信息,从而更好地理解和掌握这一重要化合物的知识。
1.3 目的本文的主要目的是通过对吡妥布替尼的化学结构、药理作用和临床应用进行深入探讨,全面了解这一药物的特性和作用机制。
同时,结合吡妥布替尼在临床上的重要性,探讨其未来发展的潜力和前景。
通过对吡妥布替尼的研究和分析,可以为其在临床应用和新药开发中提供更多的参考和指导,促进药物研究领域的发展和进步。
希望本文能够为读者对吡妥布替尼有更深入的了解,并对其在医学和药物领域的应用产生更大的兴趣。
2.正文2.1 吡妥布替尼的化学结构:吡妥布替尼是一种常用的抗癌药物,化学名称为N-[3-(3-甲氧基-4-甲基苯基)-8-(3-胺基丙基)[1,3]噁二唑-2-基]-N'-甲基胍,分子式为C23H31N7O2,分子量为437.54。
吡妥布替尼的化学结构包括一个[1,3]噁二唑环和一个甲基胍基团。
[1,3]噁二唑环是一种含氮的杂环化合物,具有环状结构和芳香性质。
肺癌精准治疗!武田强效EGFR小分子酪氨酸激酶抑制剂mobocertinib获美国FDA突破性药物资格

肺癌精准治疗!武田强效EGFR小分子酪氨酸激酶抑制剂mobocertinib获美国FDA突破性药物资格武田制药(Takeda)近日宣布,美国食品和药物管理局(FDA)已授予mobocer tinib(TAK-788)突破性药物资格(BTD),用于治疗接受含铂化疗期间或之后病情进展、携带表皮生长因子受体(EGFR)第20号外显子插入突变的转移性非小细胞肺癌(NSCLC)患者。
目前,针对这种特殊类型的NSCLC,还没有批准的治疗方案。
mobocer tinib是一种小分子酪氨酸激酶抑制剂(TKI),旨在选择性靶向EGFR和人EGFR 2(HER2)第20号外显子插入突变。
突破性药物资格(BTD)是FDA在2012年创建的一个新药评审通道,旨在加快开发及审查用于治疗严重或威及生命的疾病、并且有初步临床证据表明该药与现有治疗药物相比能够实质性改善病情的新药。
获得BTD的药物,在研发时能得到包括FDA 高层官员在内的更加密切的指导,在新药上市审查有资格进行滚动审查和优先审查,以保障在最短时间内为患者提供新的治疗选择。
FDA授予mobocertinib BTD,基于一项I/II期研究中对mobocertinib治疗有应答的患者的总缓解率(ORR)和长期生存获益。
该研究正在评估mobocertinib (160mg,每日口服一次)治疗肿瘤中携带EGFR第20号外显子插入突变、并且先前已接受过系统化疗的局部晚期或转移性NSCLC患者的疗效和安全性,这类患者目前没有靶向疗法,当前的治疗方案提供的益处有限。
该研究的数据标志着在解决这类患者需求方面的潜在重大进展。
数据显示,mobocertinib治疗的中位无进展生存期(PFS)为7.3个月、总缓解率(ORR)为43%(n=12/28)。
研究中,mobocertinib的安全性可控(n=72),最常见的治疗相关不良事件是腹泻(85%)、恶心(43%)、皮疹(36%)、呕吐(29%)和食欲下降(25%)。
常用抗肿瘤用药——伊马替尼

2.
3.
本品能通过抑制CYP3A4而使环孢素、辛伐他汀的血药浓度升高。
4.
本品与华法林合用时可导致出血危险增加。
2.
出血 治疗胃肠间质瘤最严重的并发症为腹腔或肿瘤部位的出血,发生率约为5%。出血原因可能为伊马替尼快速、强盗的治疗作用所致。
血液系统 贫血较常见,可有血小板和中性粒细胞减少。故用药期间需定期检查血象,必要时给予集落刺激因子治疗或暂停用药。
3.
其他 头痛、头晕、味觉障碍、失眠等,少数患者可有眼结膜炎。长期用药可有血压异常(高血压或低血压)。另有肝功能异常、肌酐升高的报道。有肝功能损害的患者慎用本品。
药动学
本品口服后吸收迅速,Tmax为1小时,T1/2为18~22小时。平均生物利用度为97%以上。口服剂量25~1000mg之间时,不管单次还是多次给药血药浓度为剂量成正比。进食对吸收无明显影响。血浆蛋白结合率为96%。给药7天内伊马替尼及其代谢产物68%通过粪便排泄,13%通过肾脏排泄。
相互作用
1.
药物大类
抗肿瘤药
药物小类
常用抗肿瘤用药
药物名称
伊马替尼
英文名
Imatinib
适应症
慢性粒细胞性白血病,c-kit阳性的晚期或转移性恶性胃肠间质细胞瘤。
用法用量一般CNL患者来自每次400mg,加速期患者600mg,每日1次。恶性胃肠间质瘤患者每次400~800mg,每日1次。
药理学
本品为本种苯胺嘧啶的衍生物,属新型蛋白酪氨酸激酶抑制剂。它能够选择性地抑制bcr-abl阳性克隆的特异酪氨酸激酶。此外。它还能强烈抑制血小板衍化生长因子受体(PDGF)、干细胞因子(SCF)受体、c-kit,抑制PDGF和SCF介导的生化反应。但它不影响其他刺激因子如表皮生长因子的信号传导
mvac化疗方案

mvac化疗方案MVAC化疗方案是一种常用于治疗膀胱癌的化疗方案。
下面将详细介绍MVAC方案的具体内容和临床应用。
MVAC是指甲氨蝶呤(Methotrexate)、依酮替康(Vinblastine)、阿德福韦/MVAC方案(Adriamycin)和顺铂(Platinum)四种药物的化疗方案。
它是一种组合化疗方案,通过联合使用这些药物来达到更好的治疗效果。
MVAC方案通常用于治疗晚期膀胱癌,特别是肌层侵犯或扩散到淋巴结的膀胱癌。
这种化疗方案可以通过系统性治疗来控制癌症的进展并提高患者的生存率。
MVAC方案通常包括4个疗程,每个疗程间隔为2-4周,总治疗时间为8-12周。
每个疗程的治疗方式如下:1. 甲氨蝶呤(Methotrexate):通常以100 mg/m²的剂量静脉注射,每疗程的第一天。
2. 依酮替康(Vinblastine):常常以3 mg/m²的剂量静脉注射,每疗程的第一天。
3. 阿德福韦(MVAC方案):阿德福韦使用剂量为30 mg/m²的剂量静脉注射,每疗程的第一天。
4. 顺铂(Platinum):以70-100 mg/m²的剂量静脉注射,通常在每个疗程的第二天。
在MVAC方案治疗期间,患者需要接受严密的监测和支持治疗,包括血液检查、肾功能检查和心脏功能检查等。
由于MVAC方案可能导致严重的副作用,如骨髓抑制、恶心和呕吐、口腔溃疡、肾功能损害和心脏毒性,因此在治疗期间需要密切关注患者的身体状况并及时调整剂量或给予相应的支持治疗。
除了MVAC方案,目前还有其他一些化疗方案可供选择,如GC方案(顺铂和吉西他滨)、Gem-Cis方案(顺铂和吉西他滨)、DDMVAC方案(顺铂、多柔比星、甲氨蝶呤和Cytoxan)等。
这些化疗方案在治疗膀胱癌方面都有一定的疗效,但MVAC方案仍然被广泛应用于临床实践中。
总之,MVAC化疗方案是一种常用于治疗膀胱癌的化疗方案。
伊利替康 结构式

伊利替康结构式伊利替康,也称为依西美坦,是一种利用于治疗乳腺癌和睾丸癌的化学药物。
它是一种抗肿瘤药物,通过抑制肿瘤细胞的生长和有丝分裂来起到治疗作用。
伊利替康具有独特的结构式和分子特性,下面将详细介绍它的结构式、作用机制、药代动力学和临床应用。
一、伊利替康的结构式伊利替康的化学分子式为C28H22F2N4O4,分子量为502.5克/摩尔。
其结构式如下所示:```H H| |F - C - C=C - C=C - C O| | | || || | | C N| | O // ||H - C=C-C - C=C - C-N H H/ || /C C - C//\\ / \\O O O O```图 1 伊利替康的分子结构式二、伊利替康的作用机制伊利替康的作用机制主要涉及其对肿瘤细胞的生长和分裂的抑制作用。
它是一种激酶抑制剂,通过阻断细胞周期蛋白依赖激酶(CDK)的活化而抑制细胞有丝分裂。
具体来说,伊利替康主要抑制CDK4和CDK6的活性,从而阻止细胞周期的进行,使肿瘤细胞无法继续增殖和扩散。
伊利替康也可以通过抑制转录因子E2F的活性,阻止其介导的基因转录而干扰肿瘤细胞的增殖。
这些作用机制使得伊利替康成为一种有效的抗肿瘤药物。
三、伊利替康的药代动力学1. 吸收:伊利替康口服后在胃肠道吸收迅速,生物利用度高达70%~90%。
饭后服药会延缓吸收而不影响总吸收量。
2. 分布:伊利替康主要分布在体内的肝脏、肾脏、肺和睾丸等组织中,而不会穿透血脑屏障。
3. 代谢:伊利替康主要在肝脏中代谢,由肝脏中的细胞色素P450酶系统催化酶代谢。
4. 排泄:伊利替康主要通过肝脏代谢后,以尿液和粪便的形式排泄。
伊利替康的药代动力学特点表明它在体内的吸收、分布、代谢和排泄都比较稳定,为临床应用提供了保障。
四、伊利替康的临床应用伊利替康主要用于治疗激素受体阳性的、早期和晚期乳腺癌,以及激素受体阳性的、晚期或转移性的睾丸癌。
临床研究表明,伊利替康在这些肿瘤治疗中具有显著的疗效,并且与其他治疗手段联合应用时可以进一步提高治疗效果。
近年药物研发最热门靶点汇总

近年药物研发最热门靶点汇总1.癌症2000年后肿瘤信号⽹络被逐渐阐释、完善,⼤量的分⼦靶向药物进⼊临床研究、⾛上市场,近年针对受体酪氨酸激酶靶点如Bcr-Abl(见1.1)、VEGF/VEGFRs(见1.2)、PDGF/PDGFRs(见1.3)、EGFR/HER2(见1.5)、ALk(见1.7)已有多个药物上市,me-too品种的研发逐渐放缓,但扩展适应症、克服耐药性、优化治疗⽅案的研究还没有结束。
⽬前肿瘤信号⽹络中,FGFR(见1.4)、c-Met(见1.6)、HER3(见1.5)、Hedgehog(见1.13)等靶点吸引了不少的研究,但最热的当是PI3K/Akt/mTOR (见1.15)、Raf/MEK/ERK(见1.16)两条细胞内信号通路。
2013年FDA批准了BTK抑制剂ibrutinib,对CLL 的疗效很好,吸引了⼀些药企开发me-too/me-better药物。
涉及细胞周期调控的靶点如Aurora激酶(见1.8)、CDK(见1.9)、ChK(见1.10)也有不少新药在研,最耀眼的⽆疑是CDK4/6抑制剂,已经有三个分⼦推进到后期开发,⽽Aurora激酶和ChK抑制剂则⼤多在早期临床失败。
针对DNA 损伤修复的PARP(见1.11)的药物研发也回暖,⽽针对蛋⽩-蛋⽩相互左右的新靶点如Bcl-2(见1.12)、MDM2(见1.14)、IAP也有多个分⼦进⼊临床研究。
特别值得⼀提的是表观遗传调控剂,早年发现的阿扎胞苷、地西他滨等被证明为DNA甲基转移酶抑制剂,⽬前研究得最多的是HDAC抑制剂(见1.17),表观遗传的其他靶点如组蛋⽩赖氨酸甲基转移酶EZH2、组蛋⽩H3甲基转移酶DOT1L、溴结构域蛋⽩BET等也开展了⼤量基础研究。
近来抗癌领域最耀眼的⽆疑是免疫疗法,调节CTLA4、PD1/PDL1、4-1BB、OX40、CD27等免疫检查点(见1.18)可以激活T 细胞免疫应答,⽽基因⼯程修饰的CAR、TCR T细胞的应⽤更是标志着个性化免疫治疗时代的到来。
Cobimetinib中文说明书

Cobimetinib 中文说明书【药物名】Cobimetinib【商品名】Cotellic【美国初次批准】2015年11月10日【类别】蛋白激酶抑制剂【靶点】MEK【分子结构】分子式:C46H46F6I2N6O8 (2C21H21F3IN3O2 ·C4H4O4) 分子量为:1178.71【生产公司】罗氏子公司基因泰克【适应症】Cotellic联合威罗菲尼用于BRAF V600E或V600K突变的不可切除或转移性的黑色素瘤患者的治疗。
使用限制:不适用于野生型BRAF黑色素瘤患者的治疗。
【剂量和药物管理】(1)患者选择:患者存在BRAF V600E或V600K突变。
(2)推荐剂量:Cotellic的推荐剂量是在28天-疗程的第1-21天每天1次60 mg(3片,20mg/片)口服给药给药,直至疾病进展或不耐受;可与食物同时服用或不与食物同时服用;当丢失一剂量或因为呕吐丢失一剂量,则再当天无需服用额外剂量,下一剂量照常服用。
(3)与CYP3A抑制剂同时给药的剂量调整:避免中度/强度CYP3A抑制剂和Cotellic同时给药。
如对正在服用Cotellic 60 mg患者不可避免短期(14天或更短)内与中度CYP3A抑制剂同时服用,则减少Cotellic剂量至20 mg。
当停止中度CYP3A抑制剂给药后,可Cotellic剂量到60 mg。
使用另一种中度/强度CYP3A抑制剂时,减少Cotellic剂量至40 mg 或20 mg。
(4)根据不良反应进行剂量调整。
【剂型和规格】口服片剂:20 mg(每瓶63片),白色圆形的膜包衣片剂,在一侧凹陷处标记有“COB”;NDC 50242-717-01【禁忌症】无。
【警告和注意事项】(1)原发恶性肿瘤:Cotellic可引发皮肤恶性肿瘤和非皮肤恶性肿瘤。
密切监测患者的症状。
对皮肤恶性肿瘤:在治疗开始前和治疗中每2个月进行一次皮肤学评价并及时处理;当与威罗菲尼联合给药时,暂停Cotellic给药,并进行皮肤学监测6个月。
以c-Met为肿瘤治疗靶点的受体酪氨酸激酶抑制剂的研究进展

以c-Met为肿瘤治疗靶点的受体酪氨酸激酶抑制剂的研究进展张媛;程雨兰;周金培;张惠斌【期刊名称】《中国药科大学学报》【年(卷),期】2015(46)1【摘要】受体酪氨酸激酶c-Met在细胞的增殖、代谢以及肿瘤的产生、转移、血管生成中扮演着重要角色,c-Met成为抗肿瘤治疗的重要靶点。
HGF/c-Met信号通路与VEGFR等其他通路的相互作用(cross-talk)影响了抗肿瘤药物的作用效果,产生耐药性,因此,多激酶靶点联合用药成为新的抗肿瘤治疗手段。
本文介绍了c-Met 信号通路与多种膜受体间的相互作用以及由这种相互作用引起的对激酶抑制剂的耐药性,并综述了单靶点和多靶点的小分子c-Met抑制剂的研究进展。
【总页数】12页(P16-27)【关键词】受体酪氨酸激酶;c-Met;相互作用;药物耐药性;c-Met抑制剂;抗肿瘤靶点;cabozantinib;crizotinib【作者】张媛;程雨兰;周金培;张惠斌【作者单位】中国药科大学药物化学教研室【正文语种】中文【中图分类】R914;R969.2【相关文献】1.以c-Met为靶点的酪氨酸激酶抑制剂在癌症治疗中的研究 [J], 李慧逖;韩利夫;王丹;2.C-MET在非小细胞肺癌对表皮生长因子受体酪氨酸激酶抑制剂耐药中的作用研究进展 [J], 屈晶晶;顾其华;胡成平3.酪氨酸激酶类受体C-Met抑制剂研究进展 [J], 武维斌;詹轶群;杨晓明4.以c-met为靶点的酪氨酸激酶抑制剂的研究进展 [J], 严家菊;刘靖;张首国;温晓雪;王林5.以c-Met为靶点的小分子酪氨酸激酶抑制剂的研究进展 [J], 赵锐;张喜全;孟庆义因版权原因,仅展示原文概要,查看原文内容请购买。
FDA批准罕用药Vandetanib用于治疗甲状腺髓样癌

FDA批准罕用药Vandetanib用于治疗甲状腺髓样癌
佚名
【期刊名称】《齐鲁药事》
【年(卷),期】2011(30)5
【摘要】2011年4月6日,AstraZeneca宣布美国FDA已批准其罕用药Vandetanib用于治疗那些不能进行手术切除或者持续向身体其他部位扩散的甲状腺髓样癌患者。
Vandetanib是一种合成的苯胺喹唑啉化合物,为小分子多靶点酪酸激酶抑制剂(TKI),可同时作用于肿瘤细胞EGFR、VEGFR和RET酪氨酸激酶,还可选择性的抑制其他酪氨酸激酶,以及丝氨酸/苏氨酸激酶。
它是FDA批准的唯一的用于治疗甲状腺髓样癌的药物。
【总页数】1页(P253-253)
【关键词】甲状腺髓样癌;FDA批准;罕用药;治疗;酪氨酸激酶;VEGFR;美国FDA;激酶抑制剂
【正文语种】中文
【中图分类】R736.1
【相关文献】
1.FDA拓展批准瑞戈非尼用于治疗肝癌/FDA批准midostaurin和化疗药结合治疗急性髓系白血病/FDA批准丙肝药物Sovaldi和Harvoni用于儿科患者 [J],
2.FDA批准Cometriq治疗甲状腺髓样癌 [J],
3.FDA批准Cabozantinib治疗转移性甲状腺髓样癌 [J], 张翼
4.Vandetanib获准用于治疗甲状腺髓样癌 [J],
5.美国FDA批准首种治疗甲状腺髓样癌新药Vandetanib [J], 夏训明
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
黏着斑激酶的磷酸化水平降低,肿瘤细胞的程序性
凋亡减少;15名CTCs水平可测的受试者中,有53%
的人体内CTCs水平下降30%或以上,而在所有受 试者中,有58%的人体内CECs水平下降,但几乎无
人其DCE—MRI检查发生变化;有14名受试者的病 情稳定持续4个月或更长时间,且受试的胃癌和
CAS:905854-02-6
ATP结合位点,抑制其自身磷酸化,尤其对IGF—I受 体更具特异性抑制活性。 本品化学结构式:
源二聚体便自身磷酸化,并激活下游P13K/Akt/丝
氨酸一苏氨酸蛋白激酶雷帕霉素靶蛋白(roTOR)和
Ras/有丝分裂原活化蛋白激酶(MAPK)等信号通 路,最终抑制细胞凋亡,促进细胞生长和迁移,导致
肿瘤的生长和转移。至今未发现IGF—I受体可发生 致癌性突变,但已发现,在结肠、乳腺、肺和前列腺癌
受体和IR双重抑制剂,可竞争性结合于两受体的
胰岛素样生长因子一l(IGF.I)受体是一种跨膜 受体酪氨酸激酶,与许多人类肿瘤的发生发展密切
相关,由胞外a亚单位和含跨膜及胞内区部分的
B亚单位构成,其中d亚单位负责与IGF—I和一II等 可溶性IGF.I受体配体结合,而B亚单位则发挥胞
内受体酪氨酸激酶作用。与配体结合后,IGF—I受体 的同源二聚体和IGF—I受体一受体酪氨酸激酶的异
争结合于膜受体上ATP结合口袋而是以非ATP竞 争性方式结合于未磷酸化或未激活的受体来阻抑受 体的激活及下游信号转导。
本品化学结构式:
本品对肿瘤的靶向性以及对肿瘤组织中c.Met信号
转导、细胞凋亡和血管生长的抑制作用,并评价本品 的安全性和耐受性。结果显示,经本品的1个或以 上疗程治疗后,15名受试者其肿瘤组织中c—Met和
0s中值也显著延长(7.2
3.8个月,HR为0.38,
P=0.01),且本品组中肿瘤组织内c.Met高水平 (通过免疫组化法测得)的受试者其TTP、PFS和Os 也均显著延长;本品组的不良反应发生率与安慰剂
组基本相似,只是疲劳以及中性粒细胞减少、贫血等 血液学不良事件发生率较高,但低剂量组的血液学 不良事件(尤其是中性粒细胞减少)发生率较高剂 量组有所降低。可见,本品有望成为有效的晚期
抑制剂在疗效和安全性方面要优于抗IGF-I受体抗
体,因为IGF.I受体和IR的ATP结合位点有较高的
同源性,该类抑制剂能同时结合两受体的ATP结合
显著高于erlotinib与安慰剂联用组。
位点,产生双重抑制作用,从而更有效地削弱两受体 的信号通路之间交叉协同作用;而且,一般情况下, 抑制剂的半衰期短于抗体,这样可使抑制剂的给药 剂量和频率更具灵活性,有助于控制毒副作用和提
的毒性作用。
临床研究
在38名接受标准抗癌疗法治疗无
效的转移性癌症患者中进行的旨在考察tivantinib 的安全性、耐受性、药动学、药效学以及Ⅱ期临床推 荐剂量的一项I期临床试验显示,本品(10—360 mg,bid,po,持续2周)耐受性良好,常见不良反应包
括疲劳(24%)、腹泻(21%)和便秘(2l%);33名可 评价的受试者中,1人达到部分缓解,19人病情持续 稳定达34周;基于药动学数据,确立Ⅱ期临床推荐
・热点药物追踪・
2013年第37卷第4期
182
第182页
貉学建履
2013,V01.37,No.4
Progress伽Pharmaceutical Sciences
品可通过作用于caspase依赖性凋亡途径而诱导癌 细胞死亡及抑制癌细胞生长。
此外,鉴于研究表明本品和吉西他滨可诱导癌 细胞生长停滞于细胞周期的不同时相,有研究者在 体外实验中考察了本品与吉西他滨联用时的抗癌活 性,首先将膀胱、胰腺、乳腺、卵巢、子宫等处肿瘤以
受本品(100~400 mg,bid,po)治疗,28 d为一疗程; 通过对来自受试者的活检样本进行动态对比增强磁 共振成像扫描(DCE.MRI)以及循环内皮细胞 (CECs)和循环肿瘤细胞(CTCs)的定量检测,考察
用名:ARQ-197)即为一种具口服活性的高选择性 c—Met抑制剂,具有独特的作用机制,即不同于典型 的小分子酪氨酸激酶抑制剂,其并非通过选择性竞
及NSCLC细胞分别接种于培养基中培养2 d,加入 吉西他滨共孵育24 h,随后加入培养液培养24 h,再 加入本品共孵育24 h,检测细胞活力。结果显示,本 品与吉西他滨的此联用方案明显增强了对肿瘤细胞
选择性c-Met抑制剂类抗癌药
Tivantinib
[关键词]tivantinib;c—Met抑制剂;抗癌活性 [中图分类号]R
药学进展
・热点药物追踪・
2013年937卷94期第183页
360
mg,bid)或安慰剂治疗,主要终点考察指标为治
疗后出现恶化的时间(1T11P),次级指标是无恶化生 存期(PFS)和总生存期(OS)。结果显示,与安慰剂
相邻正常组织高出9倍;此外,体内IGF—I受体水平 居高的患者发生结肠、乳腺、肺、前列腺及膀胱癌的 风险较大,而且实体瘤和急性髓细胞白血病组织中 IGF—I受体的过表达及相关信号通路的激活,往往导
致其对治疗产生抗性。同样,与IGF.I受体结构相
组相比,本品组受试者的1TrP中值显著延长『2.9
明显延长(2。4
VS
1.5个月,风险比(HR)为0.43,P=0.03],PFS中值
YS
1.5个月,HR为O.45,P=0。02),
VS
似(尤其催化区域有80%同源性)的胰岛素受体
(IR)也与人类肿瘤的发生发展相关,其在乳腺、前 列腺和卵巢癌组织中均有表达,并导致癌症预后差。 鉴于此,IGF.I受体和IR已成为令人关注的抗癌治 疗靶标。 目前,靶向IGF.I受体的肿瘤治疗策略主要包
高疗效,如临床前的动物肿瘤移植模型实验显示,在 使用多柔比星治疗时,若在给药间隙,肿瘤组织中
胰岛素样生长因子-1受体和胰岛素 受体双重抑制剂类抗癌药Linsitinib
[关键词]linsitinib;胰岛素样生长因子一1受体;胰岛素受 体;双重抑制剂;抗癌药 [中图分类号]R
979.1
IGF—I受体信号通路能短暂恢复,则其疗效要好于治 疗期间该信号通路一直处于抑制状态的肿瘤模型。 故可以推测,小分子IGF.I受体抑制剂因半衰期较 短,其用于肿瘤治疗时,较抗体更易致肿瘤组织中 IGF—I受体信号通路短暂性恢复,从而有利于疗效的 提高。由美国OSI制药公司开发的口服抗肿瘤药 linsitinib(曾用名:OSI-906)即为一种小分子IGF-I
Merkel细胞癌患者其肿瘤均略有缩小。 2012年第48届美国临床肿瘤学会(ASCO)年 会上公布了本品用于二线治疗肝细胞癌(ttCC)的 一项随机、安慰剂对照、双盲Ⅱ期临床试验阳性数 据。在该项试验中,107名无法手术且一线治疗无 效或不能耐受的HCC患者随机接受本品(240和
药理作用
体内外实验显示,tivantinib能明显
979.1
c.Met是由一种原癌基因编码的受体酪氨酸激 酶,为肝细胞生长因子(HGF)受体,仅表达于上皮 细胞,其经HGF激活后,又可激活多个信号通路,包
括Ras、P13K、Wnt和Notch通路,并产生一系列生物 学反应。已有研究表明,HGF和c—Met及其突变体 的过表达在肿瘤的生长、血管生成、侵袭和转移过程 中发挥重要作用;在乳腺、结肠、胃、肝、卵巢、肾和甲 状腺等处肿瘤以及非小细胞肺癌(NSCLC)组织中,
HCC二线治疗新药。 然而,随后的一项本品与抗癌药erlotinib联用 治疗NSCLC的Ⅲ期临床试验却未达到主要终点考 察指标的预期,即该联用组受试者的总生存率并不
括该受体的抗体及小分子抑制剂,但眼下开发的若 干抗IGF.I受体抗体在早期临床试验中尚能表现出 良好的抗癌活性,而在后期临床研究中的疗效和安 全性却并不理想。从理论上讲,小分子IGF—I受体
本文链接:/Periodical_yxjz201304007.aspx
剂量为360 mg(bid)。
均存在c—Met及其突变体的过表达和c—Met信号转
导异常,这也导致侵袭性肿瘤的发展和预后不良。 由美国ArQule公司与日本Daiichi Sankyo和Kyowa
Hakko
Kogyo公司联合开发的抗癌药tivantinib(曾
在另一项I期临床研究中,51名实体瘤患者接
抑制具c—Met表达或过表达的人结肠腺癌HT.29、 胃癌MKN一45、乳腺腺癌MDA—MB-231等细胞株的 增殖及caspase依赖性凋亡;其经口给予这些肿瘤细 胞移植模型小鼠后,可有效抑制肿瘤生长。表明,本
万方数据
2013,VoL 37.No.4
183
Progress in Pharmaceutical Sciences
及肉瘤等多种肿瘤组织中,有IGF—I受体高表达,如
结肠直肠癌(CRC)组织中IGF—I受体的表达水平较
CAS:867160-71.2
万方数据
选择性c-Met抑制剂类抗癌药Tivantinib
刊名: 英文刊名: 年,卷(期): 药学进展 Progress in Pharmaceutical Sciபைடு நூலகம்nces 2013,37(4)