有机小分子催化剂共39页文档
有机小分子催化剂ppt课件

JACS, 2005, 127, 15696.
JACS, 2006, 128, 84.
没有明确的价值取向和人生目标,实 现自我 人生价 值就无 从谈起 。人生 价值就 是人生 目标, 就是人 生责任 。每承 担一次 责任
4.4 手性磷酸—抗衡手性阴离子催化剂
JACS, 2006,
没有明确的价值取向和人生目标,实 现自我 人生价 值就无 从谈起 。人生 价值就 是人生 目标, 就是人 生责任 。每承 担一次 责任
对该类催化剂的研究近年来才得到化学家的重视,并很快有机化学的 热点和前沿,但其渊源可追溯到20世纪70年代。早在1971年Wiechert就 首次报道了L-Proline可以催化分子内不对称羟醛缩合,1974年该反应被 Hajos等优化,ee值高达94%。然而,此后该类型的催化剂没有引起人们 的重视。
没有明确的价值取向和人生目标,实 现自我 人生价 值就无 从谈起 。人生 价值就 是人生 目标, 就是人 生责任 。每承 担一次 责任
6、 金属有机化合物催化剂 6.1 以手性二胺为配体
配体
没有明确的价值取向和人生目标,实 现自我 人生价 值就无 从谈起 。人生 价值就 是人生 目标, 就是人 生责任 。每承 担一次 责任
没有明确的价值取向和人生目标,实 现自我 人生价 值就无 从谈起 。人生 价值就 是人生 目标, 就是人 生责任 。每承 担一次 责任
第一章、简 介
自用1966年第一个手性金属配合物催化剂被成功用于不对称反应至今, 不对称催化研究迄今已有40年的历史。2001年,Nobel化学奖授予了在该 领域做出突出贡献的三位化学家Knowles, Noyori和Sharpless,标志着不 对称催化研究已取得了令人瞩目的成就。
《基于可见光催化有机小分子反应的材料制备及其反应性能研究》范文

《基于可见光催化有机小分子反应的材料制备及其反应性能研究》篇一一、引言随着科技的发展,可见光催化在有机小分子反应中的应用越来越广泛。
其利用可见光作为能量来源,具有环保、高效、节能等优点。
本文旨在研究基于可见光催化的有机小分子反应的材料制备及其反应性能,以期为相关领域的研究和应用提供参考。
二、材料制备1. 材料选择本文选择了一种具有可见光响应特性的催化剂,该催化剂由多种元素组成,具有良好的稳定性和催化活性。
此外,还选择了不同种类的有机小分子作为反应物。
2. 制备方法首先,将催化剂与有机小分子混合,通过一定的搅拌速度和温度,使二者充分混合。
然后,将混合物进行热处理或光处理,使催化剂与有机小分子发生反应,生成新的化合物。
最后,对生成的化合物进行分离和提纯,得到所需的材料。
三、反应性能研究1. 反应机理研究通过光谱分析和化学分析手段,研究可见光催化下有机小分子反应的机理。
分析催化剂与有机小分子的相互作用过程,以及反应过程中产生的中间体和最终产物的结构特征。
2. 反应条件优化研究不同条件下(如温度、光照强度、催化剂浓度等)的可见光催化反应性能。
通过实验数据,找出最佳的反应条件,以提高反应的效率和产物的纯度。
3. 产物性能分析对生成的产物进行性能分析,包括产物的结构、纯度、稳定性等。
通过对比不同条件下产物的性能,评估可见光催化反应的优劣。
四、实验结果与讨论1. 实验结果通过实验,我们得到了不同条件下可见光催化有机小分子反应的产物。
通过对产物的结构和性能进行分析,我们发现,在最佳的反应条件下,产物的纯度和收率都较高。
同时,我们还发现催化剂的种类和用量对反应的性能有显著影响。
2. 实验讨论通过对实验结果的分析,我们发现在可见光催化下,催化剂的可见光响应特性是影响反应性能的关键因素。
此外,反应条件如温度、光照强度等也对反应性能有重要影响。
因此,在今后的研究中,我们需要进一步优化催化剂的制备方法和反应条件,以提高可见光催化有机小分子反应的性能。
酶蛋白的结构与功能.pptx

第22页/共93页
四、酶催化作用的特点
(一)酶与一般催化剂的共同点
1. 只能催化热力学所允许的化学反应,缩短达到化学平 衡的时间,而不改变平衡常数;
第29页/共93页
3)立体异构特异性(stereo specificity)
一些酶仅能催化一种立体异构体进行反应,或 其催化的结果只产生一种立体异构体,酶对立体异 构物的选择性称为立体异构特异性。
例如:
延胡索酸酶
延胡索酸 + H2O ======== 苹果酸
(反丁稀二酸)
顺丁稀二酸 + H2O ========
③反馈抑制调节酶活性:反应终产物反过来 抑制酶的活性。
第33页/共93页
④抑制剂和激活剂对酶活性的调节:酶受大分 子抑制剂或小分子物质抑制,从而影响酶的活性。
⑤其他调节方式:通过别构调控、酶原的激活、 酶的可逆共价修饰和同工酶来调节酶活性。
第34页/共93页
第二节 酶的结构与催化功能
一、酶蛋白的结构
3. 四级结构:除少数单体酶外,大多数酶是由多个亚基 组成的。亚基间主要靠非共价键连接;亚基的数目以2或4 居多;亚基相同或不同。在寡聚酶中,每个亚基一般无活性; 在多酶复合体中,每个亚基有一个活性部位,但各自催化不 同的生化反应。
第37页/共93页
(三)酶活性中心(部位)(active center or site)
第一节 酶的一般概念
一、酶的定义
酶是由生物活细胞所产生的,在体内外均具有 高度专一性和极高催化效率的生物大分子,包括蛋 白质和核酸。
手性有机小分子催化的不对称合成反应精品文档

Thiourea and Urea作为氢键活化的反应
R1
R1
X
R2
NN HH
R2
Etter urea catalyst: R1=NO2, R2=H, X=O Schreiner catalyst: R1=R2=CF3, X=S
OO ON
OO
ON cat.
O ON O
OO
ON cat.
Jacobsen’s Thiourea catalysts
Through Iminium strategy
MacMillan, 2019, 123, 4370
List, 2019, 127
93% ee
Works in my group: Asymmetric Direct Vinylogous Michael Addition
NC CN
+R R1 X
280 630
Ar Ar
O
OH
O
OH
Ar Ar
Rawal catalyst: R=naphthyl
T B S O
H R cat.(0.2eq) T B S O O toluene,-40--78oC
R O
ቤተ መጻሕፍቲ ባይዱ
N
N
A cC l O
R
O
up to 98% ee
Rawal et al, Nature, 2019
Primary amine as iminium catalyst
Ph
NC CN
+R
N H
Ph OH
C
X
O
NR
NC CN
C
H
O
R
H
新型手性双胍小分子催化剂
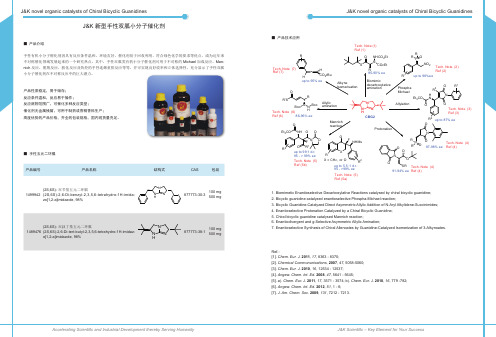
O R Allylic amination N Boc Boc N H Tech. Note (6) Ref (6) 86-96% ee O Et3 CO NH O O O NHMs
Biomimic decarboxylative amination
R3 H N
N N H
Mannich reaction
R O P NO 2 up to 96%ee O N O Tech. Note (3) Ref (3) R2 up to 87% ee R1 Tech. Note (2) Ref (2)
•
R 85-90% ee
产品性质稳定,易于储存; 反应条件温和,反应易于操作; 反应底物范围广,可催化多种反应类型; 催化剂无金属残留,可用于制药或香精香料生产; 高度优势的产品价格,齐全的包装规格,国内现货量充足。
N P O O Tech. Note (4) 87-98% ee Ref (4)
■ 手性五元二环胍
R2
N O F O R1 up to 99:1 d.r. 95 - > 99% ee Tech. Note (5) Ref (5b)
X R1 X = CH 2, or O up to 5.5 :1 d.r. 95 - >99% ee Tech. Note (5) Ref (5a)
Ref.: [1]. Chem. Eur. J. 2011, 17, 8363 - 8370; [2]. Chemical Commununications, 2007, 47, 5058-5060; [3]. Chem. Eur. J. 2010, 16, 12534 - 12537; [4]. Angew. Chem. Int. Ed. 2008, 47, 5641 - 5645; [5]. a). Chem. Eur. J. 2011, 17, 3571 - 3574; b). Chem. Eur. J. 2010, 16, 779 -782; [6]. Angew. Chem. Int. Ed. 2012, 51, 1 - 6; [7]. J. Am. Chem. Soc. 2009, 131, 7212 - 7213.
汽车燃油时代新革命-TTC燃油伴侣,小分子燃油催化剂

29010.16638/ki.1671-7988.2018.21.101汽车燃油时代新革命—TTC 燃油伴侣,小分子燃油催化剂陈 尹摘 要:TTC 创建于1996年,目前已经在德国、英格兰、美国、新加坡等三十多个国家有了业务往来。
TTC 燃油伴侣作为TTC 公司所研制的全新产品,其具备有其他燃油添加剂所不具备的功能。
文章主要介绍了TTC 燃油伴侣所具备的功能,以期可以对TTC 燃油伴侣有更加清晰的认识。
关键词:TT 燃油伴侣;燃油添加剂;积碳中图分类号:U463.1 文献标识码:A 文章编号:1671-7988(2018)21-290-02A new revolution in the automotive fuel era-TTC fuel companion,small molecule fuel catalystChen YinAbstract: TTC was founded in 1996 and has business contacts in more than 30 countries including Germany, England, the United States and Singapore. The TTC fuel companion is a new product developed by TTC, which has functions not found in other fuel additives. The article mainly introduces the functions of the TTC fuel companion, in order to have a clearer understanding of the TTC fuel companion.Keywords: TT fuel companion; fuel additive; carbon depositCLC NO.: U463.1 Document Code: A Article ID: 1671-7988(2018)21-290-02 TTC 燃油伴侣来自台湾,经过SGS 认证、MSDS 认证、国家权威检测机构认证、保险公司承保、315打假保真协会认证的一款小分子燃油催化剂,是由松树植物萃取,25L 油放入一颗,加油前放入油箱即可,使用方便、携带便捷。
有机合成中的新型催化剂官能团官能团还原方法

有机合成中的新型催化剂官能团官能团还原方法催化剂在有机合成中扮演着至关重要的角色,能够促使反应进行并提高反应效率。
传统的催化剂官能团官能团还原方法在有机合成中起到了积极的作用,但随着研究的深入,科学家们发现了一些新型催化剂,为有机合成提供了更多选择。
本文将重点介绍有机合成中的新型催化剂官能团官能团还原方法。
一、金属有机催化剂官能团官能团还原方法金属有机催化剂被广泛应用于有机合成中,其官能团官能团还原方法也随之得到研究和发展。
常见的金属有机催化剂包括铂、钯、铜等。
以铂为例,其催化剂通常采用[PtCl2(PPh3)2]和氢气的反应来实现官能团官能团还原。
这种方法适用于多种反应,如亲电加成、硝基还原等。
而钯催化剂的官能团官能团还原方法多种多样,包括催化剂为[ Pd(PPh3)4 ]、[ PdCl2(PPh3)2 ]等的氢气反应,以及钯催化下的亲电还原、硼酸盐的还原等反应。
二、非金属有机催化剂官能团官能团还原方法除了金属有机催化剂外,非金属有机催化剂在官能团官能团还原方法的研究中也发挥了重要作用。
其中,碱金属催化剂和小分子有机催化剂是常见的类型。
碱金属催化剂如钠、锂等可用于官能团官能团的还原反应。
例如,钠法罗化的反应可以将酮类化合物还原成相应的醇类产物。
小分子有机催化剂如有机亚磷酰化合物、硅氢化合物等,在官能团官能团还原方法中也展现出了良好的催化活性。
它们可以与底物发生反应,提供所需的氢原子或电子,催化官能团官能团还原反应的进行。
三、纳米催化剂官能团官能团还原方法近年来,纳米催化剂在有机合成领域引起了广泛关注。
纳米催化剂具有特殊的形貌和结构,与传统催化剂相比具有更高的催化活性和选择性。
纳米金、纳米银等纳米金属催化剂被广泛应用于官能团官能团还原方法。
其高比表面积和丰富的活性位点使得纳米催化剂具有出色的催化性能。
此外,通过调节纳米粒子的尺寸和形状,还可以进一步优化催化反应的效果。
四、生物催化剂官能团官能团还原方法生物催化剂在有机合成中表现出了独特的优势,如高催化活性、温和反应条件和对环境友好等。
有机催化

➢共轭加成反应( C=C键)
亲核取代
➢烷基化 ➢卤代
第二部分:有机催化---分子内羟醛缩合反应
---Wiechert. R. et al. , Angew. Chem. Int. Ed. Engl. 1971, 10, 496 ---Hajos. Z. G. et al., J. Org. Chem. 1974, 39, 1615
67%~92% yield
---Methot. J. L. et al., Adv. Synth. Catal. 2004,346, 1035
第一部分:背景介绍---催化剂分类及反应
❖醇、酚型催化剂:通过双氢键活化,表现出良好的催化活性和对映选择性。
❖硫脲类催化剂:由双氢键活化硝基,收率比较高。
63%~91% yield 65%~91% ee
迅速发展, 其机理方面也得到一定的阐述。
第一部分:背景介绍
Asymmetric organocatalysis VS Organometallic & Enzyme Catalysis
有机催化
有机金属催化
酶催化
优点
催化剂来源广泛 易合成,反应条件 相对易操作,价格 便宜,无毒
广泛的反应底物 配体灵活可控
第一部分:背景介绍---催化剂分类及反应
❖氮氧化物催化剂:催化醛的烯丙基化反应, 获得比较好的收率和对映选择性。
66%~96% yield 56%~94% ee
---Kina. A. et al., Adv. Synth. Catal. 2004,346, 1169
❖有机磷催化剂:烷基膦亲核性比较强,而碱性远远弱于相应的胺。
94% yield 97% ee
氮上的取代基若为酯基, 产物可以进一步发生环化反应生成产物:
催化剂在有机合成中的底物范围拓展

催化剂在有机合成中的底物范围拓展概述:有机合成是一门关键的化学领域,在制备有机化合物时起着至关重要的作用。
催化剂是有机合成中的一种有效工具,它能够加速反应速率,提高产物收率,并且在反应中可以实现高选择性。
不断探索和拓展催化剂的底物范围,对于合成更复杂和多样的有机化合物具有重要意义。
本文将讨论催化剂在有机合成中拓展底物范围的一些重要进展。
1. 金属催化剂的应用:金属催化剂被广泛用于有机合成中。
典型的金属催化反应包括金属有机配合物催化的羰基化反应、交叉偶联反应等。
近年来,研究人员一直在探索使用新型金属催化剂,以拓展其底物范围。
例如,钯催化的氯代芳烃和炔烃的Sonogashira反应,能够高效合成内炔化合物。
其他金属催化剂如铜催化剂以及贵金属催化剂等也在有机合成中得到广泛应用。
2. 有机小分子催化剂:除了金属催化剂,有机小分子催化剂也是有机合成中的重要部分。
这些有机小分子催化剂具有较低的毒性和较简单的合成,因此在实际应用中非常有潜力。
茚类化合物、吲哚类化合物和酚类化合物等从分子结构上具有良好的催化活性。
例如,茚类化合物催化的醛酮烷基化反应在药物合成中具有重要应用。
吲哚类化合物催化剂在不对称合成中也发挥着重要作用。
有机小分子催化剂的研究将进一步促进有机合成的发展。
3. 生物催化剂的应用:生物催化剂是一类新兴的催化剂,在有机合成中具有巨大潜力。
酶是生物催化剂的重要组成部分,它们可以在温和的条件下催化高效的有机合成反应。
通过对天然酶的改造和工程,研究人员可以获得具有高催化活性和高选择性的生物催化剂。
生物催化剂可以用于合成具有特殊结构和活性的有机化合物,例如药物和功能性材料等。
4. 催化剂的设计和优化:为了进一步拓展催化剂在有机合成中的底物范围,研究人员不断致力于催化剂的设计和优化。
利用计算化学和理论化学方法,可以预测和优化催化剂的结构和催化机理。
此外,通过合理的配体设计和反应溶剂的选择,也可以提高催化剂的活性和选择性。
有机小分子催化合成高聚物的研究进展

h eai s i f a y ts u tr aayi a t t, ee t t a oy r o et . t erlt n hp o mls r cu ea dc tlt cii rs lcii dp lm e rp ris o c t n c v) v yn p e
C e g u6 0 4 ) h n d 1 0 1
Ab t a t sr c :Re e r h a v n e ea p ia in o r a o a ay i o y e n e i we e s a c d a c si t p l to f g n c t l ssi p lm rs t ss r n h c o n y h
有机 催化 剂 的发展 经 历 了一个 很 长 的历史 阶段 , 早 期 的氰离 子催 化安 息香 缩合 反应 到 从
近 期 发展 起来 的不对称 有机 催化 剂 催化 立 体选择 性 反应 , 类 催化剂 均 具有 很 高的催化 效 率 这 和 选 择性 。 与传 统 的金 属有 机催 化合 成 高聚 物方 法相 比, 机催 化剂 催 化 的聚合 反应 不仅条 有
剂 ,据C n o等 道,在醇 的引发下,这类催化剂在常温时的催化L 开环聚合 的转换频 on r 报 A
率 (O ) 1 , 已经 与活 性最 高 的金 属催 化剂相 媲 美 。另 外 ,这类 催 化剂 不仅 活性 高 ,而 T F为 8S ~
且 能 得 到P < .6 L DI 11 的P A,反应 为 活 性开环 聚 合 ,可 以较 为 方便 的合 成嵌 段共 聚物 。双 功 能 硫 脲 一胺 i催化L ’ l A的开 环聚 合 可 以得 到分 子量 可控 , 分子量 分 布 , 窄 由引 发 的醇封 端 的P A。 L
新型双官能团有机小分子催化剂的设计合成

Jn2 0 a .0 8
新 型 双 官 能 团 有 机 小 分 子 催 化 剂 的 设 计 合 成
王 进 家 , 宗 旋 , 雅 文 沈 张
( 州 大 学 化 学 化 _ 学 院 , 苏 苏 州 2 52 ) 苏 T - - 江 1 13
摘
要 : 于 天 然 L 氨 基 酸 的 基 本 结 构 , 计 了一 系 列 双 官 能 团 的 有 机 小 分 子 催 化 剂 , 以 天 然 氨 基 酸 为 起 基 一 设 并
始 原 料 , 过 多 步 反 应 合 成 了 这 些 目标 化 合 物 , 物 结 构 经 核 磁 分 析 正 确 . 经 产
关 键 词 :氨 基 酸 ; 官 能 团 ; 机 小 分 子 双 有 中 图分 类 号 :0 2 . 697 文献标识码 : A 文 章 编 号 :0 0— 0 3 2 0 ) l 0 5— 3 1 0 2 7 ( 0 8 o 一0 6 0
n
H
。 H
。H
。
N2 H
R 八 o 兰 Y H
NH I
,
/ 2 H0
. H , ,
R
R
I R o - Y . /H来自,CH3 OH
——————— HF
T
u. / I M
— —
,
. h I
I r 0
HCI
O l l
1 2 氨 基 醇 3 -d的 合 成 . a3
冰 盐浴 下 , 将溶有 5 r o 氨基酸 的 甲醇悬浊液 冷却到 一I ̄ 缓慢滴加 lO m l 0 tl e o OC, O m o 二氯亚砜 , 滴加完 毕 , 回流
。 H
O R
酶生物催化剂.pptx

第33页/共58页
酶浓度对反应速度的影响
• 反应速度与酶浓度成正比:当[S][E],式中Km可
以忽略不计。
k3[E][S] v= Km + [S] =k3[E]
v
o
[S]
第34页/共58页
温度对酶促反应速度的影响
产 物 2.0 麦 芽 1.5 糖 的 1.0 毫 克 0.5 数
0 10 20 30 40 50 60 ℃ 温度对唾液淀粉酶活性的影响
例如:有机磷农药中毒 (敌百虫、敌敌畏、乐果杀虫剂1605、1059等)
RO O
P
+
RO X
有机磷化合物
E-OH 羟基酶
RO O
P
+
RO O E
磷酰化酶 (失活)
CHNOH N CH3
解磷定
RO O
P
+
HX
RO O E
磷酰化酶
(失活)
O OR P
CHNO OR + N
CH3
E-OH
乙酰胆碱酯酶是羟基酶,与有机磷农药共价结合后失活,使兴奋 性神经递质乙酰胆碱不能及时清除降解,而是过量地积累引起中毒。
▪ 活化能:在一定温度下一摩尔底物全部进入活化态所需要的
自由能,单位是KJ/mol. (增加温度、加入催化剂降低反应活化能) • 酶促反应:E + S === ES === ES* EP E + P • 非酶促反应:
催化剂的作用是降低反应活化能,从而起到提高反应速度的作用
第22页/共58页
过渡态
酶的最适温度: 酶活性最高时的温度, 也即酶的催化效率 最高, 酶促反应速度最大时的温度。
第35页/共58页
有机催化类型-概述说明以及解释

有机催化类型-概述说明以及解释1.引言1.1 概述有机催化是一种重要的催化类型,它在有机化学和有机合成领域发挥着重要的作用。
有机催化通过引入一个有机催化剂,可以促使化学反应的进行,在反应速率、选择性和产率上发挥关键作用。
有机催化剂是一种有机化合物,它能够与底物反应,并在反应过程中发生化学变化,从而促进反应进程的进行。
有机催化可以被广泛应用于各种有机合成反应中,包括碳-碳键和碳-氧键的形成、环化反应、氧化反应等。
与传统的无机催化剂相比,有机催化剂具有更好的底物兼容性、反应条件温和以及选择性高等优点。
这些优势使得有机催化成为合成有机分子的重要工具。
有机催化可以根据催化机理的不同进行分类。
常见的有机催化类型包括质子酸、质子碱、路易斯酸、路易斯碱、氢键和氢化合物等。
每种类型的有机催化剂都有其特殊的反应机制和适用范围。
因此,深入理解不同类型的有机催化对于设计和优化有机合成反应具有重要意义。
本文将进一步探讨有机催化的定义、作用以及分类,并介绍有机催化反应的机理和应用。
通过对有机催化的研究和了解,我们可以更好地利用有机催化来推动有机合成的发展,为化学领域的进一步创新和发展做出贡献。
1.2 文章结构文章结构:本文分为引言、正文和结论三个部分。
引言部分包括概述、文章结构、目的和总结四个方面。
首先,概述介绍了本文探讨的主题,即有机催化类型。
然后,文章结构说明了本文的组织结构,即引言、正文和结论三个部分。
接着,目的详细说明了本文的研究目标和意义。
最后,总结部分对整篇文章进行了简要的总结和归纳。
正文部分主要分为三个小节,分别是有机催化的定义和作用、有机催化的分类和有机催化反应机制。
首先,有机催化的定义和作用部分详细介绍了有机催化的概念和在化学反应中的作用。
其次,有机催化的分类部分将不同类型的有机催化进行了系统的分类和说明,包括金属有机催化剂、小分子有机催化剂等。
最后,有机催化反应机制部分探讨了有机催化反应的具体机制和关键步骤,为读者提供了更深入的理解。
有机小分子催化讲解

有机⼩分⼦催化讲解引⾔⾃从2000年以来,在Benjamin. List,Carlos F. Barbas III和David W. C. MacMillan 等⼈推动之下,有机催化(Organocatalysis)开始了伟⼤的复兴。
也就是从那时候开始我对这⼀领域产⽣了浓厚的兴趣,阅读了不少⽂献。
从本贴开始,将以回复的形式介绍有机催化领域的经典⽂献。
希望能对chem8er有点帮助。
本贴是为chem8⽽写,转贴请注明出处。
⾸先,罗列⼀些⽂献。
以下⽂献都是review,不是原始⽂献。
要想对此领域有深⼊的了解还是要读原始⽂献⽐较好。
专著两本:a) A. Berkessel, H. GrQger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005; b)Enantioselective Organocatalysis (Ed.: P. I. Dalko) Wiley-VCH, Weinheim, 2007。
这两本书书籍中⼼都有。
专刊两期:Acc. Chem. Res. 2004. 37, 487-621;Chem. Rev. 2007, 107, 5413-5883。
每期⼤概⼗篇⽂章,包括了organcatalyst的各个分⽀。
零散的review很多,简单罗列⼀下,不是很全。
特别是专门介绍某⼀分⽀的review 没有列出,否则太多了。
a) P. I. Dalko, L. Moisan, Angew. Chem. Int. Ed. 2001, 40, 3726-3748; b) E. R. Jarvo, S. J. Miller, Tetrahedron 2002, 58, 2481-2495; c) B. List, Tetrahedron 2002, 58, 5573-5590; d) P. I. Dalko, L. Moisan, Angew. Chem. Int. Ed. 2004, 43, 5138-5175; e) J. Seayad, B. List, Org. Biomol. Chem. 2005, 3, 719-724; f) B. List, Chem. Commun. 2006, 819-824; g) M. Marigo, K. A. J?rgensen, Chem. Commun. 2006, 2001-2011; h) F. Cozzi, Adv. Synth. Catal. 2006, 348, 1367-1390; i) M. J. Gaunt, C. C. C.Johansson, A. McNally, N. T. V o, Drug Discovery Today 2007, 12, 8-27; j) R. M. de Figueiredo, M. Christmann, Eur. J. Org. Chem. 2007, 2575-2600; k) D. Enders, C. Grondal, M. R. M. HRttl, Angew. Chem. Int. Ed. 2007, 46, 1570-1581; l) A. Ting, S.E. Schaus, Eur. J. Org. Chem. 2007, 5797-5815;m) S. B. Tsogoeva, Eur. J. Org. Chem. 2007, 1701-1716; n) A. G. Doyle, E. N. Jacobsen, Chem. Rev. 2007, 107, 5713-5743; o) C.F. Barbas III, Angew. Chem. Int. Ed. 2008, 47, 42-47; p) A. Dondoni, A. Massi, Angew.Chem. Int. Ed. 2008, 47, 4638-4660。
电催化过程PPT精选文档

2.1.2 影响电催化性能的因素
电催化剂须具备的性能:
1.催化剂有一定的电子导电性,至少与导电材料充分 混合后能为电子交换反应提供不引起严重电压降的电 子通道,即电极材料电阻要小
2.高催化活性,包括实现催化反应、抑制有害的副反 应,也包括能耐受杂质及中间产物的作用而不致较快 地中毒失活
3.催化剂的电化学稳定性,实现催化反应的电势范围 内催化表面不因电化学反应而过早的失去活性
3
2.1.1 电催化的类型及一般原理
电催化的类型:
根据电催化剂 的性质
氧化-还原电催化 非氧化还原电催化
4
氧化-还原电催化:
指在催化过程中,固定在电极表面或存在于电解液中的催化剂本身发 生了氧化还原反应,成为底物的电荷传递的媒介体,促进底物的电子传递, 这类催化作用又称为媒介体电催化。
固定于电极表面或存在于溶液中的电催化剂氧化态形式Ox在外加电场 作用下生成R,R与溶液中的底物A反应生成产物B,并且再生了催化剂的氧 化形式Ox,在外加电势作用下不断实现电催化的循环过程。
(1)N-甲基吩嗪吸附的石墨电极对葡萄糖氧 化的媒介催化
(2)麦尔多拉蓝吸附的石墨电极对还原性烟酰 胺嘌呤二核苷酸的催化氧化
(3)普鲁士兰修饰玻碳电极对维C的催化氧化
8
均相的电催化 例:Βιβλιοθήκη 苯氧化成苯甲醛,已知甲苯氧化在高超电势
下只以低速率发生,向溶液中加入某些金属离子Mn+, 从而进行催化:
使反应在Mn+/M(n+1)电对的电势下发生
9
修饰电极
异相电催化的优点:
(1)催化反应发生在氧化-还原媒介体的式电位附近,通 常只涉及简单电子转移反应 (2)催化剂用量少,可在反应层内提供高浓度的催化剂 (3)从理论上预测,对反应速度的提高要远超过均相催 化剂 (4)不需要分离产物和催化剂
有机催化简介

有机催化简介一、什么是有机催化呀?哎呀,有机催化呢,就像是化学反应里的小魔法师。
它是一种在有机化学领域超级重要的东西。
简单说呢,就是用有机小分子来促使化学反应发生的过程。
你可以想象啊,那些有机小分子就像一个个热情的小助手,拉着反应物的手,说:“来呀,咱们一起变个魔术呀。
”然后就把反应物变成了新的产物。
二、有机催化的类型1. 手性有机催化这个可就很有趣啦。
手性就像是我们的左右手,看起来一样,但是却不能完全重合。
手性有机催化就是专门来处理这种手性相关的反应的。
比如说,在合成一些药物的时候,手性就特别重要。
因为不同手性的分子可能在生物体内有完全不同的效果。
就像双胞胎,虽然长得很像,但是性格可能完全不同呢。
这种催化方式能够精准地合成出我们想要的手性分子,厉害吧!2. 酸碱有机催化这就和我们中学学过的酸碱有点关系啦。
不过这里的酸碱是有机小分子哦。
酸型的有机催化剂可以提供质子,碱型的呢就可以接受质子。
它们就通过这种给质子或者拿质子的方式,来推动化学反应的进行。
就好像在一场接力赛里,酸先把质子这个“接力棒”交出去,碱再把它接过来,然后反应就顺利往前跑啦。
三、有机催化的重要性1. 在药物合成中的作用好多好多的药物合成可离不开有机催化呢。
因为药物分子往往都比较复杂,结构要求很精确。
有机催化就像是一个超级精密的工匠,能够按照我们的要求,一块一块地把药物分子搭建起来。
比如说,治疗疟疾的青蒿素,它的合成过程可能就会用到有机催化的方法,这样才能高效、准确地得到我们需要的青蒿素分子,然后去拯救那些被疟疾困扰的人呀。
2. 对材料科学的影响在材料科学里,有机催化也有很大的功劳。
现在我们用的很多新型材料,像一些高性能的塑料、特殊的纤维等等,它们的合成过程中可能就用到了有机催化。
有机催化可以让这些材料具有更好的性能,比如更强的韧性、更高的耐热性之类的。
就像给材料穿上了一层超级铠甲,让它们变得更厉害啦。
四、有机催化的发展历程最开始呢,人们可能没有意识到有机小分子可以有这么大的催化作用。
有机小分子催化的不对称羟醛缩合反应

2006年第26卷有机化学V ol. 26, 2006第5期, 618~626 Chinese Journal of Organic Chemistry No. 5, 618~626* E-mail: zhaoguo@Scheme 1色化学要求. 本文就对近年来发展的有机小分子催化的不对称羟醛缩合反应予以介绍.No. 5姜丽娟等:有机小分子催化的不对称羟醛缩合反应6191 非水相中的不对称羟醛缩合反应1.1 脯氨酸及其衍生物 1.1.1 L -脯氨酸早在20世纪70年代, Hajos [3a]和Eder [3b]就发现脯氨 酸能够催化分子内的羟醛缩合反应, 并具有高度的对映异构体选择性和较高的化学产率. 该反应还被人们用来合成许多有用的化合物[4], 特别是用于类固醇和许多天然产物的合成中[5]. 后来有关这方面的研究鲜有报道. 直到2000年, Barbas 和List 等[6]报道了脯氨酸催化的分子间不对称羟醛缩合反应, 并进行了深入的研究(Eq. 1), 大大拓宽了这一反应的应用前景.Barbas 研究小组[6a]首先以丙酮和对硝基苯甲醛作为反应物, 研究了各种氨基酸对此反应的催化能力(表1). 研究结果表明: 五元环效果最好, 四元环次之, 六元环活性很低, 而非环状结构的普通氨基酸几乎没有催化活性. 把羧基变成酰胺也不发生反应, 这说明羧基的质子在催化反应中也起了关键作用.作者由此对脯氨酸催化反应的机理进行了假设(Scheme 2), 认为脯氨酸类似于醛缩酶(micro-aldo-ase)的功能, 它不仅提供亲核的氨基基团, 而且羧基可作为一种酸/碱助催化剂, 可以促进机理中每一个单独步骤, 包括: (a)氨基的亲核进攻, (b)醇氨中间体的脱水, (c)亚胺的脱质子化作用, (d)碳—碳键形成, (e, f)亚胺-醛中间表1 氨基酸催化的丙酮和对硝基苯甲醛的羟醛缩合反应 Table 1 Amino acids catalyzed asymmetric aldol reaction of acetone with 4-nitrobenzaldehyde Entry Catalyst Yield/% ee /%1(L )-His, (L )-Val (L )-Tyr, (L )-Phe<10N.d.2<10 N.d.355 40468 765<10N.d.6<10 n.d.767 73物的水解. 近年来陆续有文献对此反应机理进行报道[7], 通过计算等多方面研究, 证实了这种独特新颖的不对称羟醛缩合反应可能是通过上述烯胺机理进行的.2002年, MacMillan 等[8]进一步研究了醛与α-位无取代的醛进行的分子间缩合反应, 也得到较好的结果. 从表2中, 我们可以看出, 产物的ee 值普遍较高, 但反应的非对映选择性受作为电子受体的醛的影响较大,用Scheme 2620有 机 化 学 V ol. 26, 2006表2 脯氨酸催化的醛与醛的分子间羟醛缩合反应Table 2 The proline-catalyzed cross-aldol addition reaction ofaldehydesEntry R R' Yield/% Anti ∶Syn ee /% (anti )1 Me Et 80 4∶1 99 2 Me t -Bu 88 3∶1 97 3 Me c-C 6H 11 87 14∶1 99 4 Me Ph 81 3∶1 99 5 n -Bu i -Pr 82 24∶1>996n -Bui -Pr 80 24∶1 987 Bn i -Pr 75 19∶1 91异丁醛作为电子受体时, 反应的非对映选择性明显要比其它醛作为电子受体时高.近年来有关脯氨酸催化的反应的报道更是层出不穷[9,10], 在多种不对称催化反应中都表现出非常好的催化性能. 值得一提的是, 最近MacMillan 研究小组[11]把它用在了高对映选择性地合成六碳糖中. 在生物体的各种复杂过程(如信号转换、识别及免疫响应等)中, 六碳糖都起了非常重要的作用, 但是目前还很少有化学方法能够有效合成六碳糖和各种多聚糖, 特别是如何选择性地构建这些单糖中的五个羟基成为合成化学的一个挑战. 作者应用脯氨酸不对称催化羟醛缩合反应, 不仅两步合成六碳糖(Scheme 3), 而且通过选择不同的Lewis 酸和溶剂, 可以有选择地合成不同的糖类(Scheme 4).这类aldol 反应的应用使糖类合成的步骤大大简化, 将极大地促进药物化学和生命科学的研究.(A) Step 1: Proline-catalyzed enantioselective dimerization ofα-oxyaldehydes; (B) Step 2: Mukaiyama aldol-carbohydrate cycli-zation.Scheme 3(A) Step 1: The enantioselective dimerization of α-oxyaldehydes; (B) Step 2: The Lewis acid-mediated Mukaiyama aldol-carbohydrate cycli-zation.Scheme 4No. 5姜丽娟等:有机小分子催化的不对称羟醛缩合反应6211.1.2 脯氨酸衍生物张雅文等[12,13]在研究Aldol 反应中发现, 若在四氢吡咯环的3-位引入取代基, 对催化性能影响不大, 而5-位取代会使催化能力大大降低, 乃至丧失. 而研究4-位取代的脯氨酸的催化活性(Scheme 5), 取得了一些很好的结果. 由于取代基的引入使催化剂的溶解性增强, 催化剂的用量明显降低(2 mol%). 温度在-25 , ℃可以得到较高的产率(81.3%)和较好的ee 值(89.7%). 同时由于溶剂是丙酮, 后处理也更为方便.Scheme 5除了手性氨基酸外, 还有其它一些对脯氨酸进行修饰的有机小分子也能够催化不对称羟醛缩合反应. Berkessel 等[14]根据苯磺酰胺基团在药物化学中被称为羧酸基的生物等价体(bio-isoster), 合成了如表3所示的催化剂, 并得到较好的催化效果, 在 5 mol%的催化量下, 反应可以达到98%的ee 值. 并且此类催化剂的磺酰胺基团具有很好的可调节性, 可以通过改变苯环上的取代基来改变催化剂的电子效应和空间效应, 调节反应的对映选择性. 作者还通过X 射线结构分析, 得出此羟醛缩合反应可能的过渡态, 如Scheme 6.Scheme 6龚流柱等[15]报道了由脯氨酸和β-氨基醇合成的酰胺可以高效地催化丙酮和芳香醛以及脂肪醛的羟醛缩合反应. 对于丙酮和硝基苯甲醛的反应(表4), 在室温下, 丙酮为溶剂, 可得到89%的产率, ee 值为69%. 在 -25 ℃下, 虽然产率有所下降(66%), 但ee 值可以提高到93%. 而与脂肪醛反应能达到高于99%的ee 值, 这说明该催化剂与脯氨酸相比, 具有更高的活性和更好的对映选择性. 作者还通过理论计算对其过渡态进行研究, 说明了它所催化的Aldol 反应具有的高度对映选择性原因: 催化剂6在上述反应过程所形成的过渡态与脯氨酸催化的过渡态基本一致, 所不同的是催化剂6中的氨基表 3 不同溶剂中磺酰胺催化的丙酮和对硝基苯甲醛的羟醛缩合反应Table 3 Sulfamides catalyzed direct aldol reaction between acetone and p-nitrobenzaldehydeCatalyst and ee (Yield)/% of Products Solvent3 4 5 L -proline DMSO 93 (98)92 (98) 92 (73) 72 ( 68) Methanol 54 (62)70 (97) 69 (40) 37 (87) THF 93 (73)90 (62) 86 (98) 69 (92) Acetone 93 (98)71 (98) 90 (98) 67 (97) Chloroform85 (58)78 (95)88 (54)59 (97)表 4 手性有机催化剂6催化的丙酮和醛的羟醛缩合反应Table 4 Direct aldol reactions of acetone catalyzed by chiral organic catalyst 6Entry Aldehyde Yield/%ee /%1 4-NO 2C 6H 4CHO 66 932 3-NO 2C 6H 4CHO 63 87 3 4-BrC 6H 4CHO 77 904 PhCHO 51 835 c -C 6H 11CHO 85 976 (CH 3)2CHCHO 43 98 7t -BuCHO 51 >998 CH 3CH 2CHO1787键较短, 起了主要作用.作者[16]又对不同的手性β-氨基醇所形成的酰胺进行尝试, 发现具有吸电子基团的亚胺比具有供电子基团的亚胺催化效果更好, 只需2 mol%的催化量, ee 值都在96%以上. 例如以丁酮为底物, 虽然有两个产物生成, 但ee 值都达到98%以上(Eq. 2).622有 机 化 学 V ol. 26, 2006肖文精等[17]报道了另一种酰胺8对环戊酮和各种芳香醛进行不对称催化, 优化反应条件, 能以较高产率和ee 值得到反式产物(Eq. 3).1.2 手性二胺-质子酸催化剂脯氨酸在催化羟醛缩合反应时, 由于脱水产物不可避免, 所以产率不高. 为了提高反应的有效性, Yama-moto 等[18]研究了另一种催化羟醛缩合反应的环境友好的有机小分子催化剂: 手性二胺-质子酸催化剂. 作者对此类催化剂进行了深入研究, 首先确定二胺和质子酸的比例, 实验表明1∶1是最好的, 大于或小于都会大幅度地降低反应速率. 接着在对质子酸的研究中发现反应速率随质子酸的酸性增强而加快, 丙酮是最好的反应溶剂. 通过对不同类型的二胺进行考察, 认为二级和三级胺是效果最好的(Scheme 7). 作者先考察了丙酮和对硝基苯甲醛的羟醛缩合反应, 发现反应2 h 即达到60%的产率和88%的ee 值. 接着发现环酮与对硝基苯甲醛的反应产率也很高(88%~97%).该反应的机理还在研究中, 分析过渡态的可能构型如Scheme 8所示.1.3 非天然手性仲胺催化剂以往的有机小分子催化剂都是建立在天然产物, 如氨基酸、生物碱的基础上的, 在结构的改变上有一定的局限性, 并且在活性和选择性方面都有一定局限性. 例如脯氨酸及其衍生物在与缺电子的芳香醛反应时易形Scheme 7Scheme 8成亚氨盐, 使得所需催化剂的用量大增. 为了解决这些问题, Maruokat 等[19]设计并合成了一类完全不同的新的催化剂: 手性仲胺氨基酸(Scheme 9), 这类高效的催化剂为人们研究Aldol 反应开创了一种新的思路.Scheme 9在5 mol% 催化量下, 这类催化剂催化丙酮和对硝基苯甲醛反应的产率为70%, ee 值为93%, 而同样条件下, 脯氨酸催化下的产率很低(18%), ee 值也不高(71%)(表5). 反应也可以扩展到各种不同的芳香醛和脂肪醛, 而且它们产物的ee 值都非常高(>95%). 作者还通过计算说明了催化剂(S )-16中氨基和羰基之间的距离比脯氨酸的长, 过渡态中的氢键较易形成, 这可能决定了它的高活性和高对映选择性.表5 手性氨基酸催化的直接不对称羟醛缩合反应Table 5Direct asymmetric aldol reaction catalyzed by chiral amino acidsEntry Catalyst Solvent Yield/% ee /% (Conf.)1 (S )-16 DMSO 70 93 (R ) 2L -Proline DMSO 1871 (R )3 (S )-16 CH 3CN 32 95 (R )4 (S )-16 NMP 78 94 (R )5 (S )-16 DMF 82 95 (R )No. 5 姜丽娟等:有机小分子催化的不对称羟醛缩合反应6232 水相中的不对称羟醛缩合反应2.1 吡咯烷-四唑Yamamoto[20]报道了一种能催化水溶性醛如三氯乙醛与酮的Aldol反应的催化剂(Eq. 4), 得到85%的产率和84%的ee值. 当催化剂17用量为5 mol%, 并加入100 mol%水时, 反应的产率为85%, ee值为84%, 而无水条件下, 经过60 h, 反应基本不发生. 更为有趣的是作者还发现当含水量超过100 mol%时, 水含量增加(200 mol%或500 mol%), 产物的ee值也随之增加(92% ee或94% ee), 而脯氨酸的反应就很慢(46 h反应产率约为10%). 底物酮还可以进一步扩展(表6).表6 催化剂17催化的各种酮与三氯乙醛-水化物的羟醛缩合反应Table 6Reaction of various ketones with chloral monohydrate in the presence of 17Entry Ketone Temp./℃t/h Yield/%ee%1 40 24 79 972 30 24 93 823 30 36 91 824 30 24 55 865 40 48 72 886 40 96 76 917 40 96 83912.2 小肽与脯氨酸相比, 小肽中更多的有效原子决定了其结构和功能的多样性. 实验表明[21], 表7中所示的小肽是一个非常高效的催化剂. 用 1 mol%的催化剂催化丙酮和对硝基苯甲醛的反应, 在室温下4 h就能得到99%的产率和80% ee, 而同样条件下脯氨酸为68%(产率)和76% (ee值). 并且反应在-20 ℃时, ee值还可以提高10%. 龚流柱等[22]也考察了小肽用于羟基酮和醛的催化反应, 通过进一步优化条件而得到以手性1,4-二醇为主要产物(Eq. 5), 这是其它催化剂无法得到的. 其反应机理还在研究中.表7 小肽催化的羟醛缩合反应Table 7Aldol reactions catalyzed by peptides1 mol% catalyst 30 mol% proline Entry RYield/%ee/% (Conf.) Yield/%ee/% (Conf.)1 4-NO2C6H499 80(S) 68 76(R)2 Ph 69 78 (S) 62 60(R)3 c-Hexyl 66 82(S) 63 84(R)4 i-Pr 7979(S)97 96(R)624有 机 化 学 V ol. 26, 20062.3 吡咯烷-咪唑Vincent 等[23]开发了一种能催化羟醛缩合反应的吡咯烷-咪唑催化剂(BIP). 如表8所示, 与脯氨酸催化相同的条件下, 仅得到40%的产率和44%的ee 值, 但当有酸加入时, 反应的速率和产率大幅度提高(Entries 2, 3 vs 1). 溶剂、温度及所加的质子酸都是影响反应的重要因素, 如Entry 11, 2 mol%的BIP/TFA 催化剂用量, 丙酮为溶剂, 可以得到87%的产率和82%的ee 值. 它的另一个显著优点是反应只需用等量的酮和醛, 而在脯氨酸催化中, 酮需大大过量. 另外, 作者还发现配体BIP 与Lewis 酸如Zn(OTf)2也能够有效催化Aldol 反应(Entry 15), 由此说明此类配体也可以作为金属不对称催化的潜在的双齿配体. 与脯氨酸相比, BIP-TFA 催化剂的高效性主要是由于具有两个潜在的亲核位置, 分析可能的过渡态构型(Scheme 10). 这类催化剂的应用使我们意识到: 可以通过对配体结构的修饰来不断提高不对称催化反应的产率和对映选择性. 2.4 生物碱Janda 研究小组[24]发现烟碱代谢物(nornicotine)与脯氨酸在结构上具有相似性(Scheme 11), 因此开始研究Scheme 10Scheme 11它对羟醛缩合反应的催化作用. 实验表明: 在与脯氨酸催化丙酮和对硝基苯甲醛反应相同的条件下并未得到产物, 可是在磷酸盐水溶液中, 产率达81%, 反应没有明显的脱水产物和其它副产物. 更有趣的是, 在接近生理pH 7.5~8才能表现最好的催化活性. 后来, Noodle-man 等[25]运用动力学和计算机技术对其催化机理进行深入研究, 它与脯氨酸催化有明显不同(Scheme 12). 通常在分析这类反应的烯胺机理时, 都认为反应过程中形表8 BIP 催化的丙酮和对硝基苯甲醛的羟醛缩合反应Table 8 BIP catalyzed direct aldol reaction between acetone and p-nitrobenzaldehydeEntry BIP/mol% Acid (mol%) T /℃ Solvent Time/h Yield/% ee /%1 30— 20 DMSO 8 40 442 20 AcOH (100) 20 DMSO 0.5 65 503 20 AcOH (30) 20 DMSO 1 65 464 20 AcOH (20) -20 DMSO 4 65 54 5 20 AcOH (20) -20 DMF 24 61 756 20 AcOH (20) -20 THF 8 70 647 20 AcOH (20) -20 CH 2Cl 2 18 92 36 8 20 AcOH (20) -20 Acetone478 62 9 20 TFA (20) -20 Acetone 18 86 82 10 10 TFA (10) -20 Acetone 24 95 80 11 2 TFA (2) -5 Acetone 24 87 82 12 20 TfOH (20) -20 Acetone 24 84 80 13 20 TFA (20) -5 THF 24 67 82 14 5 TFA (5) -5 THF4869 80 15 20 Zn(OTf)2 (20) 20 Acetone 1787 74No. 5姜丽娟等:有机小分子催化的不对称羟醛缩合反应625Scheme 12成的烯胺中间体在水相中会马上水解, 而不宜在水相中进行. 而此催化反应机理的提出是对这种传统观念的挑战, 它也为以后设计更优的水相催化剂开辟了广阔的空间.3 展望越来越多的有机小分子催化剂涌现出来, 催化活性和有效性都在不断提高, 有些报道还应用了离子溶液[26]或高压[27]等方法来扩大其合成应用范围. 由于它们的显著优点: 反应条件温和, 副反应少, 反应的立体和区域选择性高, 可以迅速实现反应规模的放大, 引起了化学工作者极大的关注. 但是这类催化剂在适用性和通用性方面都有一定的局限性, 底物范围较窄, 通用性也还不够. 因此, 设计并合成能广泛应用于不对称催化有机小分子催化剂应成为今后的发展趋势.References1 For an overview of total syntheses involving the aldol bondformation, see:(a) Mukaiyama, T. Tetrahedron 1999, 55, 8609.(b) Nicolaou, K. C.; Vourloumis, D.; Winssinger, N.; Baran, P. S. Angew . Chem ., Int . Ed . 2000, 39, 44. 2 Reviews:(a) Nelson, S. G. Tetrahedron : Asymmetry 1998, 9, 357. (b) Gröger, H.; Vogl, E. M.; Shibasaki, M. Chem . Eur . J . 1998, 4, 1137.(c) Bach, T. Angew . Chem ., Int . Ed . Engl . 1994, 33, 417. (e) Denmark, S. E.; Stavenger, R. A.; Wong, K.-T. J . Org . Chem . 1998, 63, 918.3 (a) Hajos, Z. G.; Parrish, D. R. J . Org . Chem . 1974, 39,1615.(b) Eder, U.; Sauer, G.; wiechert, R. Angew . Chem ., Int . Ed .Engl . 1971, 10, 496. 4 Cohen, N. Acc . Chem . Res . 1976, 9, 412.5 (a) Danishefsky, S.; Cain, P. J . Am . Chem . Soc . 1976, 98,4975.(b) Kwiatkowski, S.; Syed, A.; Brock, C. P.; Watt, D. S. Synthesis 1989, 818.(c) Ramamurthi, N.; Swaminathan, S. Indian J . Chem ., Sect . B 1990, 29, 401.(d) Przezdziecka, A.; Stepanenko, W.; Wicha, J. Tetrahedron : Asymmetry 1999, 10, 15896 (a) List, B.; Lerner, R. A.; Barbas, III, C. F. J . Am . Chem .Soc . 2000, 122, 2395.(b) List, B.; Notz, W. J . Am . Chem . Soc . 2000, 122, 7386. (c) Sakthivel, K.; Notz, W.; Bui, T.; Barbas, III, C. F. J . Am . Chem . Soc . 2001, 123, 5260.7 (a) Bahmanyar, S.; Houk, K. N. J . Am . Chem . Soc . 2001,123, 12911.(b) Rankin, K. N.; Gauld, J. W.; Boyd, R. J. J . Phys . Chem . A 2002, 106, 5155.(c) Domingo, M. A. R. Theor . Chem . Acc . 2002, 108, 232. (d) Hoang, L.; Bahmanyar, S.; Houk, K. N.; List, B. J . Am . Chem . Soc . 2003, 125, 16.(e) Bahmanyar, S.; Houk, K. N.; Martin, H. J.; List, B. J . Am . Chem . Soc . 2003, 125, 2475.8 Northrup, A. B.; MacMillan, D. W. C. J . Am . Chem . Soc .2002, 124, 6798.9 For selected studies of proline-catalyzed aldol reaction see:(a) List, B.; Pojarliev, P.; Castello C. Org . Lett . 2001, 3, 573.(b) Chowdari, N. S.; Ramachary, D. B.; Córdova, A.; Bar-bas, III, C. F. T etrahedron Lett . 2002, 43, 9591.(c) Pidathala, C.; Hoang, L.; Vignola N.; List, B. Angew . Chem ., Int . Ed . 2003, 42, 2785.(d) Bahmanyar, S.; Houk, K. N.; Martin, H. J.; List, B. J . Am . Chem . Soc . 2003, 125, 2475.(e) Storer, R. I.; MacMillan, D. W. C. Tetrahedron 2004, 60, 7705.626有机化学V ol. 26, 2006(f) Thayumanavan, R.; Tanaka, F.; Barbas III, C. F. Org.Lett. 2004, 6, 3541.(g) Pan, Q.-B.; Zou, B.-L.; Wang, Y.-J.; Ma, D.-W. Org.Lett. 2004, 6, 1009.(h) Ward, D. E.; Jheengut, V. Tetrahedron Lett. 2004, 45,8347.(i) Nothrup, A. B.; Mangion, F.; Hettche, I. K.; MacMillan,D. W. C. Angew. Chem., Int. Ed. 2004, 43, 2152.(j) Kazmaier, U. Angew. Chem., Int. Ed. 2005, 44, 2186.(k) Enders, D.; Grondal, C. Angew. Chem., Int. Ed. 2005, 44, 1210.(l) Casas, J.; Engqvist, M.; Ibrahem, I.; Kaynak, B.; Crdova,A. Angew. Chem., Int. Ed. 2005, 44, 1343.(m) Ward, D. E.; Jheengut, V.; Akinnusi, O. T. Org. Lett.2005, 7, 1181.(n) Suri, J. T.; Ramachary, D. B.; Barbas, III, C. F. Org.Lett. 2005, 7, 1383.10 Reviews see:(a) Gröger, H.; Wilken, J. Angew. Chem., Int. Ed. 2001, 40,529.(b) Jarvo, E. R.; Miller, S. J. Tetrohedron 2002, 58, 2481.(c) List, B. Tetrohedron 2002, 58, 5573.(d) List, B. Acc. Chem. Res. 2004, 37, 548.(e) Rdova, A. C. Acc. Chem. Res. 2004, 37, 102.11 Northrup, A. B.; MacMillan, D. W. C. Science 2004, 305,1752.12 Shen, Z.-X.; Zhou, H.; Ma, J.-M.; Liu, Y.-H.; Zhang, Y.-W.Chin. J. Org. Chem. 2004, 24, 1213 (in Chinese).(沈宗旋, 周华, 马济美, 刘艳华, 张雅文, 有机化学, 2004, 24, 1213. )13 Shen, Z.-X.; Chen, W.-H.; Zhang, Y.-W. Chirality 2005, 17,119.14 Berkessel, A.; Koch, B.; Lex, J. Adv. Synth. Catal. 2004,346, 1141.15 Tang, Z.; Jiang, F.; Yu, L.-T.; Cui, X.; Gong, L.-Z.; Mi,A.-Q.; Jiang, Y.-Z.; Wu, Y.-D. J. Am. Chem. Soc. 2003,125, 5262.16 Tang, Z.; Yang, Z.-H.; Chen, X.-H.; Cun, L.-F.; Mi, A.-Q.;Jiang, Y.-Z.; Gong, L.-Z.J. Am. Chem. Soc. 2005, 127,9285.17 Chen, J.-R.; Lu, H.-H.; Li, X.-Y.; Cheng, L.; Wan, J.; Xiao,W.-J. Org. Lett. 2005, 7, 4543.18 Nakadai, M.; Saito, S.; Yamamoto, H. Tetrahedron2002,58, 8167.19 Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Angew.Chem., Int. Ed. 2005, 44, 3055.20 Torii, H.; Nakadai, M.; Ishihara, K.; Saito, S.; Yamamoto,H. Angew. Chem., Int. Ed. 2004, 43, 1983.21 Krattiger, P.; Kovasy, R.; Revell, J. D.; Ivan, S.;Wennemers, H. Org. Lett. 2005, 7, 1101.22 Tang, Z.; Yang, Z.-H.; Cun, L.-F.; Gong, L.-Z.; Mi, A.-Q.;Jiang Y.-Z. Org. Lett. 2004, 6, 2285.23 Lacoste, E.; Landais, Y.; Schenk, K.; Verlhaca, J.-B.Vincent, J.-M. Tetrahedron Lett. 2004, 45, 8035.24 Dickerson, T. J.; Janda, K. D. J. Am. Chem. Soc. 2002, 124,3220.25 Dickerson, T. J.; Lovell, T.; Meijler, M. M.; Noodleman, L.;Janda, K. D. J. Org. Chem. 2004, 69, 6603.26 Loh, T.-P.; Feng, L.-C.; Yang, H.-Y.; Yang, J.-Y.Tetrahedron Lett. 2002, 43, 8741.27 Hayashi, Y.; Tsuboi, W.; Shojia, M.; Suzuki, N.Tetrahedron Lett. 2004, 45, 4353.(Y0509133 QIN, X. Q.)。
光致变色与电致变色材料演示文稿

当前41页,共46页,星期一。
五.电致变色器件
电致变色器件(ECD)就是利用物质的 电致变色效应,以电致变色层为基础,
辅以其它相关层和结构而构成的器件。其具
有视角宽、驱动电压低、无功耗记忆等独特 优点 。有固态,半固态,液体三种形式。
目前,已经产业化的电致变色器件有以下几
如:二噻吩乙烯衍生物,以开环异构体为“关”的状态
,闭环异构体为“开”的状态,通过光子调控( 300nm光波和白光),可以可逆实现开环和关环异构 体间转换。
当前26页,共46页,星期一。
当前27页,共46页,星期一。
2.光信息存储
信息存储包括将信息在介质上“写入”和“读出
”这两个功能。同样让双稳态结构分别对应一种功 能,可以实现信息的写入与读出。
四.电致变色材料
电致变色(EO)是指在电场作用下发生稳 定,可逆的氧化还原反应,外观上表现为颜
色或透明度可逆变化的现象。具有电致变色 特性的材料称为电致变色材料,由电致变色 材料制备的器件称为电致变色器件。
电致变色的工作原理:
电致变色材料在外加电场作用下发生电化 学氧化还原反应,得失电子,使材料的颜色 发生变化。
当前14页,共46页,星期一。
稳定性-化合物a<化合物b<化合物c
当前15页,共46页,星期一。
2.3 二芳基乙烯类衍生物
(1)结构特征 二芳基乙烯类衍生物是在乙烯基的1, 2-位上连有芳香 环(Ar)的一类化合物,具有一个共轭6 ∏ 电子的己三烯 母体结构。如图3-7所示,其中芳基(Ar)可为苯环、 五元杂环或稠杂环等,取代基R可为氢原子、烷基、 脂环烃、芳香烃及卤原子等。尤其是,当芳香环(Ar
当前11页,共46页,星期一。