高分子前药
5-氨基水杨酸-聚乙二醇高分子前药的制备及其释放行为

5氨 基 水 杨 酸 ( - n s l yi ai ,一 A, 一 5Amioai l c 5AS 又 c c d 名美 沙拉 嗪 ) 治 疗 溃 疡 性 结 肠 炎 (B 和节 段 性 回 是 I D) 肠 炎等 炎症性肠 道 疾 病 的主 要 药 物 之一 , 国外 已 有 多 种制 剂上 市 , 国 内 目前 尚无 生 产[ 。 口服 5氨基 水 但 1 ] 一
5 基 水杨 酸 的前 药研 究 主要 有 : 5 基 水 杨 酸 与 嘻 用 嘻 二元 羧 酸缩 聚L 用 乙二 醇 或 乙 二 胺 连 接 5A A 的 3 ; “ 一s
羧基 和 甲基丙 烯酰 基 , 后再 自由基 聚合 ; 5 基 然 在 嘻
纯, 除氯仿 和 DMF外 , 余 均直接 使用 。 其
摘
要: 用羧 基 活化 法 , 以聚 乙二 醇 ( E 、 P G) 丁二 酸 酐 为 原 料 , 备 了 5氨 基 水 杨 酸 (- S 的 高 分 子 前 药 。 傅 立 叶 制 一 5A A)
红 外 分 析 表 明 5AS 已经成 功 与 P G 共 价 键 连 接 , 溶 性 实验 表 明前 药 的 水 溶 性 比 5AS 的 明显 增 大 。 对 高 分 子 前 r A E 水 - A 药在 磷 酸 缓 冲 溶 液 中进 行 释 放 研 究 , 果 表 明 :H 值 为 20和 5 4时 5AS 的 释 放 量 较 少 ; p 值 为 7 2时 ,- S 结 p . . - A 而 H 的体 积 比为 1 先 水
: 0 搅 拌 3 n 重 复 3次 , 后倒 人 无水 氯化 钙 中 1, 0mi , 然
干 燥 2 , 4h 最后 再加 人适 量五 氧化 二磷 ,O 6 ℃回流 1h ,
高分子药物

第 七章 高分子药物
• 7.2.2 高分子络合物药物
• 一些含有给电子基团的高分子可与金属离子或小分子药物生成络 合物。因药物与高分子间化学平衡, 可保持原化合物的生理活性, 降低其毒性和刺激性, 还能保持一定的浓度,从而达到低毒、高效 和缓释的作用。 • 如水溶性较高的水杨醛羧甲基壳聚糖衍生物( RS-CMC),将碘与 RS-CMC 络合制备成RS-CMC-I2,作为抑菌药物。
• 由微胶囊包裹的高分子药物可以控制药物的释放时间,降低药物 毒副作用,增加药物的稳定性和有效利用率,实现药物的靶向输 送,将药物用高分子材料包囊,使药物进入到人体后在一定的时 间内释放,达到对药物治疗剂量进行有效控制的目的。
第 七章 高分子药物
• 7.2.4 高分子微胶囊药物
•
•
天然高分子包囊材料为可胶凝的胶体材料,如明胶、阿拉伯胶及淀粉等, 这类包囊材料无毒、成膜性好、稳定性好。
4、抗血栓剂:葡聚糖硫酸钠、聚乙烯硫酸钠 5、凝血剂:聚己烷二甲胺基丙基溴、羟乙基淀粉 6、抗病毒药物: 顺丁烯二酸酐聚合物 7、抗菌药物:青霉素聚合物 8、肠道药:聚丙烯酸、聚马来酸酐 •
第五 聚合 物的化学反应 第章 七章 高分子药物
7.4 具有药理活性高分子
具有药理活性的高分子如酶制剂、多糖、多肽、蛋白质、激素等天然药理活性 高分子, 以及一些具有药理活性的化学合成高分子都可用作药物。这类高分子本身 可与人体生理组织发生一定的物理和化学的反应, 具有治疗病变的作用。一些具有 药理活性的高分子,可以直接作为药物,当它被降解为小分子后就不再具有药性, 是真正意义上的高分子药物。
•
第 七章 高分子药物
7.4.2 具有高分子链的天然药物
1、 天然植物多糖如灵芝多糖可以激活机体免疫系统, 提高细胞中白细胞介 素、干扰素和肿瘤坏死因子的水平。 2、壳寡糖可抑制肿瘤组织内新生血管生长, 切断肿瘤细胞营养来源和转移 途径,以抑制某些炎症性疾病。 3、蛋白质和多肽药物 在抗肿瘤、疫苗、抗菌等方面展现出诱人的前景,用 于预防艾滋病的多肽疫苗, 目前正在进行临床实验, 实验证实两种多肽能 刺激机体产生特异性抗体和特异性细胞免疫, 并有良好的安全性。 4、淀粉衍生物如羟乙基淀粉、抗性淀粉、慢消化淀粉等作为凝血剂、降血 脂、降血糖等药物在开发利用。 5、天然膳食纤维预防及治疗肠道疾病等。
含有环丙沙星高分子前药物的合成

约 占院 内感染 的 4 %;- O []心脏 外科 生物材 料植 入 1 2 术 合 并 感 染 发 生 率 为 3 -% ;9 33 9 %的 机 械 通 气 患 者 气管 插管 处有 细菌 定 植 并 反复 发 生 感染 ;大静 脉 导管 , 口引流管 , 工 关节 置 入 、 用缝 合 线 、 伤 人 医 医用敷料 、医 用介 入诊 疗 导 管等 相 关 感染 的发生
法有如下几种: ) 1 将抗菌药物经离子键结合在医
用植 入件— — 导 尿管 、 血管 支架 表面 ;) 过物 理 2通 作 用 将抗 菌药 物 包 埋 在 聚 合物 基 质 里圆; ) 天 3将
然 的抗 菌 剂—— 银 涂 覆在 导 尿管 表面 阻 止细 菌感
心 的感染 和 并发 症 问题 越 来越 严 重 。根 据 流行 病
( . hmir 1C e sy&C e cl n ier gD pr n, i owa ct n l eh i l ol eP t nFj t hmiaE gn ei e at tMe h u n a o a &T cnc l g , ui ui n me z Vo i aC e a 姐
文 献标 识 码 : A
含 环 沙高子 药 的成 有 丙 星分前 物 合
连鸿丹 ・ 叶艳秋 董声雄 , ,
(. 1湄洲湾职业技 术学院 化 学工程 系,福建 莆 田 3 15 ; . 州大学 化 学化 工学院,福建 福 州 3 0 0 52 4 2 福 50 2)
摘 要 : 以抗茵药物 . 盐酸环丙沙星( I O) CP R 代替传统的聚氨酯扩链剂, 采用六亚甲基二异氰酸酯( D ) H I、
含喹诺酮类药物的高分子前药的合成及表征的初步研究

多 ,主要用 于人工心脏 ,辅 助人工 心脏 ,人 工血管 ,心 导管 ,人工心脏 瓣膜及人 工软骨 等【。 2 】
预聚 。随后 ,计 量 的盐 酸环丙沙 星溶于溶 剂 D O中 , MS
加热 至 7 ℃ ,加入等 摩尔 的三 乙胺 ( T 0 E A)助溶 。将该 溶液加 到预聚溶液 中 ,最后再加 计量催 化剂 ,反应继 续 进行 2 h 4 。反应 完成后 用 甲醇终止 反应 。 C P法将 P C法 中 P L和盐 酸环丙沙星 的相关步 D D C 骤顺序对 换 ,各步反应 条件 相同 。 用 去离 子水 多 次洗 涤 沉淀 ,在蒸 馏水 中连 续搅 拌
用前景 ,是 近几年 生物医学 材料领域 的一个研 究热 点。 它具有低毒 、 效、 高 缓释和 长 效等特点 , 生物相 容性好 ,
P C法 :在干燥 N 的氛 围下 ,于三 口瓶 中加入计 D 2
量 的溶剂 D O和 P L,搅拌至 P L溶解 ,然 后再用 MS C C
l 注 射器加入 H 和催化 剂二 月桂酸二丁基锡 , ml DI 加热
下,药物可 以从 高分子 中控 制释放 出来 。 关键词: 高 分子前药 ;环 丙沙 星;聚氨酯 中图分类号 : T 3 4 B 4 B 2 ;T 3 文献标 识码 :A
文章编 号: 10 .7 12 0 ) 0 19 3 (0 7增刊.9 1 3 16 . 0
21 原 料与试剂 . 聚 己内酯 ( C P L,15 ) 2 0 ,分析 纯 ;六 亚 甲基 二异
境下才释放 出药物显 示其药 理活性 , 决 了过 去药物一 解 旦释放就无法停 止 的不 可控 问题 , 避免 了药物 过量和 产
生抗药性。
高分子材料 中聚氨 酯 ( U)材料 的微 相分离 结构… P
高分子药物

(2)羟乙基淀粉
一、多糖类高分子药物
(3)双醛淀粉
(4)甲壳质衍生物 与 抗癌药物偶联,包括 5-氟尿嘧啶、甲氨蝶 呤、丝裂霉素等
5-氟尿嘧啶与6-O-羧甲 基甲壳素连接,实现 5-氟尿嘧啶的缓释, 对P-388淋巴细胞白血 病有明显抑制作用
Chem. Pharm. Bull. 1992, 40, 559-561
三硫酸双糖是肝素的主要双糖单元。
肝素含有硫酸基与羧基,呈强酸性,为聚
硫酸酯
阴离子,与阳离子反应成盐。
肝素的糖苷键不易酸解;O-硫酸基对酸水 解相当稳定;N-硫酸基对酸水解敏感;
磺酰胺与氧化剂反应,降解成酸性产物,还原剂 的存在,基本不影响肝素的活性。
硫酸化程度高的肝素具有较高的降脂和抗 凝活性;高度乙酰化的肝素,抗凝活性降 低甚至完全消失,而降脂活性不变
几乎所有的线粒体和许多细菌(如大肠杆菌) 含有结合锰的超氧化物歧化酶(Mn-SOD);
大肠杆菌和其他一些细菌还含有结合铁的超氧 化物歧化酶(Fe-SOD);
超氧化物歧化酶结构示意图 His-组氨酸;Asp-天冬氨酸
在高等植物中,不同形式的超氧化物歧化酶定 位于不同的细胞区室中。Mn-SOD存在于线粒体 和过氧化物酶体;Fe-SOD主要位于叶绿体; CuZn-SOD则定位于原生质、叶绿体、过氧化物酶 体和质外体(apoplast)中
一、多糖类高分子药物
(2)多糖的无机酸酯 具有药物活性的原因:硫酸根等聚阴离子具有强 负电荷,能与病毒分子结合而阻断病毒对细胞的吸附,从而抑制病毒的 反向转录酶;能与受体细胞表面的正电荷分子结合,干扰病毒对受体细 胞的吸附,消除病毒引起的细胞病变。
壳聚糖硫酸酯、硫酸软骨素、纤维素硫酸酯
2、多糖醚及其苷键, 并含有支化的β-1,6-糖苷键;
生物医用高分子知识点总结

载体材料要求(1)一定机械强度,加工性能(2)生物相容性与生物降解性(3)根据使用要求,其它一些特殊性能,例如pH 响应性、靶向性等载体材料分类根据材料的来源:天然与合成高分子材料根据材料的降解性能:降解与非生物降解高分子材料根据材料在水中的溶解性:疏水与水溶性高分子材料根据材料对外界信号响应性:刺激响应与惰性材料根据材料化学结构分类(以疏水性生物降解材料为例)(1)聚酯:PLGA PCL聚碳酸酯等(2)聚酸酐(3)聚氨基酸(4)聚膦腈(5)聚磷酸脂典型非生物降解疏水性高分子:聚氨酯典型疏水性生物降解材料-1. 聚酯聚(丙交酯-材料,这主要归co-乙交酯)(PLGA是一类应用最早、最为广泛的合成疏水性生物生物降解聚酯的合成开环聚合(以聚乳酸PLA的合成为例)1.分子量可从几千到几百万广泛可调2.采用手性单体聚合可得到旋光性聚合物,具有很高的机械强度,可作为骨替代材料3.其它生物降解聚酯如聚已内酯、聚(三甲基碳酸酯)等均采用类似的合成路线缩合聚合:合成可生物降解塑料的方法,但目前还仅能得到低分子量的产物生物降解聚酯研究进展:PEG-b-PLA合成PEGb-PLA的性质:温敏性避免网状内皮系统的吞噬持续释放药物典型疏水性生物降解材料-2. 聚酸酐类开环聚合仅适用于环己二酸酐的聚合,其它环状酸酐例如琥珀酸酐、戊二酸酐由于环较稳定,还不能通过开环聚合得到聚酸酐。
所得聚已二酸酐分子量较低,一般在5,000 以下。
熔融缩聚1.除热不稳定、易成环二酸单体外,其它所有二酸均可通过这种方法聚合2.聚酸酐分子量与二酸结构以及聚合条件有关,单体柔性越大、真空度越高,聚合物分子量越大脱三甲基氯硅烷法溶液缩聚1.适用于热不稳定二酸单体2.聚酸酐分子量在几千左右微波聚合合成聚酸酐优点1 )无需真空2)反应时间短3)预聚-缩合一步完成聚(酰亚胺-酸酐)提高常规聚酸酐力学强度,有可能用作骨替代材料主链型高分子前体药物:水杨酸光交联聚酸酐单体与预聚物观察不到荧光主链中仅当亚甲基数为2的聚酸酐可发荧光苯环上取代基团对荧光激发与发射波长有影响荧光强度基本随聚合物分子量增大线性增强与其它二酸单体共聚,荧光波长会发生偏移,其中脂肪族二酸发生蓝移,而芳香二酸发生红移共聚物序列结构对荧光性质影响不大疏水性生物降解高分子材料的两种降解方式:本体降解表面降解降解方式的决定因素聚合物基质的水化速度(V hydr。
氟尿嘧啶大分子前体药物肿瘤靶向的研究进展

氟尿嘧啶大分子前体药物肿瘤靶向的研究进展【关键词】大分子前体药;5氟尿嘧啶;肿瘤靶向;缓释;控释5氟尿嘧啶(5fluorouracil,5FU) 作为首选抗代谢药用于临床医治结直肠癌、胃癌、乳腺癌等多种癌症。
但由于首过代谢显著及亲脂性较低, 5氟尿嘧啶的生物利费用低, 阻碍抗肿瘤疗效, 且其医治剂量与中毒剂量接近。
为克服临床应历时存在的恶心、呕吐、腹泻、脱发、体重减轻、白细胞与血小板下降等副作用, 对5氟尿嘧啶的衍生物的合成、药理、毒理、代谢等进行了大量的研究, 以寻求5FU的前体药物。
已有研究[1]说明,将5FU制成大分子前体药能较好的解决这种问题。
5FU的大分子前体药与5FU相较较,具有缓释、长效、利用率高、不良反映小和靶向性强等优势。
随着现代医学的进展,大分子前药(macromolecular prodrugs) 的研究和应用日趋受到人们的重视。
1975 年Ringsdorf[2]第一次提出了大分子前药的一样模型(图1) 。
那个模型包括了大分子载体、与载体相连接的小分子活性药物、定位基,有时也有效于连接载体和小分子药物的连接基。
定位基团的目的是引导大分子前药抵达人体中特定的组织及细胞,小分子药物和载体一样以共价键相连接,但必需可通过体液水解、酶解而断裂,释放出具有医治作用的原药。
因此,大分子前药具有两个很突出的特点:控释作用和靶向性。
小分子药物尽管疗效高,但其中的许多品种却同时存在着专门大的副作用。
在它们进入人体后的短时刻内,血液中药物的浓度远远超过医治所需的浓度,太高的浓度可能引发很多副作用,同时它们在生物体内新陈代谢速度快,半衰期短,易排泄,故随着时刻的推延,药物的浓度降低较快,阻碍疗效;另一方面,小分子药物对进入体内指定部位也缺乏选择性,这也是给药量增多、疗效较低、副作用大的缘故之一[3]。
基于这些缘故,最近几年来药物释放体系(DDS) 已成为热点研究领域,大分子前药在其中占据了很重要的位置。
淀粉支载阿司匹林前药的合成及其体外释放

b y u l t r a v i o l e t s p e c t r o p h o om t e t e r . A n d v / t r o d r u g r e l e a s ei n v e s i t g a i t o nw a r c a r d e d o u t i n a r t i i f c i a l g st a r i c j u i e c a n d a r i t i f c i a l i n t e s t i ・ n a l j ic u e .T he r e s lt u s c l e a r l y s h o w d e ha t t sp a i r i n W s a c o u p l e d it w h he t s t a r c h m o l e c u l e s u c es c s f u l l y nd a he t d r u g r e l e a s e r a t e o f h t e
s t a r c h nd a a c e t o x y b e n z o y l c h l o r i d e .I t s s t r u c t u e r wa s c h a r a c t e r i z e d w i h H- t NMR. T h e d r I I g - l o a d i n g r a i t o o f p r o d u c t W s a me a s u r e d
福建农林大学学报 ( 自然科 学版)
J o u r n a l o f F u j i a n A g r i c u l t u r e a n d F o r e s t r y U n i v e r s i t y( N a t u r a l S c i e n c e E d i t i o n )
透明质酸在紫杉醇新型给药系统中的应用

透明质酸在紫杉醇新型给药系统中的应用武玉敏【摘要】透明质酸是一种酸性黏多糖,其作为优良的药物载体,具有生物相容性良好,生物可降解的特性,同时可与肿瘤细胞表面富含的CD44、RHAMM等受体结合,目前已成为抗肿瘤药物递送载体研究的主要热点。
本综述主要通过查阅近年文献,对透明质酸在紫杉醇新型给药系统中的应用进行综述。
%Hyaluronic acid is an acid mucoPolysaccharide,as a drug carrier is excellent,with good biocomPatibility,bio-degradable ProPerties,and can be combined with CD44 ,RHAMM recePtor in the surface of tumor cells. Referring to recent literatures,the aPPlication of hyaluronic acid in Paclitaxel new tyPe delivery systems was reviewed in this article.【期刊名称】《药学研究》【年(卷),期】2014(000)008【总页数】3页(P475-477)【关键词】透明质酸;紫杉醇;给药系统【作者】武玉敏【作者单位】山东大学药学院,山东济南250012【正文语种】中文【中图分类】R944透明质酸(hyaluronic acid,HA)是以D-葡萄糖醛酸-N-乙酰氨基葡萄糖为双糖单位组成的线性大分子酸性黏多糖,可与肿瘤细胞表面富含的CD44、RHAMM等受体结合,激活针对HA的细胞内信号通路或激活HA的内化作用,而调节细胞的运动等行为[1,2]。
因其良好的生物相容性和可降解性,以及对肿瘤细胞的高亲和性和抗肿瘤辅助治疗作用,透明质酸已经用于多种抗肿瘤药物的递送,包括紫杉醇,多西他赛,多柔比星等,成为目前抗肿瘤药物递送载体研究的主要热点[3,4]。
新药研究中的前药原理

无法想象的。
医院在目前“内忧外患”的情况下,维持现状,固步自封,不思进取显然是行不通的。
要想在竞争中不被对手击倒,就应该具有“忧患意识”。
首先,在管理上要科学化。
即科学地制定一套长远的、可持续发展的战略方案。
其次,要树立竞争意识。
人员管理是核心,积极培养员工竞争意识非常重要。
第三,要强化服务意识。
医院药房作为具有一定特殊性的服务行业,应加强服务营销的理论培训,提高员工的服务意识和专业素质。
21采取联合或合作策略。
合作(或联合)的目的就是融集资金和资源互补。
上海、苏州等地在2002年率先提出了“托管药房”的创新模式,奏响了我国医药分业的前奏曲。
所谓“托管”,即是医院与第三方合作,在药房所有权不变的情况下,原职工编制不变,只是返还一定比例利润并承担职工工资、奖金、社会福利等一切费用的前提下委托第三方来进行经营管理。
以上海公惠医院为例,托管前药房药品的月销售额为150~160多万元,托管后为300多万元,足足翻了一番,药房的利润也在原来的基础上至少提高了近3%。
经过近两年的实践证明,经过合作双方的优势互补,以经济作为杠杆整合了用人机制,充分调动了员工的工作积极性,最大限度的避免了人浮于事的现象,提高了药房的服务质量,大大节约了医院的管理成本,使医院的收益得到明显提高。
由此可见,医院在市场化经营理念下的效益竞争也能形成良性循环。
・综 述・新药研究中的前药原理刘剑峰,徐文方(山东大学药学院 济南 250012)摘要:前药具有改变药物的理化性质,提高生物利用度,增加水溶性,减少不良反应,定位到达靶器官等特性。
前药保持或增强原药的药效,同时又克服了原药的缺点。
本文主要综合前药原理在新药设计中的典型实例,介绍前药原理的应用规律。
关键词:前药 前药原理 新药研究 靶向药物 生物利用度中图分类号:R91412 文献标识码:B 文章编号:1672-7738(2005)02-0099-06The principle of prodrugs in new drugs designLiu Jian 2feng ,Xu Wen 2fang(Pharmacy School of Shandong University ,Ji ’nan ,250012)Abstract :The prodrugs can change the physico 2chemical property of drugs ,increase bioavailability ,enhance water 2solubility ,re 2duce side effects or attach to the target 1The prodrugs can retain or improve effects of the parent drug ,meanwhile ,overcome the draw 2back of the parent drug 1The paper introduced and exemplified the principle of the prodrugs used as a method in new drug design 1K ey words :prodrugs ;principle of prodrugs ;new drug design ;targeting drugs ;bioavailability 保持药物的基本结构不变,仅在某些功能基上做一定的结构修饰得到的化合物,在体内又转化为原来的药物而发挥疗效时,称原来的药物(原药)为母体药物(parent drugs ),结构修饰后的化合物为前体药物(prodrugs ),简称前药。
药用高分子材料试题库

第一题、名词解释1、药用辅料2、高分子药物p193、全同立构4、均聚物5、端基6、聚合反应7、压敏胶8、玻璃化转变温度9、单体10、智能水凝胶第二题、填空题11、依据药用高分子材料的用途一般可分为三大类:,和包装用的材料。
12、淀粉是天然存在的糖类,由两种多糖分子组成,分别是和,它们的结构单元是。
13、控释、缓释给药的机制一般可分为五类:p84 扩散、、离子交换和高分子挂接。
14、含A、B两种单体的共聚物分子链的结构单元有四种典型的排列方式:、、、。
p715、自由基聚合反应的特征可概括为慢引发、快增长、速终止。
16、高分子的溶解是一个缓慢过程,其过程课分为两个阶段:一是,二是。
p2917、非晶态聚合物的物理状态分为玻璃态、和,这些状态通称为力学状态。
P2318、热致凝胶化和是水溶液非离子型衍生物的重要特征,这种特征表现为聚合物溶解度不随温度升高而升高。
19、聚乙烯醇PV A17-88的聚合度为,醇解度为。
20、淀粉形成均匀糊状溶液的现象称为糊化;而淀粉凝胶经长期放置会变成不透明的沉淀现象则称为老化。
21、纤维素都是由D-葡萄糖单体缩聚而成的一个直链高分子,而且都是以 -1,4-葡萄糖苷键的形式连结起来的。
22、药用明胶按制法分为酸法明胶(A型明胶)和碱法明胶(B型明胶),在等电点时,明胶的粘度、溶解度、透明度、溶胀度最小,但两种明胶在使用上无区别。
23、黄原胶是新型多糖类发酵产品,被广泛应用于食品、石油、地矿、陶瓷、纺织、印染、医药、造纸、灭火、涂料、化妆品等20多个行业,因此被誉为“工业味精”,是目前世界上生产规模最大且用途极为广泛的微生物多糖。
24、透明质酸分子中含有大量的羧基和羟基,其具有强大的保水作用,可结合自身400倍以上的水,是目前发现的自然界中保湿性最好的物质,被称为理想的天然保湿因子。
25、脱乙酰甲壳质又称为壳聚糖,是一种白色、五臭的新型药用辅料。
26、丙烯酸树脂实际上是甲基丙烯酸酯、丙烯酸酯、甲基丙烯酸单体按不同比例共聚而成的一大类聚合物,其中丙烯酸树脂Ⅱ、Ⅲ、Ⅳ已载入中国药典。
2.1 纳米药物靶向给药类型

纳米药物靶向给药类型水溶性聚合物现在通常用于延长药物循环和停留时间,提高药物的溶解度,降低药物的全身毒性。
早在20世纪80年代,Maeda等人和Jain观察到水溶性聚合物与细胞毒性药物的共价缀合物更有效地靶向肿瘤组织而不是其游离形式的细胞毒性药物。
这种高渗透性允许大分子选择性地外渗入肿瘤和不良的淋巴引流,并由此增加肿瘤中大分子的滞留。
为了开发EPR效果,需要适当考虑聚合物输送系统的尺寸和其他物理化学特性。
下面的例子将说明这个观点。
Abraxane和Doxil是FDA批准用于癌症治疗的第一批纳米载体。
鉴于它们的尺寸相对较大(分别为130和150纳米),这些纳米粒子不太可能深入到肿瘤组织中。
因此,这些纳米载体的尺寸需要进行严格的优化。
事实上,在最近的一项研究中,Sisson等证明直径在25-50nm范围内的聚甘油(PG)微凝胶颗粒非常有效,能够非破坏性的被癌细胞摄取。
这些研究强调至少一个“最优”尺寸的重要性聚合物能够有效的输送并聚集到疾病或陷入困境的组织。
肿瘤侵袭,转移,对化疗药物的耐药性以及辐射是成功治疗肿瘤的主要障碍。
有些限制可以通过治疗策略来克服,这些策略可以增加特异性和功效,同时降低抗癌药物的毒性。
其中一种方法包括将聚合物递送系统专门针对癌细胞。
通过采用以下两种方法之一可以实现纳米递送系统的聚合形式对癌细胞和肿瘤的靶向性:被动靶向和主动靶向。
1 被动靶向NDS的被动靶向作用主要依赖于其纳米尺寸与肿瘤部位的微血管结构。
与正常组织相比,绝大部分肿瘤组织由于生长代谢旺盛,其血管重塑不完全,血管内皮之间具有10-1000nm 空隙,且肿瘤部位淋巴管缺乏,淋巴回流受阻。
因此相应尺寸的 NDS可经血液循环达到肿瘤组织通过增强渗透滞留(Enhanced permeability and retention,EPR)效应富集于肿瘤组织,这是被动靶向效应的基础(图 1-1)。
一般地,10-100 nm大小的NDS具有较好的 EPR 效应;此外,NDS的物理化学性质,包括表面电荷、粒子形态等均对EPR效应具有显著影响;另一方面,肿瘤区域血管内皮的生长状态及血管密集程度也可影响EPR效应的发挥。
5_氟尿嘧啶大分子前体药物肿瘤靶向的研究进展

5-氟尿嘧啶大分子前体药物肿瘤靶向的研究进展何练芹,朱亮(广东药学院药科学院,广东广州510006)关键词:大分子前体药;5-氟尿嘧啶;肿瘤靶向;缓释;控释中图分类号:R 979 文献标识码:A 文章编号:1006-8783(2008)05-0536-04作者简介:何练芹(1983-),女,在读硕士研究生;通讯作者:朱亮,男,教授,硕士研究生导师,从事药物靶向制剂研究,E m a i:l li ang z99@ 。
5-氟尿嘧啶(5-fl u orourac i,l 5-FU )作为首选抗代谢药用于临床治疗结直肠癌、胃癌、乳腺癌等多种癌症。
但由于首过代谢显著及亲脂性较低,5-氟尿嘧啶的生物利用度低,影响抗肿瘤疗效,且其治疗剂量与中毒剂量接近。
为克服临床应用时存在的恶心、呕吐、腹泻、脱发、体重减轻、白细胞与血小板下降等副作用,对5-氟尿嘧啶的衍生物的合成、药理、毒理、代谢等进行了大量的研究,以寻求5-FU 的前体药物。
已有研究[1]表明,将5-F U 制成大分子前体药能较好的解决这类问题。
5-FU 的大分子前体药与5-FU 相比较,具有缓释、长效、利用率高、不良反应小和靶向性强等优点。
随着现代医学的发展,大分子前药(m acro m olecu lar pr odr ugs)的研究和应用日益受到人们的重视。
1975年R ingsdo rf [2]首次提出了大分子前药的一般模型(图1)。
图1 大分子前药的一般模型 F i g .1Sch e mat i c representati on of a m acro m olecu lar p rodrug这个模型包含了大分子载体、与载体相连接的小分子活性药物、定位基,有时也有用于连接载体和小分子药物的连接基。
定位基团的目的是引导大分子前药到达人体中特定的组织及细胞,小分子药物和载体一般以共价键相连接,但必须可通过体液水解、酶解而断裂,释放出具有治疗作用的原药。
氟尿嘧啶大分子前体药物肿瘤靶向的研究进展研究

氟尿嘧啶大分子前体药物肿瘤靶向的研究进展研究【摘要】目的:讨论氟尿嘧啶大分子前体药物肿瘤靶向的研究进展。
方法:分析近几年关于氟尿嘧啶大分子前体药物肿瘤靶向的研究进展,并以载体为分类展开讨论。
最后提出大分体前体药物研究进一步发展面临的问题和挑战。
结论:目前已有多种大分子载体可应用于氟尿嘧啶大分子前体药物,且具有良好的性质,但为了能在临床上得到更广泛的应用,还需要近一步的药代研究。
【关键词】氟尿嘧啶,前体药物,靶向前言氟尿嘧啶作为最早的抗癌药物之一,最早被从海参中提取[1],在临床应用上主要用于治疗胃癌和乳腺癌等多种癌症[2-3]。
它能够在细胞内转化为有效的氟尿嘧啶脱氧核苷酸,通过阻断脱氧核糖尿苷酸受细胞内胸苷酸合成酶转化为胸苷酸,而干扰DNA的合成。
氟尿嘧啶同样可以干扰RNA的合成。
1大分子前体药物靶向研究的必要性由于氟尿嘧啶的治疗剂量与中毒剂量比较接近,临床应用时很容易出现呕吐、腹泻以及白细胞下降等副作用,将氟尿嘧啶制成大分子前体药物能够很好的解决这一问题[4]。
氟尿嘧啶大分子前体药物的设计模型包括大分子载体、与载体相连接的小分子活性药物、定位基(有时也用到连接基)。
定为基主要作用是引导该该前体药物达到靶向部位。
而小分子药物和载体必须断裂(酶解作用等),才能释放出具有治疗作用的氟尿嘧啶。
因此,氟尿嘧啶大分子前体药物的设计具有两个特点:具有控释左右和靶向作用。
本文以氟尿嘧啶大分子前体药物的载体为分类,研究了氟尿嘧啶大分子前体药物肿瘤靶向的进展。
2载体分类2.1聚乙烯和聚丙烯类聚合物该类聚合物是最早应用于抗肿瘤药物的高分子载体的,近年来[5],有研究表明:将氟尿嘧啶制成甲基丙烯酸衍生物,将该衍生物与乳糖混合后,进行机械化学固态聚合反应制备氟尿嘧啶的大分子前体药物。
此种方法制备的高分子前体药物,既有利于操作(制备过程干燥状态下进行,产物不需后处理工作),又便于控制药物剂量,具有较高的临床应用价值。
2.2磷酸酯类聚合物该类聚合物的特点是与人体内环境具有优秀的生物相容性,主要原因是人体内的甘油酸酯和核酸等与磷酸酯类聚合物组成结果类似[6]。
高分子前体药物分子设计结构
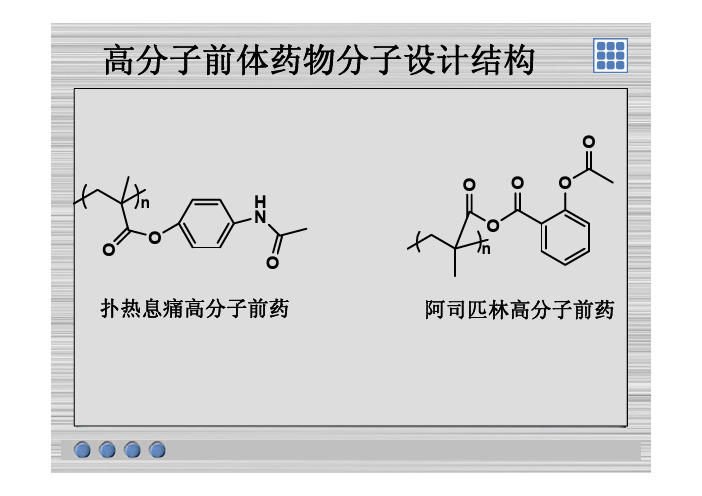
高分子前体药物分子设计结构O O OOO H NnOOOn扑热息痛高分子前药阿司匹林高分子前药扑热息痛在pH=2的磷酸盐缓冲溶液中紫外扫描图243nm扑热息痛高分子前药扑热息痛在pH=7.8的磷酸盐缓冲溶液中紫外扫描图243nm扑热息痛高分子前药甲基丙烯酸-扑热息痛单体的红外扫描图扑热息痛高分子前药-1H-NMR 甲基丙烯酸扑热息痛单体的N 图O OH NO扑热息痛高分子前药聚(甲基丙烯酸-扑热息痛)的红外扫描图扑热息痛高分子前药聚(甲基丙烯酸-扑热息痛)的1H-NMR图O H Nn没有烯氢OO扑热息痛高分子前药pH=2阿司匹林在p 的磷酸盐缓冲溶液中紫外扫描图276nm阿司匹林高分子前药pH=7.8阿司匹林在p 的磷酸盐缓冲溶液中紫外扫描图296nm阿司匹林高分子前药甲基丙烯酸-阿司匹林单体的红外扫描图阿司匹林高分子前药甲基丙烯酸-阿司匹林单体的LC-MS图M+Na271.12M+1249.23阿司匹林高分子前药聚(甲基丙烯酸-阿司匹林)的红外扫描图阿司匹林高分子前药目标分子结构O OnNH OHO高分子负载手性催化剂N-BOC-羟基脯氨酸(Ⅰ)的红外扫描图高分子负载手性催化剂N-BOC-羟基脯氨酸(Ⅰ)的MS 图M 1M+1M-1230.12232.26高分子负载手性催化剂N-BOC-羟基脯氨酸对甲氧基苄酯的红外扫描图高分子负载手性催化剂N-BOC-羟基脯氨酸对甲氧基苄酯的LC-MS 图M+1352.13M+Na37407374.07高分子负载手性催化剂对甲氧基苄酯的红外扫描图高分子负载手性催化剂对甲氧基苄酯的MS图M+1420.22高分子负载手性催化剂。
医用高分子材料总论

医用高分子材料公管学院信息管理与信息系统专业涂佳琪20091020053文摘:生物体是有机高分子存在的最基本形式,有机高分子是生命的基础。
动物体与植物体组成中最重要的物质——蛋白质、肌肉、纤维素、淀粉、生物酶和果胶等都是高分子化合物。
因此,可以说,生物界是天然高分子的巨大产地。
高分子化合物在生物界的普遍存在,决定了它们在医学领域中的特殊地位。
由于高分子材料的分子结构、化学组成和理化性质与生物体组织最为接近,因而决定了它最有可能作为医用材料,应用于人工器官、外科修复、理疗康复、诊断检查、患疾治疗等医疗领域。
一、医用高分子材料的基本要求由于高分子科学和医学的日益发展及相互渗透,使人类有可能逐步实现修补人体缺损,增进健康,延长寿命。
另一方面愈来愈多的医疗器械也以高分子作为原材料。
因此从质量和数量两个方面对医用高分子材料提出了更高的要求。
医用高分子材料是一类特殊用途的材料。
它们在使用过程中,常需与生物肌体、血液、体液等接触,有些还须长期植入体内。
由于医用高分子与人们的健康密切相关,因此对进入临床使用阶段的医用高分子材料具有严格的要求,要求有十分优良的特性。
归纳起来,一个具备了以下七个方面性能的材料,可以考虑用作医用材料。
一般满足下列几个基本条件:(1)在化学上是不活泼的,不会因与体液或血液接触而发生变化人体环境对高分子材料主要有以下一些影响:1)体液引起聚合物的降解、交联和相变化;2)体内的自由基引起材料的氧化降解反应;3)生物酶引起的聚合物分解反应;4)在体液作用下材料中添加剂的溶出;5)血液、体液中的类脂质、类固醇及脂肪等物质渗入高分子材料,使材料增塑,强度下降。
但对医用高分子来说,在某些情况下,“老化”并不一定都是贬意的,有时甚至还有积极的意义。
如作为医用粘合剂用于组织粘合,或作为医用手术缝合线时,在发挥了相应的效用后,反倒不希望它们有太好的化学稳定性,而是希望它们尽快地被组织所分解、吸收或迅速排出体外。
药物化学论文-5-氟尿嘧啶

成绩中国农业大学课程论文(2011-2012学年秋季学期)论文题目:5-氟尿嘧啶肿瘤细胞靶向性前药的研究进展课程名称:药物化学任课教师:班级:学号:姓名:5-氟尿嘧啶肿瘤细胞靶向性前药的研究新进展摘要:5-氟尿嘧啶(5-fluorouracil,5-FU),是目前临床上广泛应用的抗代谢类抗肿瘤药物之一,通过干扰核酸的合成抑制癌细胞的增生,对结肠癌、乳腺癌、胃癌等癌症显示有良好的治疗效果[1]。
但同时也伴随着很强的毒副作用。
因此,对5-氟尿嘧啶进行有效的分子修饰,以克服其缺点,提高其靶向性和选择性,最大程度地发挥其活性作用和减轻毒副作用,已成为当今抗癌药物研究的热点。
本文根据不同靶向载体,对近年来国内外各种修饰的5-氟尿嘧啶肿瘤细胞靶向性前药及其抗肿瘤效果进行综述。
关键词:5-氟尿嘧啶前药、肿瘤细胞、靶向性、新进展综述:5-氟尿嘧啶(5-fluorouracil,5-FU)是目前临床上广泛应用的代谢类抗肿瘤药物之一,通过干扰核酸的合成抑制癌细胞的增生,主要用于结直肠癌、乳腺癌、胃癌等[1].但由于首过代谢显著且亲脂性较低,5-FU的生物利用度低,影响抗肿瘤疗效,其治疗剂量与中毒剂量接近,临床应用时引起严重的消化道反应和骨髓抑制等不良反应。
为了降低5-FU 严重的不良反应,提高其中肿瘤靶向性,国内外学者对5-FU进行了大量的化学修饰工作,合成了多种5-氟尿嘧啶衍生物。
其中替加氟(tegafur)、卡培他滨(caoecitabine)最具代表性,虽然它们毒性均5-氟尿嘧啶低,化疗指数较5-FU,但对肿瘤细胞无选择性,故不能避免对正常组织的损害。
利用某些结构对肿瘤细胞的特殊亲和力作为靶向药物的载体,已成为当今靶向药物研究的热点。
目前,肿瘤细胞靶向性药物可根据靶向部位不同分为肿瘤细胞靶向性前药、肝靶向性前药、直肠靶向性前药、骨靶向性前药等。
现根据不同靶向载体,对5-FU肿瘤细胞靶向性前药进行综述和介绍。
肿瘤细胞是分裂增殖失去调控的细胞,要抑制其生长而又尽量不影响正常细胞的功能,关键在于利用肿瘤细胞和正常细胞的区别,增加药物对肿瘤细胞的选择性。
[设计]前药设计的主要思想
![[设计]前药设计的主要思想](https://img.taocdn.com/s3/m/30a4fe2482c4bb4cf7ec4afe04a1b0717fd5b373.png)
前药(Prodrug)是一类体外活性较小或无活性,在体内经酶或非酶作用,释放出活性物质而发挥药理作用的化合物”’。
前药目的主要在于提高药物生物利用度,增加药物稳定性,减小毒副作用,促使药物长效化等。
随着科技的不断发展,前药设计在新药研究中越来越受到人们的重视,它在现有药物的基础上进行结构修饰,相对风险性较小,投资少,见效快,非常适合我国药品研发的实际。
1 改善药代动力学性质1.1抗肿瘤药物依托泊苷其水溶性磷酸酯前药在体内经内源性磷酸酶作用转化为原药,生物利用度可由原药的0.04%提高到50%。
1.2安瑞那韦的磷酸酯前药夫沙那韦,该药具有高水溶性和固态稳定性。
将该前药制成钙盐片剂溶解度显著提高(100 mg/mL),口服后,提高了生物利用度,降低用药量,减轻了患者的用药负担。
1.3蛋白酶抑制剂如沙奎那韦,印地那韦,分别制成前药。
在体内通过细胞内酶的水解作用在细胞内释放出原药,从而提高了蛋白酶抑制剂对血脑屏障的渗透率和生物利用度。
2改善溶解性2.1考布他汀的甘氨酸氨基甲酸酯前药,溶解度提高到5.0mg/mL。
2.2左旋多巴的乙酯的水溶性得到很大提高,临床实验表明口服左旋多巴乙酯的溶液起效快。
2.3喜树碱的水溶性前药伊立替康。
伊立替康本身没有抗癌活性,主要在肝脏经酯键断裂代谢成7一乙基一10一羟基喜树碱而起作用,具有水溶性好,抗癌谱广等特点。
3消除不适宜的制剂性质3.1药物不适宜的制剂性质常常影响患者的口服用药。
将克林霉素制成克林霉素磷酸酯(clindamycin phosphate),可增加其水溶性,通过静脉注射或肌内注射途径给药。
在血液中碱性磷酸酯酶作用下很快水解为克林霉素,避免了口服用药引起的味道不适和胃肠道反应。
3.2甘油酯前药有利于减轻口服抗炎药对胃肠道的刺激4延长药物作用时间4.1前药也能用来延长药物作用时间,例如,氟奋乃静的癸酸酯前药注射24h一72h 后开始起作用,可持续作用l一8周,平均作用时间3—4周。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
空间位阻的减小
• 当活性药物或靶向配体偶联到 高分子载体上时,彼此基团的 接近而引起的空间阻碍和分子 内张力,使形成的共价键不稳 定,特别是一些多肽和蛋白质 大分子药物会引起较大的空间 位阻。 在设计多肽和蛋白质类前 药时需要在载体与药物之间增 加一个连接臂,用来降低空间 位阻的影响,增加载药的稳定 性。特异性配体用连接臂接枝 到载体上后,也会大大增加配 体与受体蛋白质的结合力度。 常用的一些连接臂有氨基酸、 小分子肽链、双功能试剂等, 这些物质具有多活性基团,形 成的价键可以在体内特定位置 酶解
。
靶向配体的引入 •
一般来说 ,合成 聚合物 、多糖等载体材 料都属于靶非定向型载 体 ,大多通过EPR 效 应提高药物的作用效果 , 靶向性不强 。有必要将 具有靶向性的生物活性 分子 ,如酯类 、乳糖 残基、叶酸、糖蛋白、 抗原、凝集素等引入到 高分子前药中,可以使 高分子药物具有良好的 靶向性和生物相容性。
•
•
•
•
•
实例
置的物保高 使血将护分 靶药缓,子 向浓慢通的 性度释过链 和变放内上 选化,吞, 择。避进可 。同免入以
第
设计合成
三
节
高分子载体材料的选择
1
具有活性功能基团,如羧基、羟基、巯基、氨基等
2
生物相容性好,无毒性,无免疫原性
3
良好的生物降解能力和分子量低于肾排泄值
4
实用性,原料容易得到
合成高分子载体材料
大分子前药的转运路线图
正常组织内毛细血管内皮细 胞排列紧密,高分子载体难以像小 时了细 得活 分子药物透过毛细血管。而在类似 ,间胞 到性 于肿瘤等病变组织,新生毛细血管 靶歇后 高药 丰富,而且血管内皮细胞彼此排列 向给, 分物 疏松,间隙较大,人体结肠腺癌血 引药活子接 管内皮细胞间隙达 400nm,所以高 导造性 链枝 分子前药很容易透过并进入病变组 装成药 的在 织,产生肿瘤被动靶向性
2
降低小分子药物的毒副作用
减少抗药性
3
4
增强靶向性和选择性
提高稳定性和药物利用率
5
第
ቤተ መጻሕፍቲ ባይዱ
药物结构
一
节
药物结构
大分子药物载体
小分子活性药物
可溶性基团
可断裂连接基
靶向基团
第
作用机理
二
节
合成大分子前药所应具备的基本要求
聚合物载体既要满足生物相容性,也要满足生物可降解性
药物和大分子的复合物能改变前药的生物相容性 小分子药物可以连接到大分子药物载体上,也可重新断裂下来 靶向基团引导大分子前药到达人体中特定的组织及细胞
普通药物药效不佳
毒副作用
在体内不稳定
1
水溶性差
2
3
循环半衰期短
4
什么是高分子前药?
• 高分子前药是指本身没有药理作用、也不与药物发生化学反
应的高分子作为药物的载体,依靠两者间微弱的氢键结合形成或
者通过缩聚反应将低分子药物连接到聚合物主链上而得到的一类
药物。
优
控制药物释放速度 点
1
advantage
目前 已 有 聚 乙 二 醇-腺 苷 脱 氨 酶 (PEG-adenosine deaminase)、 聚 乙 二 醇-天 冬 酰 胺 酶 ( PEG-asparaginase)、线性聚 乙 二 醇 -干 扰 素 α2 b (Linear PEG-interferon α2 b )、支 化 聚 乙 二 醇 -抗 VEGF 适 体 (Branched PEG-anti-VEGF aptamer)等十多种 PEG 前药进入市场或临床 试验阶段
用氨基酸对 PEG改性后,活性基团比原有的PEG多 1 倍 ,氨基酸的存在还避免了阿糖胞苷在体内迅速酶解。 将活性茶碱分子与 PEG 衍生物结合,改性后的PEG 载药 量是未改性时的 6倍,并且明显延长了药物在体内的药效持续 时间。
高分子前药的化学合成
载体或药物的末端 修饰
• 将低分子药物与高 分子载体结合的方法有 共聚 、嵌段和接枝等 。 二者通过在体内 p H 下具有较高生物降解 活性的酯键或酰胺键等 相联,在前药合成前需 要对载体或药物进行酰 化修饰。 羧基具有很强的亲 核性,在合成蛋白质或 多肽类前药时首先对载 体进行羧基化,羧基化 的载体容易与多肽和蛋 白质中的氨基或羟基亲 和。
天然高分子载体材料
天然高分子载体材料比合成高分子载体材料具有更高的生物活性 ,壳 聚糖(CTS)可以选择性地向肿瘤细胞聚集,并具有免疫调节和抗肿瘤 作用,CTS 在体内可被溶菌酶水解生成低分子物质,从而被人体吸收。
高分子载体材料的改性
无论是合成高分子载体还是天然高分子载体都会或多或少 存在一定的缺陷,如 PEG 活性基团少,CTS、PGA、PLA 水 溶性差等 。为了克服这些不足 ,通常用小分子如氨基酸 、多 功能团活性分子 、有机酸的衍生物等对高分子载体进行修饰。