基因组重测序mutmap分析
全基因组重测序数据分析详细说明

全基因组重测序数据分析详细说明全基因组重测序(whole genome sequencing, WGS)是一种高通量测序技术,用于获取个体的整个基因组信息。
全基因组重测序数据分析是指对这些数据进行处理、分析和解读,以获得有关个体的遗传变异、基因型、表达和功能等信息。
下面详细说明全基因组重测序数据分析的过程和方法。
首先,全基因组重测序数据的质量控制是必不可少的。
这一步骤包括对测序数据进行质量评估、剔除低质量序列,并进行去除接头序列和过滤序列等预处理操作,以确保后续分析的准确性和可靠性。
接下来,需要对全基因组重测序数据进行序列比对,将读取序列与参考基因组进行比对,以确定每个读取序列在参考基因组上的位置。
常用的比对工具包括Bowtie、BWA、BLAST等。
比对的结果将提供每个读取序列的基因组位置信息。
在序列比对完成后,就可以进行个体的变异检测。
变异检测的目的是识别个体的单核苷酸多态性(single nucleotide polymorphisms, SNPs)、插入缺失变异(insertions/deletions, indels)和结构变异(structural variations, SVs)等基因组变异。
通常,变异检测分为两个步骤:变异发现和变异筛选。
变异发现即根据比对结果,通过一定的算法和统计学原理,找到潜在的变异位点。
然后,利用临床数据库、已知变异数据库和基因功能注释数据库等,进行变异筛选,剔除假阳性和无功能变异,筛选出最有可能的致病变异。
接着,对筛选出的变异位点进行基因型確定。
基因型的确定可以通过直接从比对结果中读取碱基信息,或者通过再次测序来获取高度精确的基因型,以获得更可靠的变异信息。
随后,对变异位点进行注释和功能预测。
注释是指对变异位点进行功能和可能影响的基因、基因组区域和调控元件等进行注释。
常用的注释工具包括ANNOVAR、SnpEff、VEP等。
功能预测则是根据变异位点的位置和可能影响的功能进行预测,如是否影响蛋白质功能、是否在编码序列、是否在启动子或增强子区域等。
全基因组重测序数据分析

全基因组重测序数据分析1. 数据质量控制:对测序数据进行质量控制,包括去除低质量的碱基、过滤含有接头序列和接头污染的序列等。
这一步骤可以使用各种质控工具,例如FastQC、Trim Galore等。
2. 比对到参考基因组:将经过质控的测序数据与参考基因组进行比对。
参考基因组一般是已知的物种的基因组序列,在人类研究中通常使用人类参考基因组。
比对工具主要有BWA、Bowtie等。
3. 变异检测:从比对结果中检测出样本与参考基因组之间的差异,称为变异检测。
这包括单核苷酸变异(SNV)、插入/缺失(Indel)、结构变异(SV)等。
常用的变异检测工具有GATK、SAMtools、CNVnator等。
4. 注释和解读:对检测到的变异进行注释和解读,以确定其对基因功能和疾病相关性的影响。
注释可以包括基因、转录本、蛋白质功能、通路、疾病关联等信息。
常用的注释工具包括ANNOVAR、Variant Effect Predictor等。
5.结果可视化:将分析结果以图表或图形的形式展示出来,以便研究人员更好地理解和解释结果。
常用的可视化工具包括IGV、R软件等。
除了上述步骤,全基因组重测序数据分析还可以应用于其他研究领域,例如种群遗传学、复杂疾病研究、药物研发等。
在进行这些研究时,可能还需要其他分析方法和工具来完成特定的研究目标。
总之,全基因组重测序数据分析是一个复杂而关键的过程,它可以帮助研究人员了解个体的基因组特征,并揭示与疾病发生和发展相关的重要信息。
在不断发展的测序技术和分析方法的推动下,全基因组重测序数据分析将在基因组学领域中发挥越来越重要的作用。
如何应用生物大数据技术进行基因组重测序数据分析

如何应用生物大数据技术进行基因组重测序数据分析生物大数据技术是近年来迅速发展的一种先进工具,它可以处理庞大的基因组重测序数据,并从中获取有关基因组和生物进化等方面的重要信息。
本文将介绍如何应用生物大数据技术进行基因组重测序数据分析,以帮助读者了解如何应用这一技术来解决生物学研究中的问题。
首先,对于基因组重测序数据分析,我们需要了解数据的来源和类型。
基因组重测序是指通过测序技术对基因组进行全面的、高通量的测序,并生成大量的测序数据。
这些数据包括原始测序数据、清洗后的测序数据以及从测序数据中推断得出的基因组序列信息。
因此,准确理解数据类型对于正确分析和解读数据至关重要。
接下来,生物大数据技术可以应用于基因组重测序数据的多个方面,包括基因组组装、SNP鉴定、基因表达分析和功能注释等。
基因组组装是一项重要的任务,它旨在将碎片化的测序数据组装成完整的基因组序列。
生物大数据技术可以通过采用多种算法和工具来提高组装的准确性和效率。
此外,生物大数据技术可以用于鉴定单核苷酸多态性(SNP)和变异位点。
这些变异位点在个体间的差异可以揭示不同物种之间的进化关系,以及复杂疾病等的遗传基础。
通过使用生物大数据技术,我们可以从基因组重测序数据中识别SNP,并进一步分析这些变异位点的功能和相关性。
另外,基因表达分析是生物大数据技术在生物学研究中的重要应用之一。
基因表达是指基因转录和翻译为蛋白质的过程,通过测定不同条件下的基因表达水平,可以揭示基因功能与生物学过程之间的联系。
生物大数据技术可以通过分析基因组重测序数据中的转录本信息和表达量,帮助研究人员了解基因在不同组织和环境条件下的表达模式。
最后,生物大数据技术还可以为基因组重测序数据提供功能注释。
基因功能注释是指通过将重测序数据与已知的生物信息数据库进行比对和注释,来研究基因功能和作用机制。
通过生物大数据技术,我们可以利用大规模的生物信息数据库,为基因组重测序数据提供功能注释,从而更深入地研究基因组的结构和功能。
如何使用生物大数据技术进行基因组均衡重测序分析

如何使用生物大数据技术进行基因组均衡重测序分析基因组均衡重测序分析是一种利用生物大数据技术的高级测序技术,可以为基因组研究提供深入的洞察力。
通过分析基因组的均衡重测序数据,我们可以揭示基因组的变异、功能元件和表达模式等信息。
在本文中,我们将探讨如何使用生物大数据技术进行基因组均衡重测序分析,以加深我们对基因组的理解。
首先,我们需要了解什么是基因组均衡重测序。
基因组均衡重测序是指将同一样本的DNA分成许多相同大小的片段,并使用高通量测序技术对这些片段进行测序。
这样做的好处是可以提高测序的覆盖度和准确性,并降低测序中的错误率。
在进行基因组均衡重测序分析之前,我们需要准备样本的DNA。
首先,选择合适的生物材料,如血液、组织或细胞。
然后使用基因组DNA提取试剂盒进行DNA的提取。
提取后的DNA需要经过质量检测,以确保其质量合格。
接下来是建立DNA文库,这是基因组均衡重测序的关键步骤。
通过亚洲一种叫做“bridge PCR”的技术,将DNA片段连接到测序平台上,形成DNA文库。
文库建立完毕后,可以进行下一步的均衡重测序。
在进行基因组均衡重测序分析之前,我们需要选择合适的测序平台和相应的测序方法。
当前常用的测序平台有Illumina、PacBio和Oxford Nanopore等。
Illumina 主要用于高通量测序,PacBio和Oxford Nanopore则适用于长读长测序。
根据实验设计的要求和测序平台的性能,选择合适的测序平台。
此外,还需要选择合适的测序方法,如全基因组测序(WGS)、全外显子测序(WES)或甲基化测序等。
在进行基因组均衡重测序分析之前,我们需要对测序数据进行质量控制。
这是非常重要的步骤,可以确保测序数据的质量和准确性。
常用的质量控制工具有FastQC和Trimmomatic等。
使用这些工具,我们可以对测序数据进行质量评估、去除低质量的序列和剪切适配序列。
经过质量控制后,我们可以进行基因组均衡重测序分析了。
全基因组重测序大数据分析报告

全基因组重测序数据分析1. 简介(Introduction)通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation产生对应的易感机制和功能。
我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。
实验设计与样本(1)Case-Control 对照组设计;(2)家庭成员组设计:父母-子女组(4人、3人组或多人);初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。
全基因组重测序数据分析

全基因组重测序数据分析全基因组重测序是一种高通量测序技术,可以获取一个个体的整个基因组的序列信息。
全基因组重测序数据分析是从这些序列数据中提取有用信息的过程,包括基因组装、变异检测和功能注释等。
本文将详细介绍全基因组重测序数据分析的步骤和一些常用的分析方法。
全基因组重测序数据分析的第一步是基因组装。
基因组装是指将测序得到的片段序列根据其重叠关系拼接成连续的序列。
目前有许多基因组装软件可供选择,如SOAPdenovo和SPAdes等。
这些软件会将测序片段根据其序列重叠情况进行集成,以获取最长的连续序列。
基因组装后,下一步是进行变异检测。
变异是指个体基因组与参考基因组之间的差异,可以分为单核苷酸变异(SNV)和结构变异(SV)两种类型。
SNV是指个体基因组中的单个碱基发生改变,包括单碱基插入、缺失和替换等。
SV则是指较大的基因组片段发生改变,包括插入、缺失、倒位和重组等。
变异检测的主要目标是通过比对个体的测序数据与参考基因组的序列,识别和注释这些变异。
为了提高变异检测的准确性,通常需要进行数据预处理和质量控制。
数据预处理包括去除接头序列、低质量序列和重复序列等,以提高后续分析的准确性和效率。
质量控制则是评估测序数据的质量,如测序深度、覆盖度和错误率等,以保证分析结果的可靠性。
除了变异检测,全基因组重测序数据还可以用于其他类型的分析,如基因表达分析和基因组结构分析。
基因表达分析可以通过比对测序数据和转录组数据库,识别并定量基因的表达水平。
基因组结构分析可以揭示染色体水平的变异和基因组结构的演化。
这些分析可以帮助研究人员研究基因组的功能和进化等问题。
总之,全基因组重测序数据分析是一个复杂的过程,涉及到多个步骤和分析方法。
通过对测序数据的组装、变异检测和功能注释等分析,可以获得有关个体基因组的详细信息,为基因功能研究和遗传疾病诊断提供重要参考。
随着测序技术的不断发展,全基因组重测序数据分析将会变得更加高效和准确。
mutmap基础知识

1、2、3、内容简介:本研究介绍了一种应用重测序方法进行数量性状基因座(QTL)定位的方法,该方法选用的研究对象可以是具有极端性状的一对品种杂交后获得的近交重组系群体(RILs)或F2代群体,选择群体中具有两种不同极端性状的20-50个个体分别构建DNA混池后进行重测序,通过对比两个混池的SNP位点的测序深度相关的一个参数(SNP-index)来定位QTL。
该方法能用于群体遗传学研究,能快速识别人工选育及自然选择发生的染色体区域。
文章解读:本研究选用两个在同一性状上表型相反的两个品种作为亲本,杂交后获得F2代,再通过单粒传自交至F7代,获得近交重组系群体(RILs)。
如果群体中此性状的符合正态分布,则说明此性状关联的基因座是数量性状的,可以进行QTL分析。
这里,我们选择这个群体中此性状表型最明显和最不明显的个体作为研究对象,一般选择20-50个个体分别混池,产生两个池,分别为最强表型池和最弱表型池。
然后对两个混池分别进行重测序,采用的测序平台为Illumina Genome Analyzer IIx,测序深度一般要大于6×,这两个池所代表的的应该是某个基因组区域的两个等位基因各自所对应的表型。
由此,我们观测来自两个亲本的基因组中的不平等表现,以此来识别导致两个池性状差异的含有QTL的基因组区域所在。
本研究引入了一个SNP位点的测序深度相关的参数SNP-index,该参数是指某个位点含有SNP的reads数与测到该位点的总reads数的比值,大小范围为0-1。
若该参数为0,代表所有测到的reads都来自被用作参考基因组的那个亲本的基因组;该参数为1,代表所有reads都来自另一个亲本;该参数为0.5,则代表此混池中SNP来自两个亲本的基因组的频率一致。
将两个池观测到的SNP都计算出SNP-index,然后将两个池的SNP-index值相减后得到Δ(SNP-index),将Δ(SNP-index)对应该SNP所在染色体位置作图,Δ(SNP-index)比较大的区域即可作为QTL的候选区域。
全基因组重测序基础及高级分析知识汇总

全基因组重测序基础及高级分析知识汇总借着2月8号,刚刚在Nature上发表柑橘的遗传进化的文章,小编来讲述一下全基因组重测序基础知识,以及常见的分析思路及软件,帮助大家迅速入门。
全基因组重测序是通过对已有参考序列(Reference Sequence)的物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的遗传差异性分析。
通过全基因组重测序,研究者可以找到大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异位点。
基于以上变异位点作为分子遗传标记,在人类复杂疾病、动植物经济性状和育种研究及物种起源、驯化、群体历史动态等方面具有重大的指导意义(Bentley2006; Casillas& Barbadilla 2017)。
一、基础理论知识全基因组重测序研究主要是依据在全基因组水平发现的分子遗传标记进行物种的群体遗传学研究,进一步的利用统计方法进行影响表型和经济性状候选基因和功能突变的研究。
分子群体遗传学研究的理论基础知识及统计分析方法日趋完善和呈现多样性,作为初学者,有必要对其中的一些基础概念有一定的了解,才能为后续的深入学习、研究提供基石。
以下基础知识主要参考国内动物遗传学书籍和最新的一篇关于分子群体遗传学方面的综述改变而成(吴仲贤编1961; 李宁2011; 吴常信2015; Casillas & Barbadilla 2017)。
高通量测序技术作为分子群体遗传学研究的有力工具,在科学研究、生产及疾病诊断治疗中起到原来越重要的作用,对关于高通量测序相关的理论基础知识进行一定程度的了解,也有助于文献阅读和。
1. 群体遗传学基础知识群体(Polulation):是指生活在一定空间范围内,能够相互交配并生育具有正常生殖能力后代的同种个体群。
使用MutMap快速定位突变基因流程篇

使用MutMap快速定位突变基因流程篇在上一篇文章《使用MutMap快速定位突变基因:原理篇》中,我对MutMap的原理进行的简单的介绍。
在本篇教程中,我会对MutMap分析的流程及分析过程中使用到的软件及其参数的设置进行逐一的分解,以求对MutMap的分析流程有更深入的理解。
1.对重测序数据进行质控,挑出高质量的reads(1)使用软件:FASTX-Toolkit① fastq_quality_filter:过滤掉低质量的reads使用说明:fastq_quality_filter [-h] [-v] [-q N] [-p N] [-z] [-i INFILE] [-o OUTFILE]过滤低质量序列[-q N] = 最小的需要留下的质量值[-p N] = 每个reads中最少有百分之多少的碱基需要有-q的质量值[-z] =压缩输出[-v] =详细-报告序列编号,如果使用了-o则报告会直接在STDOUT,如果没有则输入到STDERR在MutMap中,使用q30p90对数据进行质控。
【特别说明】:对于 Illumina 1.8+ Phred+33规则进行质量打分的测序数据,需要添加-Q33选项,否则程序不能识别打分字符,会报错。
② fastx_quality_stats:序列的基本信息,如GC含量等使用说明:fastx_quality_stats [-h] [-N] [-i INFILE] [-o OUTFILE][-h] = 获得帮助信息[-i] = FASTA/Q格式的输入文件[-o] = 输出路径,如果不设置会直接输出到屏幕(2)结果输出① 过滤后的高质量reads结果输出到每个样本下的q30p90/sep_pair/*1.RnFq30p90.1或2.gz,这些reads为质控后的高质量的reads,用于构建参考基因组和后续的比对分析。
② 过滤后reads的统计信息统计结果输出到每个样本下的q30p90/sep_pair/*1.RnFq30p90.1或 2.gz_stats.txt,这些txt格式的文件中存储着过滤后的reads的统计信息。
动植物重测序
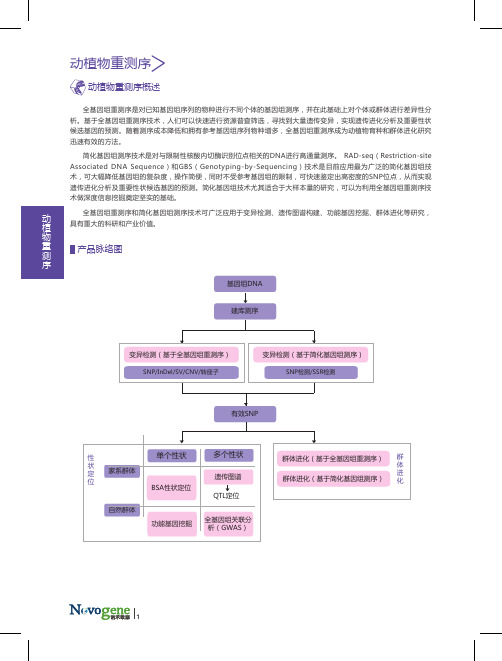
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS(Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图动植物重测序建库测序单个性状家系群体自然群体SNP/InDel/SV/CNV/转座子基因组DNA有效SNP性状定位群体进化群体进化(基于简化基因组测序) 群体进化(基于全基因组重测序) 变异检测(基于简化基因组测序)SNP检测/SSR检测遗传图谱全基因组关联分析(GWAS)功能基因挖掘变异检测(基于全基因组重测序) QTL定位BSA性状定位多个性状动植物重测序动植物重测序概述SNP检测、注释及统计基因组DNA350 bp小片段文库HiSeq PE150测序数据质控与参考基因组比对利用全基因组重测序技术对某一物种个体或群体的基因组进行测序及差异分析,可获得SNP、InDel、SV、CNV、PAV、转座子等大量的遗传多态性信息,建立遗传多态性数据库,为后续揭示进化关系、功能基因挖掘等奠定基础。
如何应用生物大数据技术进行基因组重测序数据分析

如何应用生物大数据技术进行基因组重测序数据分析生物大数据技术的快速发展为基因组重测序数据分析提供了更加精准和高效的解决方案。
基因组重测序数据分析是基于DNA或RNA样品的测序数据,用于揭示基因组结构和功能,以及与疾病相关的变异等信息。
本文将详细介绍如何应用生物大数据技术进行基因组重测序数据分析。
一、数据质控基因组重测序数据分析的第一步是对原始数据进行质控,以确保实验结果的准确性和可靠性。
数据质控涉及到对测序数据进行去除低质量碱基、去除接头序列、去除污染序列等步骤。
可以使用适当的工具如FastQC、Trimmomatic等对数据进行处理,以获得高质量的测序数据。
二、序列比对在完成数据质控后,下一步是将测序数据与参考基因组进行比对。
参考基因组是一个已经测序和注释过的基因组,用作分析的背景。
比对的目的是找到测序数据在参考基因组上的位置,以便进一步的功能注释和变异检测。
在比对过程中,可以选择合适的比对工具如Bowtie、BWA、STAR等,根据不同的需求和研究目的进行选择。
三、变异检测变异检测是基因组重测序数据分析的一个重要环节,旨在发现与疾病或表型相关的基因突变。
在基因组重测序数据中,常见的变异类型包括单核苷酸多态性(SNP)、结构变异和基因剪接变异等。
变异检测需要使用适当的工具如GATK、SAMtools、VarScan等,根据不同的变异类型进行分析和筛选。
四、功能注释功能注释是对基因组中的变异进行识别和解释的过程,旨在揭示变异对基因功能和表达的影响。
常见的功能注释方法包括基于基因本体论(GO)、Pathway和基因集富集分析等。
这些方法可以帮助研究人员理解基因在细胞过程和信号通路中的作用,从而对疾病机理有进一步的认识。
五、数据可视化数据可视化是基因组重测序数据分析的最后一步,旨在以图形化的方式呈现结果。
通过数据可视化,研究人员可以更直观地理解数据的特征和分布,从而得出更有意义的结论。
常用的数据可视化工具包括R语言中的ggplot2和Python中的matplotlib等。
全基因组重测序数据分析

全基1. 简通过变(d 的功况,dise 比较实验(1)(2)基因组重测序简介(Introduc 过高通量测序识deletioin, du 功能性进行综合杂合性缺失ease (cance 较基因组学,群验设计与样本Case-Contr)家庭成员组序数据分析ction)识别发现de plication 以及合分析;我们(LOH )以及r )genome 中群体遗传学综ol 对照组设计组设计:父母novo 的som 及copy numb 们将分析基因及进化选择与中的mutation 综合层面上深计 ;-子女组(4人matic 和germ ber variation 因功能(包括与mutation 之n 产生对应的深入探索疾病基人、3人组或m line 突变,)以及SNP miRNA ),重之间的关系;以的易感机制和基因组和癌症多人);结构变异-SN 的座位;针对重组率(Rec 以及这些关系功能。
我们将症基因组。
NV ,包括重排对重排突变和combination )系将怎样使得将在基因组学排突SNP)情在学以及初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
全基因组重测序数据分析详细说明

全基因组重测序数据分析1. 简介(Introduction)通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation 产生对应的易感机制和功能。
我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。
实验设计与样本(1)Case-Control 对照组设计;(2)家庭成员组设计:父母-子女组(4人、3人组或多人);初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。
使用生物大数据技术进行基因组重测序分析的方法

使用生物大数据技术进行基因组重测序分析的方法基因组重测序是通过高通量测序技术对生物体的基因组进行测序的过程。
随着生物大数据技术的不断发展,基因组重测序分析已经成为研究生物学领域的重要手段之一。
本文将介绍使用生物大数据技术进行基因组重测序分析的方法和流程。
首先,进行基因组重测序分析的第一步是准备样本和测序文库。
样本可以是细胞、组织或直接从环境中采集的样本。
测序文库是用于测序的DNA片段的集合,可以通过PCR扩增或其他方法制备。
通过采用适当的方法提取DNA,并根据测序目的选择合适的文库制备方法,可以保证测序的质量和准确性。
第二步是进行高通量测序。
高通量测序技术,如Illumina HiSeq、PacBio、ONT等,可以同时测序大量的DNA分子,大大提高了测序效率和产出。
在测序过程中,测序仪会生成大量的短序列读段,也即测序reads。
这些reads包含了样本中DNA分子的片段信息。
第三步是对测序数据进行质量控制和预处理。
测序数据通常存在测序错误、质量不均匀以及过度表示等问题。
因此,需要对测序reads进行去除低质量碱基、去除接头序列、去除重复reads等处理,以减少后续分析中的误差和干扰。
常用的质控工具有TrimGalore、FastQC等。
第四步是将预处理后的reads进行比对到参考基因组上。
参考基因组是已经测序和注释完善的一个生物种群的基因组序列。
比对的目的是确定样本中每个read 对应参考基因组中的位置,从而了解样本的基因组结构和基因组重排等相关信息。
常用的比对工具有Bowtie、BWA、STAR等。
第五步是进行基因组注释。
基因组注释是将比对到参考基因组上的reads与相应的基因、转录本、蛋白质等功能元件进行关联,以分析样本中的基因组变异、表达水平和基因功能等。
常用的注释工具有GATK、Cufflinks等。
第六步是进行变异检测和功能分析。
通过对比参考基因组和样本基因组的差异,可以检测到SNP(单核苷酸多态性)、Indel(插入缺失)、CNV(拷贝数变异)等多种变异类型。
生物大数据技术中的基因组重测序方法与分析

生物大数据技术中的基因组重测序方法与分析基因组重测序是生物大数据技术中的一项核心方法,可用于揭示个体、物种及种群的遗传变异,有助于了解基因组的结构和功能。
本文将介绍基因组重测序的方法和分析流程,以及其在生物研究和医学领域的应用。
基因组重测序是指对一个生物个体的基因组进行全面的测序,包括编码基因、非编码区域以及整个基因组的变异信息。
目前常用的基因组重测序方法主要有两种:全基因组测序(Whole Genome Sequencing,简称WGS)和外显子组测序(Exome Sequencing)。
WGS是对整个基因组进行测序,包括编码基因和非编码区域,能够提供全面的基因组变异信息。
它通过将DNA样品切割成小片段,使用高通量测序技术对这些片段进行测序,再通过基因组拼接算法将这些碎片拼接成完整的基因组序列。
WGS广泛应用于种群遗传学、人类基因组计划等研究项目中,可以发现个体间和物种间的遗传变异。
而外显子组测序则只对编码基因进行测序,这些编码基因是构成蛋白质的重要组成部分。
人类基因组中,编码基因仅占据整个基因组的一小部分,但大部分疾病相关变异发生在编码基因区域。
外显子组测序通过选择富集编码区域的方法,可以更高效地测序这些关键基因,并且产生更少的数据量,降低测序成本。
在基因组重测序完成后,需要进行一系列的分析流程来解读测序数据。
首先,对测序数据进行质控与预处理,去除低质量序列、去除仪器测序误差及接头序列。
然后,将剩余的高质量序列与参考基因组进行比对,以识别个体或物种的特定变异。
从比对结果中,我们可以获得每个个体的SNP(Single Nucleotide Polymorphism,单核苷酸多态性)和Indel(Insertion/Deletion,插入/缺失)等遗传变异信息。
这些变异信息对于研究遗传疾病、物种起源和进化等具有重要意义。
此外,还可以通过对基因组重测序数据进行拼接分析,将碎片序列拼接成完整的基因组序列,进一步了解个体或物种的基因组结构和功能。
1_Mutmap项目结题报告模板

Mutmap项目结题报告客户单位:报告单位:联系人:联系电话:传真:报告日期:项目负责人:审核人:目录目录 (1)1 项目概况 (1)1.1 合同关键指标 (1)1.2 项目基本信息 (1)1.3 项目执行情况 (2)1.4 分析结果概述 (2)2 项目流程 (3)2.1 实验流程 (3)2.2 信息分析流程 (3)3 生物信息学分析方法和结果 (5)3.1 测序数据质控 (5)3.1.1 原始数据介绍 (5)3.1.2 碱基测序质量分布 (7)3.1.3 碱基类型分布 (9)3.1.4 低质量数据过滤 (10)3.1.5 数据质量统计 (10)3.2 与参考基因组比对统计 (11)3.2.1 比对结果统计 (11)3.2.2 插入片段分布统计 (11)3.2.3 深度分布统计 (12)3.3 SNP检测与注释 (14)3.3.1 样品与参考基因组间SNP的检测 (14)3.3.2 样品之间SNP的检测 (17)3.3.3 SNP结果注释 (19)3.4 Small InDel检测与注释 (22)3.4.1 样品与参考基因组间Small InDel的检测 (22)3.4.2 样品之间Small InDel检测 (22)3.4.3 Small InDel的注释 (23)3.5关联分析 (26)3.5.1 高质量SNP筛选 (26)3.5.2 关联分析 (26)3.5.3 候选区域SNP注释 .................................................. 错误!未定义书签。
3.5.4候选区域基因注释..................................................... 错误!未定义书签。
3.6结果可视化 (36)4 数据下载 (37)4.1 结果文件查看说明 (37)参考文献.............................................................................................. 错误!未定义书签。
单细胞测序umap降维原理

单细胞测序umap降维原理
单细胞测序UMAP降维是一种新型的数据降维技术,其核心原理是将高维的单细胞转录组数据映射到一个低维空间中。
在UMAP降维过程中,单细胞数据样本被视作高维空间中的点,算法会针对这些点之间的距离进行计算,并将它们映射到一个低维空间中,从而达到降维的目的。
UMAP降维的优点在于,它能够准确地反映单细胞数据样本中点之间的相对距离信息,同时还能够保留原始数据样本的结构。
此外,UMAP降维计算速度快,能够处理大规模数据集。
总之,单细胞测序UMAP降维技术是一种功能强大的数据降维方法,为研究者们提供了一种高效、准确、实用的数据分析工具。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
在MutMap中,一个突变体与其野生型品系杂交后自交,在F2群体中可以明确观察到表型差异的分离情况。
这一方法尤其适用于作物物种,可以使遗传杂交次数最小化,而可得到突变表型的F2子代是必须的。
我们将MutMap应用于一个日本骨干水稻栽品种的7个突变体,鉴定出来包含了淡绿色叶片和半矮生突变表型
相关突变位点的唯一基因组区域。这些结果显示MutMap可以加速水稻和其它作物的遗传改良。
MutMap:即突变位点图谱,针对有参考基因组的突变体物种,通过目标性状差异构建两个极端的子代DNA池,检测功能性突变位点。诺禾致源已做过多种动、植物突变检测项目。
主要的农艺性状是由多基因控制的,而单个基因仅引起较小的表型效应,故而对其鉴定和克隆非常困难。
我们在此介绍MutMap,该方法基于对一个分离群体中呈现有用表型植株的DNA混合后而进行的全基因组重测序。
单个性状定位主要针对有参考基因组的物种,通过混合分组分析(BSA)的手段,利用二代高通量测序得到数据与参考序列比对开发分子标记,进行标记与性状的共分离分析,检测变异并进行性状相关区域的基因结构及功能的注释,分析基因控制目标性状的机制。单个性状定位主要分为以下3个产品:MutMap、QTL-seq以及功能基因挖掘。
日本水稻骨干栽培种Hitomebore(一目惣),EMS处理得1200个M1系,自交3-4代,得M3-M4。
F2群体,群体大于200子代,
野生型与突变型分离比为3:1,被选用于MutMap分析。
选择20个突变性状的F2子代进行DNA提取,混合后全基因组重测序(10X以上),