化学原料药的含量测定常用方法
药物的含量测定方法ppt课件
23
(2)测含量(用样品与内标物)
含量(Cx
)
f
Ax AS' / CS'
As’:内标物质峰面积或者峰高 Ax: 供试品的峰面积或者峰高 Cs’: 内标物质浓度 f:校正因子
24
示例一 炔诺酮的含量测定
照高效液相色谱法(附录ⅤD测定)
色谱条件与系统适用性试验 用十八烷基硅烷键 合硅胶为填充剂;以甲醇-水(65:35)为流动相, 检测波长为244nm。理论塔板数按炔诺酮峰计 算不低于1500,炔诺酮峰与内标物质峰的分离 度及主成分峰后一杂质峰与主成分峰的分离度均 应符合要求。 内标溶液的制备 取黄体酮约25mg,精密称定, 置25ml量瓶中,加甲醇溶解并稀释至刻度。
12
二、光谱分析法
红外分光 度法
光谱分析法
药典 收载
火焰光度 紫外可见 荧光分析
法
分光度法
法
原子吸收分 光光度法
13
紫外可 见分光
度法
基于物质对紫外区 (200~400nm)和可见光 区(400~760nm)的单色 光辐射建立起来的光谱
分析法
基 础
朗伯-比尔定律
A lg 1 Ecl T
c(g
W测 0.13263 VitC% = W样 = 0.1332 =99.57%
10
又例 非那西丁含量测定
精密称取本品0.3630g加稀盐酸回流1小时后,放冷, 用亚硝酸钠滴定液(0.1010mol/L)滴定,用去 20.00ml。每1ml亚硝酸钠液(0.1mol/L)相当于 17.92mg的C10H13O2N,请计算非那西丁的含量。
25
测定法 取本品约25mg,精密称定,置25ml量瓶
中,加甲醇溶解并稀释至刻度,摇匀,精密量取
药物的含量测定方法与验证-样品的前处理方法

R-NH2
H2SO4 K2SO4
(NH4)2SO4 NaOH (NH4)HSO4
NH3↑
NH3↑+H3BO3→NH4BO2 →H2SO4滴定
13
前处理方法-湿法破坏
பைடு நூலகம்
凯氏定氮法(kjeldahl nitrogen determination)
间接法 水-NaOH溶液 硫 浓H2O2溶液与水 磷水 硒 硝酸溶液(1→30)
22
前处理方法-干法破坏
氧瓶燃烧法(oxygen flask combustion method ) 3.样品制备
固体样品(研细) 无灰滤纸 液体样品 纸袋
样品研细后放中间,按 虚线折叠 固定于铂丝下端螺旋 处.
23
前处理方法-干法破坏
氧瓶燃烧法(oxygen flask combustion method )
4.实例-碘苯酯的含量测定(P163)
I
CH3
CH (CH2)8 COOCH2CH3
24
例1. 氧瓶燃烧法中的装置有 A. 磨口硬质玻璃锥形瓶 B. 磨口软质玻璃锥形瓶 C. 铂丝 D. 铁丝 E. 铝丝
25
例2.选择氧瓶燃烧法所必备的实验 物品 A. 磨口硬质玻璃锥形瓶 B. 铂丝 C. 普通滤纸 D. 氢气 E. 无灰滤纸
一方法测定
回收率%= 测定对照品量平均结值果比×较100%
加入对照品的量
33
分析方法验证的效能指标
—— 1.准确度 (accuracy)
(一)含量测定方法的准确度
2.制剂准确度
用本法测定 结果与已知
简述化学原料药含量检查内容和要求

简述化学原料药含量检查内容和要求化学原料药含量检查是指对化学原料药中的有效成分进行检测,以确定其含量的方法。
化学原料药是制药工业中重要的原材料,其含量的准确性对于药品的质量和疗效至关重要。
下面将详细介绍化学原料药含量检查的内容和要求。
一、检查内容化学原料药含量检查通常包括以下方面:1.物质鉴定:对化学原料药进行物质鉴定,确保所使用的原料为正确的物质,无杂质。
常用的物质鉴定方法包括红外光谱法、紫外光谱法、核磁共振法等。
2.含量测定:对化学原料药中的有效成分进行测定,确定其含量是否符合规定的标准。
具体测定方法根据化学原料药的性质和要求不同而有所不同,常用的方法包括高效液相色谱法、比色法、滴定法、光度法等。
3.杂质分析:对化学原料药中的杂质进行定性和定量分析,以确保杂质的含量在规定范围内。
常用的杂质分析方法包括气相色谱法、液相色谱法、电泳法等。
4.含量相对标准偏差测定:含量相对标准偏差是指对同一批次化学原料药进行多次测定,计算其含量值的离散程度。
该指标反映了含量的稳定性和准确性。
常用的统计学方法包括方差分析、t检验、偏度和峰度分析等。
5.溶出度测定:对固体药物的溶出度进行测定,以评估药物在体内溶出的速度和程度。
常用的溶出度测定方法包括离子强度法、流动取样法、旋转篮法等。
二、检查要求化学原料药含量检查的要求如下:1.仪器设备:使用经过校准和验证的仪器设备进行检测,确保测定结果准确可靠。
仪器设备应定期维护、清洁和检查,确保其工作状态良好。
2.标准物质:使用纯净的标准物质进行浓度测定和校准。
标准物质的纯度应高于待测物质,以确保测定结果的准确性。
3.样品制备:样品制备过程应注意避免污染和损失,确保所测定的是样品中的有效成分。
对于固体样品,需要进行粉碎和均匀混合。
4.检测条件:在检测中需要控制好温度、湿度、pH值等条件,以确保测定结果的准确性和可重复性。
5.检测方法验证:建立合适的检测方法,并进行方法验证,包括选择性、灵敏度、精密度、准确度等方面的验证,确保方法的可行性和可靠性。
原料药含量测定方法

原料药含量测定方法原料药是指制药过程中使用的原始材料,它们是制药产品的组成部分。
对原料药进行准确测定是制药过程中至关重要的环节,它直接关系到最终药物的质量和安全性。
因此,制定可行且准确的原料药含量测定方法对于保证药物质量至关重要。
原料药含量测定方法通常包括物质定量法和物质辨认法。
物质定量法是通过测定原料药中目标成分的定量浓度来评估其含量。
物质辨认法则是通过与已知物质进行比较来判断原料药中是否含有目标成分。
下面将介绍几种常用的原料药含量测定方法。
首先,重量法是一种常见的物质定量方法,它是利用原料药中目标成分的质量来评估其含量。
该方法的操作简单,适用范围广。
具体操作步骤为:首先,取一定量的原料药样品,用天平准确称取质量。
然后,将样品溶解或稀释至适当的浓度,加入适量的溶剂,并摇匀混合。
随后,使用合适的分析仪器如高效液相色谱仪或气相色谱仪测定样品中目标成分的质量。
最后,根据测定结果计算出目标成分的含量。
其次,滴定法也是一种常用的物质定量方法。
滴定法是通过滴定试剂来确定原料药中目标成分的浓度。
该方法具有高灵敏度和较高的准确性,能够在较短的时间内得出准确的结果。
具体操作步骤为:首先,取适量的原料药样品,加入适量的溶剂进行溶解。
然后,将溶液滴入滴定管中,加入一定量的滴定试剂,反应使其发生定量反应。
当滴定试剂完全反应完毕时,滴定终点发生变化,可以通过指示剂或指示电极反应颜色变化或电动势变化来判断滴定终点。
最后,根据滴定试剂的用量计算出目标成分的含量。
此外,色谱法也是一种常用的物质定量方法,常见的有气相色谱法和高效液相色谱法。
色谱法通过成分在固定相和流动相之间的分配行为来实现成分分离和定量。
具体操作步骤为:首先,将样品通过适当的前处理方法如提取、过滤等制备成测试液,然后通过色谱柱分离目标成分,最后使用检测器测定目标成分的峰面积或峰高,根据峰面积或峰高计算出目标成分的含量。
此外,在实际的原料药含量测定过程中,还需要考虑一些因素,如选择适当的分析方法、准备好校准曲线、验证方法的准确性和精密度等。
药物分析 药物的含量测定方法——光谱分析法

示例:氟康唑片溶出度检查
• 规格:100mg ➢ 盐酸溶液(9→1000)1000ml为溶出介质, 45分钟取 样, 滤过; 取续滤液, 在 261nm波长处测定吸光度; 氟康唑对照品, 用溶出介质制成每1ml中含0.1mg 的 溶液, 同法测定, 计算每片的溶出量
➢ 标示量(%)= AX cR D 100 AR B
原理:
A = lg 1 = Ec(l 朗伯-比尔定律) T
E 1% 1cm
c(g
A /100m l)
1. 特点与适用范围 (1) 方法简便易行 (2) 方法灵敏度高 (3) 结果准确度较高 (4) 方法专属性较差
• 制剂含量测定与定量检查
2. 仪器校正和检定 (1) 波长 (2) 吸光度的准确度
测定用波长/nm 220 340
透光率/% <0.8% <0.8%
3. 对溶剂的要求
当含有杂原子的有机试剂做溶剂时,它们的使用范围均不能 小于截止使用波长,如甲醇、乙醇的截止使用波长为205nm;
在测定供试品之前,应先检查所用溶剂在测定波长附近是否 有干扰,要求溶剂与吸收池的吸光度在200-400nm范围内不得超 过0.40,在241-250nm范围内不得超过0.20;在251-300nm范围内 不得超过0.10;在300nm以上不得超过0.05。
• 比色法——为避免干扰或提高灵敏度, 向体系中加入 适当的显色剂, 使产物最大吸收波长移至可见光区后 测定的方法
• 适用范围:紫外光区无强吸收或有干扰药物的测定
(二) 荧光分光光度法
Fluor
• 物质受UV或Vis照射后能发射出比激发光 波长更长的荧光; 激发光停止照射,荧 光随之消失;激发和发射光谱用于定性, 荧光强度用于定量
药物分析技术 第五章 药物含量测定

2.滴定液的配制和标定
配制方法
▪ 1.直接法:准确称取--溶解--定容 ▪ 2.间接法:配制---用基准物质标定 ▪ 标定:用基准物质或标准溶液准确测定滴
定液浓度的过程 浓度校正因子F:比值应该在0.95-1.05之间
加催化剂来加速; ④必须有适当简便的方法确定滴定终点。
容量分析的计算问题
应该将滴定度(T )乘以滴定液的浓度校
正因数(F),换算成实际的滴定度(T'
=T*F)。其中, 滴定液实际配制浓度
F=
滴定液规定的浓度
加上,有时溶液需要稀释,所以被测药物 的含量公式可表示为:
含量%= F T V D 100% W
▪ 4.方法:具有一定的分辨力、专属性、稳定性和 灵敏度,其准确度和精密度均高。
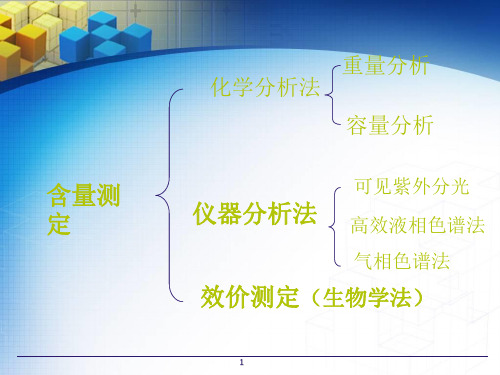
重量分析
化学分析法
容量分析(滴定)
含量测定
仪器分析法
光谱法
色谱法 电泳法
效价测定(生物学法)
重量分析法
▪ 以质量为测量值的分析方法。 称取一定重量的供试品,用适当的方法
将被测组分与试样中其它组分分离,根据 被测组分和供试品的重量以计算组分的含 量的百分数的定量方法。
注意事项
▪参考课本
五、碘量法
以碘为氧化剂,或以碘化物为还 原剂的滴定法。
直接碘量法 间接碘量法
剩余碘量法 置换碘量法
碘滴定液的配制:加大量碘化钾
增加I- 浓度 A、增加 I2溶解度,并减少 I2 挥发
KI
I2
KI
2 3
B、降低 I2/I -氧化电位,反应完全
滴定液的标定 碘滴定液
药物的含量测定方法与验证

9
(1) 直接滴定法
解:精密称取供试品Ws=0.4018g
NaOH实际浓度0.1002mol/L,则F=0.1002/0.1=1.002
消耗的NaOH滴定液体积:V样=22.20ml
V样 F T 100% 阿司匹林含量= WS
22.20 1.002 18.20 103 100% 0.4018
三 色谱分析法
1.特点与适用范围
特点: 高灵敏度:10-15~ 10-12g/ml, 高专属性: 高效能与高速度: 适用范围:广泛应用于药物制剂的含量测定,尤其 是复方制剂含量测定的首选方法。
37
(二)荧光分析法
2.干扰及排除
(1)溶剂——空白试验 (2)溶液: 药物浓度不宜过高, 悬浮物——过滤, 溶氧——通惰性气体除去 溶液pH。 (3)玻璃仪器及测定池——高度洁净 (4) 温度:控制测定温度
38
(二)荧光分析法
3.测定方法与含量计算
在一定实验条件时,在较稀浓度范围内药物的荧 光强度与溶液中该物质的浓度成正比。
1% 1cm
品相当于标示量的百分含量
34
A D V 1% C X D V E1cm L 100 标示量% = 100% V B V B 0.521 200 100 2 1000 5 4211100 100% 99.0% 50 2
35
(二)荧光分析法
20
二、光谱分析法
(2)吸光度的准确度校正 60mg重铬酸钾用0.005mol/L的硫酸溶液稀释至 1000ml,在规定波长处测定并计算吸收系数,与 规定值比较。 规定值:235nm 123.0~126.0
257nm
313nm
142.8~146.2
药物分析中的药物含量分析方法

药物分析中的药物含量分析方法药物含量分析是药物分析领域中一项十分重要的技术手段。
药物的含量分析主要用于确定药物制剂中活性成分的含量,以保证药物的质量和疗效。
本文将介绍常见的药物含量分析方法,包括定量分析法、滴定分析法、色谱分析法和光谱分析法。
1. 定量分析法定量分析法是药物含量分析的基础方法之一。
它基于物质的定量分析原理,通过实验测定药物含量的多少。
常用的定量分析方法有重量法、容量法和电位滴定法。
(1)重量法:将一定质量的药物样品称取,并进行溶解、稀释等处理后,通过质量差计算出药物的含量。
(2)容量法:通过向药物样品中滴加标准溶液,使溶液达到等量点(终点),从而推算出药物的含量。
(3)电位滴定法:利用反应溶液中的特定药物含量与溶液电压的关系,通过电位滴定仪进行电位滴定,从而确定药物的含量。
2. 滴定分析法滴定分析法是一种通过滴定试剂与药物样品反应来确定药物含量的方法。
常用的滴定法有酸碱滴定法、氧化还原滴定法和络合滴定法。
(1)酸碱滴定法:根据药物样品的酸碱性质,采用适当的滴定试剂进行滴定,并通过滴定量计算出药物的含量。
(2)氧化还原滴定法:利用药物与氧化剂或还原剂反应的氧化还原过程,通过滴定试剂的耗量推算出药物含量。
(3)络合滴定法:利用药物与滴定试剂之间形成络合物的特性,通过滴定试剂的耗量计算出药物的含量。
3. 色谱分析法色谱分析法是一种基于化学试剂在固定相上的吸附、分离和检测的方法。
常用的色谱法有气相色谱法(GC)、液相色谱法(HPLC)和薄层色谱法(TLC)。
(1)气相色谱法(GC):将药物样品挥发成气态,通过在固定相上的分离和检测,确定药物的含量。
(2)液相色谱法(HPLC):将药物样品溶解在溶剂中,通过在固定相上的分离和检测,确定药物的含量。
(3)薄层色谱法(TLC):将药物样品涂抹在薄层板上,通过吸附、分离和检测,确定药物的含量。
4. 光谱分析法光谱分析法是一种根据药物与光的相互作用,通过测量药物对光的吸收、散射和发射等光学性质,来确定药物含量的方法。
药物的含量测定方法与验证-定量分析方法的分类与特点

T=m×a/b×M=0.05×1/1×260.23 =13.01(mg/ml)
含量(%) =[(V0-VS)B× FB×TA ]/W×100% =[(23.21-15.73)×1.038×13.01]
×1000/0.1022 ×100% =98.8%
14
二、光谱分析法
(一)紫外-可见分光光度法
可以避免样品前处理及 进样体积误差对结果的影响
28Βιβλιοθήκη 地塞米松的HPLC法测定:取本品精密称 定,质量为0.0697g,置50ml量瓶中, 加30ml乙醇溶解,用水稀释至刻度,摇 匀,精密量取5ml,置25ml量瓶中,加 0.2mg/ml甲基睾丸素液5.0ml,用流动 相稀释至刻度,摇匀,取20µl进样, 测得地塞米松的峰面积为1313,甲基睾 丸素的峰面积为1032,质量校正因子为 1.05,求其含量。
29
地塞米松% =(f×AX )/AS×Cs × D/W× 100% =(1.05×1313) /1032×0.2×250/69.7×100%
=96.8%
30
三、色谱分析法—HPLC
(2)外标法 含量:
C供=(A供/ A对) C对
31
用外标法测定某胺类药物的含量。对照品溶液的制备: 精密称取对照品50.0mg,置50mL量瓶中, 加甲醇溶解并稀释至刻度,精密量取上述溶液5mL, 置25mL量瓶中,加流动相稀释至刻度。 供试品溶液的制备:精密称取供试品50.5mg, 照对照品溶液制备方法制备。 取对照品溶液和供试品溶液各10μL进样, 测得对照品溶液中,对照品的峰面积分别为1350, 供试品溶液的峰面积分别为1313。 试计算样品的百分含量
12
已知:司可巴比妥钠的摩尔质量M=260.23, 司可巴比妥钠与溴反应的摩尔比为1:1; 供 试 品 的 量 W=0.1022g , 硫 代 硫 酸 钠 滴 定液(0.1mol/L) 浓度校正因数F=1.038; 供试品滴定消耗硫代硫酸钠滴定液 15.73ml , 空 白 试 验 消 耗 硫 代 硫 酸 钠 滴 定液23.21ml。
药物的含量测定方法与验证

A为吸收度,l为液层厚度,c为溶液浓度,E为吸收系数。
A Ecl
– E:单位液层厚度时的吸收度。有两种表示法
摩尔吸收系数:在一定波长下,溶液浓度为1mol/L, 厚度为1cm时的吸收度。用ε表示。 百分吸收系数:在一定波长下,溶液浓度为1% 1% E (W/V),厚度为1cm时的吸收度。用 1cm 表示。
三、色谱分析法
• 特点与适用范围
– – – – 高灵敏度 高专属性 高效能与高速度 适用于药物制剂,尤其复方制剂的含量测定。
• 分类
– 依据分离原理:吸附色谱、分配色谱、离子交换色谱、 分子排阻色潽等 – 依据分离方式:纸色谱法,薄层色谱法,柱色谱法, 气相色谱法,高效液相色谱法等
三、色谱分析法
一、容量分析法(滴定法)
• 有关计算
– 含量的计算
直接滴定法
V T 含量 % 100 % W
所配制滴定液浓度与规定浓度不一致时:
T T F
含量 %
F
实际摩尔浓度 规定摩尔浓度
V T V T F 100 % 100 % W W
一、容量分析法(滴定法)
一、容量分析法(滴定法)
• 特点
– 方法简便易行 – 方法耐用性高 – 测定结果准确 – 方法专属性差
• 适用范围:较多用于化学原料药物的含量 测定
一、容量分析法(滴定法)
• 有关计算
– 滴定度:每1ml某摩尔浓度的滴定液所相当的 被测药物的重量。中国药典用mg表示。 – 滴定度的计算
b T (mg / ml ) m M a
– 阴性对照 – 阳性对照 – 纯度测试
四、检测限/定量限
• 检测限常用方法
药物的含量测定方法和验证1

一、容量分析法(滴定分析法)
含量的计算: (2)制剂标示量百分含量的计算
VTF 平均片重 标示量% W 100% 标示量
例:取司可巴比妥钠胶囊(标示量为0.1g)20粒, 除去胶囊后测得内容物总重为3.0780g,称取 0.1536g,按药典规定用溴量法测定。加入溴液 (0.1 mol/L) 25ml,剩余的溴液用硫代硫酸钠液 (0.1025mol/L)滴定到终点时,用去17.94ml。 空白试验用去硫代硫酸钠液25.00ml。按每1ml 溴 液(0.1mol/L)相当13.01mg的司可巴比妥钠,计 算该胶囊中按标示量表示的百分含量。
二、光谱分析法
(二)荧光分析法 某些物质受到光照射时,除吸收某种波长的光 之外还会发射出比原来所吸收的光的波长更长 的光;当激发光停止照射后,这种光线也随之 消失,这种光称为荧光。 从样品池出来的荧光是各方向发射的,在与激 发光光源排成直角的方向测荧光强度可以避免 透射的激发光的干扰。
三、色谱分析法
一、容量分析法(滴定分析法)
例题:滴定分析中,一般利用指示剂的突变来判断化 学计量点的到达,在指示剂变色时停止滴定,这一点 为: A.化学计量点 B.滴定分析 C.滴定等当点 D.滴定终点 E.滴定误差 答案:D
一、容量分析法(滴定分析法)
酸碱滴定 氧化还原滴定 滴定分析法 配位滴定 沉淀滴定 非水滴定
三、色谱分析法
例题:HPLC法与GC法用于药物复方制剂的 分析时,其系统适用性试验系指 A.测定拖尾因子 B.测定回收率 C.测定保留体积 D.测定分离度 E.测定柱的理论板数
化学原料药含量测定

化学原料药含量测定
一、原料药物的含量用什么表示
原料药物的含量限度一般以百分比来表示。
原料药物指用于生产各类制剂的原料药物,是制剂中的有效成份。
由化学合成、植物提取或者生物技术所制备的各种用来作为药用的粉末、结晶、浸膏等,但病人无法直接服用的物质。
一般含量限度唯一范围即有上限和下限;若为仿制药含量限度不得超出原研产品含量限度;若为新药,则需要做临床实验(药效学,药理毒理实验等)及处方工艺开发所确定的规格来修订。
二、原料药物质量研究的一般内容有哪些
1、性状
(1)外观、色泽、臭、味、结晶性等外观、色泽、臭、味,结晶性等为药物的一般性状,应予以考察,并应注意在贮存期内是否发生变化,如有变化,应如实描述,如遇光变色、易吸湿、风化、挥发等情况。
(2)溶解度通常考察药物在水及常用溶剂(与该药物溶解特性密切相关的、配制制剂、制备溶液或精制操作所需用的溶剂等)中的溶解度。
(3)熔点或熔距
熔点或熔距是已知结构的化学原料药的重要物理常数之一,熔点或熔距数据是鉴别和检查该原料药的纯度指标之一。
常温下呈固体状态的原料药应考察其熔点或受热后的熔融、分解、软化等情况。
结晶性原料药一般应有明确的熔点,对熔点难以判断或熔融同时分解的品
种应同时采用热分析方法进行比较研究。
(4)旋光度或比旋度
旋光度或比旋度是反映具光学活性化合物固有特性及其纯度的指标。
对这类药物,应考察其性质(采用不同的溶剂),并测定旋光度或比旋度。
2、鉴别
原料药的鉴别试验要采用专属性强,灵敏度高、重复性好,操作简便的方法,常用的方法有化学反应法、色谱法和光谱法。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
化学原料药的含量测定常用方法
化学原料药的含量测定常用方法有以下几种:
1. 酸碱滴定法:通过酸碱反应的滴定实验,测定原料药中含有的酸或碱的含量,进而推算出原料药的含量。
2. 比色法:通过物质在溶液中的吸收特性,利用分光光度计或比色计测定原料药中某种物质的含量。
3. 重量法:通过称量原料药和标准品的质量差异,计算原料药中某种物质的含量。
4. 气相色谱法:将原料药样品进样至气相色谱仪中,利用样品中不同组分在色谱柱中的保持时间差异,来分析并测定原料药中各组分的含量。
5. 高效液相色谱法:原料药样品与参比物同时进样至高效液相色谱仪中,通过检测不同组分在色谱柱中的保持时间差异,推算出原料药中各组分的含量。
6. 紫外-可见光谱法:通过测量原料药样品在紫外或可见光波长范围内的吸光度,计算原料药中某种物质的含量。
7. 氧化还原滴定法:通过氧化还原反应滴定法,测定原料药中含有的氧化剂或还原剂的含量。
这些方法都有其适用范围和特点,实际应用时需要根据具体情况选择合适的测定方法。