HPGD基因缺失突变致厚皮性骨膜病一例
厚皮性骨膜病患者HPGD及SLCO2A1基因变异

主要内容
基本概念 临床诊断 基因诊断 治疗进展
1.基本概念
厚皮性骨膜病(Pachydermoperiostosis,PDP)
厚皮性骨膜病是一种以杵状指、皮肤增粗肥厚、骨骼改变为 典型临床特点的常染色体隐性疾病,患者可伴有多汗、关节
肿胀及疼痛等其他临床表现。目前研究发现HPGD与SLCO2A1变
LLB临床特点
突变筛查
IV-1,先证者母,表型正常 IV-2,先证者父亲,表型正常 V-1,先证者,男,32岁 V-4,先证者弟弟,27岁,患者
此后收集的另外6个PDP家系
7个家系中8名PDP患者临床特点总结
患者
性别 年龄,岁 发病年龄,岁 骨骼改变
PDP-P1
男 31 19
PDP-P2
男 27 18
Sandeep Uppal, et al NATURE GENETICS 2008
2)PDP与SLCO2A1
SLCO2A1编码前列腺素转运体(PGT),介导细胞外PGE2再摄取入细胞内 进一步代谢。 2012年,Zhenlin Zhang等在一个近亲结婚家系及两个非近亲结婚家系筛查到 SLCO2A1变异,将其明确为PDP的第二个致病基因。
++ + ++ + + ++
+
+ - + + +
++ + ++ + + ++
+
+
+
+
+
+
+
+
-
-
-
-
+
++
+
+
+
+
+
+
+
++
+
+
+
++
+
-
-
+
+
HPGD突变导致罕见的以杵状指趾为主要表现的原发性肥厚性骨关节病家系研究

中华骨质疏松和骨矿盐疾病杂志 2018 年 5 月第 11 卷第 3 期 CHIN J OSTEOPOROSIS & BONE MINER RES Vol. 11 No. 3 May 10ꎬ 2018
DOI: 10������ 3969 / j.issn.1674 ̄2591������ 2018������ 03������ 004
基金资助: 国家自然科学基金面上项目 (81570802) ꎻ 中国医学科学院医学与健康科技创新工程项目 ( 2016 ̄I2M ̄ 3 ̄ 003) ꎻ 国家重点研发计划 (2016YFC0901501)
[Abstract] Objective To investigate the phenotypes of two girls with main manifestation of unexplained digital clubbing and to detecte its pathogenic mutations in their family. Methods Two girls who suffered from digital clubbing were included. Clinical featuresꎬ serum levels of bone turnover biomarkersꎬ bone radiographyꎬ and bone mineral density ( BMD) were evaluated. Secondary causes of digital clubbing were screened. Sanger sequencing was performed to detect mutation in HPGD gene in these patients and their parents. Results The proband and her sister suffered from digital clubbingꎬ palmar and plantar hyperhidrosis since 6 ̄month ̄old. The proband had a medical history of patent ductus arterio ̄ sus. Pachydermia and joint swelling were not found in 2 patients. X ̄ray films did not show signs of pachydermia. Bone re ̄ sorption biomarker and BMD were in age and gender matched normal range. Secondary causes of digital clubbing were not identified. The compound heterozygous mutations of c. 310 _ 311delCT in exon 3 and c. 324 + 5G > A in intron 3 were
成骨不全1例报道并文献复习

成骨不全1例报道并文献复习张鹏燕;许莉军;李珊;郑丽丽【期刊名称】《河南医学研究》【年(卷),期】2018(027)008【总页数】2页(P1421-1422)【关键词】成骨不全;骨折;基因【作者】张鹏燕;许莉军;李珊;郑丽丽【作者单位】郑州大学第一附属医院内分泌科河南郑州 450052;郑州大学第一附属医院内分泌科河南郑州 450052;郑州大学第一附属医院内分泌科河南郑州450052;郑州大学第一附属医院内分泌科河南郑州 450052【正文语种】中文【中图分类】R681.1成骨不全(osteogenesis imperfecta,OI)又称脆骨症,是一组以骨骼脆性增加及胶原代谢紊乱为特征的常染色体显性或隐性遗传性结缔组织病。
1788年Ekman 首次报道此病,称其为先天性骨软化症;1844年Vrolik从病理学角度将其命名为成骨不全症[1]。
其病变不仅限于骨骼,还常累及眼、耳、皮肤、牙齿等其他结缔组织,通常以多发性骨折、蓝巩膜、牙齿发育不良、头面部畸形、听力损失、关节松弛和皮肤异常等为特点,具有遗传异质性[2]。
本文对郑州大学第一附属医院收治的1例OI患者的临床资料进行分析,探讨分析该病的临床特征及诊疗特点。
1 病例介绍1.1 病史患者男,23岁,以“反复骨折10 a余,右耳听力下降3 a”为主诉于2017年5月就诊于郑州大学第一附属医院。
患者为足月剖宫产,出生时体质量为3.15 kg,身长不详,出生时即为蓝色巩膜,三角脸形,生长发育过程与同龄人相当,身高及体质量低于同龄人。
10 a前因摔倒出现左侧胫骨粉碎性骨折,就诊于当地医院,给予钢板内固定术治疗,术后0.5 a后骨折处愈合;后至北京积水潭医院就诊,行相关检查(具体不详),疑诊为“成骨不全?”,给予口服药物治疗(具体不详),服药1 a后自行停药;8 a前因手部轻微外伤出现左肘关节骨折,就诊于当地医院,给予钢板内固定术治疗,术后0.5 a后骨折处愈合,因摔倒再次出现左侧胫骨及左侧腓骨骨折,继续接受钢板内固定术治疗,术后1 a后骨折处愈合;3 a前无明显诱因出现右耳听力下降;1 a前至新乡市人民医院就诊,行听力测试及左手腕关节正位片检查,诊断不详,给予利塞膦酸钠片(5 mg/次,1次/d)、仙灵骨葆胶囊(1.5 g/次,2次/d)口服治疗,服药2周后自行停用,同时给予维生素D2注射液(15 mg/次,2次/月)肌内注射治疗,1个月后自行停用;20 d前至中国人民解放军总医院就诊,检测甲状旁腺激素、25羟维生素D3、血钙、血磷水平正常,髋关节正位片及胸腰椎正侧位片检查未见明显异常,骨密度检查示骨质疏松,未治疗。
Madehmg病1例及文献回顾

]
Ma d e l u n g病 1例 及 文献 回顾
羊 书勇 , 郑 维银 , 李晨 军 , 李
中图分类号 R 7 3 0 . 2 6 1
浩
文献 标 识 码 B
[ 关键 词] Ma d e l u n g 病; 脂肪瘤病 ; 肥颈综合征 文章 编号 1 0 0 4 - 0 1 8 8 ( 2 0 1 3 ) 1 1 — 1 1 6 4 01 -
m a t o s i s it w h Ma d e l u n g g s y n d r o me s y mp t o m s [ J ] . L  ̄y ng l a R h i n o l
O t o l ( s t u t t g ) , 1 9 8 0 , 5 9 ( 1 1 ) : 7 4 9 - 7 5 8 .
膨隆 , 皮肤色泽正常 , 皮 温不 高 , 触诊 时皮 下有 结节 状包 块 ,
大小不一 , 质地韧且不均 , 与皮肤无 粘连 , 无压痛 。颈 部运动 受限, 偶有呼吸 困难 。B超显示脂 肪肝 。血 、 尿、 粪、 心 电图 、 胸 片等其他 常规 检查 未见明显异常 。C T显示颈部 肌 肉间隙 内脂肪组织 大量 蓄积浸润并包绕颈鞘 , 向下突入胸腔顶部 。 因患者颏下 、 颌 下和颈部包 块 巨大 , 已影 响患者 仰 卧时 通气, 患者要求手术 治疗 。局麻 下行 颈部 包块切 取 活检 , 报 告为“ 脂肪瘤 ” ; 后再局麻下行 气管切 开 , 插 管全 麻后行 双侧 颏下 、 颌下、 颈部 、 锁骨上窝脂 肪瘤 切除术 , 采 用颈 部顺 皮纹 的大弧形切 口。术 中见病 变与皮肤边界清楚 , 为无 明确包膜
病例 男, 4 3岁 , 因“ 颌下、 颈 部 快 速生 长 无 痛肿 物 2 解组织 内脂肪浸润情 况外 , 还可 以显示 气管 受压 的情 况 、 纵 隔是否受 累 , 以及其他脏器损 害 。病理 检查 病变主要表现
PHIP基因突变所致Chung-Jansen综合征一例
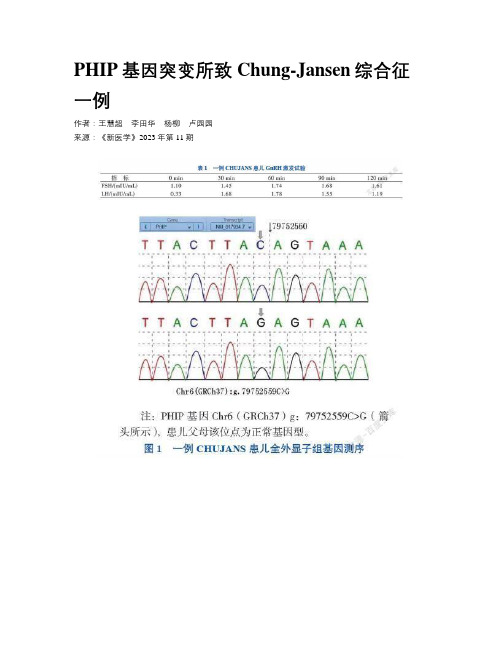
PHIP基因突变所致Chung-Jansen综合征一例作者:王慧超李田华杨柳卢园园来源:《新医学》2023年第11期【摘要】 Chung-Jansen综合征(CHUJANS)是一种常染色体显性遗传病,是新近发现的罕见肥胖综合征,主要表现为发育迟缓、智力障碍、肥胖和畸形。
该文报道1例以肥胖、睾丸小为主要表现的CHUJANS患儿,该患儿发育迟缓、智力障碍,伴有左肾缺如及低促性腺激素性性功能减退,基因检测结果提示PHIP基因突变,突变位点c.600+1G>C,最终诊断为CHUJANS。
经过长期综合性治疗,患儿远期生活质量获得极大改善。
CHUJANS发病率低,且累及多系统,该例扩展了CHUJANS的基因突变谱,有助于提高临床医师对该疾病的认识水平,及早识别并给予干预将有助于改善患者预后。
【关键词】 Chung-Jansen综合征;PHIP基因;杂合突变;儿童A case of Chung-Jansen syndrome caused by PHIP gene mutation Wang Huichao△, Li Tianhua, Yang Liu, Lu Yuanyuan. △970 Hospital of the PLA Joint Logistic Support Force,Weihai 264299, ChinaCorresponding author, Lu Yuanyuan, E-mail:*******************【Abstract】 Chung-Jansen syndrome (CHUJANS), an autosomal dominant genetic disorder, is a newly discovered rare obesity syndrome, mainly manifesting as developmental delay, mental retardation, obesity and dysmorphism. We reported one CHUJANS child with obesity and small testes as the main manifestations. The patient had developmental delay, mental retardation, complicated with left renal agenesis and hypogonadotropic hypogonadism. Genetic testing prompted PHIP gene mutation at c.600+1G>C. The child was diagnosed with CHUJANS. After long-term comprehensive treatment, the long-term quality of life was significantly improved. As Chung-Jansen syndrome is low in prevalence and multi-systemic, this case report expands the spectrum of mutations in CHUJANS,which can deepen clinicians’ understanding of this disease. Early diagnosis and intervention contribute to enhancing clinical prognosis.【Key words】 Chung-Jansen syndrome; PHIP gene; Heterozygous mutation; ChildrenChung-Jansen综合征(CHUJANS,OMIM#617991)是一种以发育迟缓、智力障碍、肥胖和畸形为特征的常染色体显性遗传病,由位于6q14染色体上的PHIP基因(OMIM#612870)中的杂合突变所致,可在婴儿期发病,大多为基因突变从头合成,少数为家族遗传[1]。
骨膜增生厚皮症一例报告

骨膜增生厚皮症一例报告向鹏月;高琳;冷启书【期刊名称】《遵义医学院学报》【年(卷),期】2012(035)004【总页数】2页(P340-341)【关键词】骨膜增生厚皮症;治疗【作者】向鹏月;高琳;冷启书【作者单位】遵义医学院附属医院内分泌科,贵州遵义 563099;遵义医学院附属医院内分泌科,贵州遵义 563099;遵义医学院附属医院内分泌科,贵州遵义 563099【正文语种】中文【中图分类】R596骨膜增生厚皮症又称皮肤骨膜增厚症,是以皮肤和四肢远端骨组织以肥厚改变为主要特征的一种罕见遗传病,但明确的发病机制尚无文献报道。
该病于1868年由Friedreich首先发现。
1935年由Touraine、Solente及Gole正式确立此病为一种独立的疾病。
现将我院近期收治的1例报告如下。
1 病例介绍1.1 临床资料患者,男,21岁。
以“头面部皮肤增厚3年”来院就诊。
于2012年3月8日在我科就诊。
3年前无明显诱因出现头面部皮肤逐渐增厚,并出现明显皱褶及回状沟纹,伴有面部皮肤针刺样疼痛、皮脂分泌增多,手足指(趾)明显增粗。
发病来,一般情况可,既往无特殊病史。
父母非近亲结婚,家族无类似病史。
查体:体温、脉搏、呼吸、血压均正常。
智力正常。
忧郁面容,头皮、面部皮肤呈沟回纹征(见图1),皮肤增厚、皮脂溢出明显。
全身皮肤粗糙,背部可见散在痤疮样皮疹,浅表淋巴结不大,甲状腺不大,心、肺、腹体格检查未见明显异常,杵状指(见图2),手足明显增粗。
双下肢无水肿。
生理反射存在,病理反射未引出。
1.2 辅助检查血、尿、大便常规正常。
肝肾功能、电解质正常。
血脂示高密度脂蛋白胆固醇0.82 mmol/L,余正常。
空腹静脉血糖4.87 mmol/L,餐后2 h 静脉血糖5.93 mmol/l。
0点生长激素2.5 mIU/L,中午12点生长激素0.4 mIU/L(参考值0.5-13 mIU/L)。
免疫球蛋白正常。
皮质醇晨8时 49.7 nmol/L、午夜0 时48.3 nmol/L(参考值上午:138-690 nmol/L,下午69-350 nmol/L)。
厚皮性骨膜病1例报告

[1 静百鹏 骨 的软骨粘液样 纤维瘤 【1 中华骨科 杂志, 2 J
.
18 8 1 : 98 ( )3
.
【 参考文献 1
【 收稿日 】 20一 .5 期 0 l 51 0
●
维普资讯
罕少疾病 杂志 20 0 2年第 9卷第 1 期
后囟 门异 常 宽大 ,且 延迟 闭 合为 本病 早期 特 征性之 。 。 Frc aah曾报道 可有智 力减退 。其 x线表现主要是 长短 管状 骨 的对称性 骨膜增 生 , 以后有 骨膜下 新骨彤 成 ,致使皮 质增 厚呈分 层状 , 常见 于胫 ,腓 、桡 ,尺骨 ,次 为股 、 肱 ,手 、 足 各短 状 骨 ,偶 见于 锁骨 、脊 椎 、 肩 日 等 甲骨
有骨 膜 反应 , 双 手 ,双 足 呈 杵状 指 c ) c 2) 趾 图 ,积 手末节指 有骨吸 收。 头颅前 后囱未 闭,颅 缝极 宽,双侗 颞骨 鳞 部骨 化不 良,项 枕 问有 多数缝 间骨 。 临床 诊断 : 厚 皮 性骨 噗病 。 2 讨论
i ,剖 1 3 芏 9 5平 T u an 等 确 立 为 独 立 瘴 o rie
_
●
●
_
_
-
●
_
_
无粘滑感。镜 F所见肿瘤 由粘 液样 和软骨样两 部分组 成。 电镜下 可见纤 维样细 胞和软骨样细 胞两种成分。 眶 粘液样纤维瘤属 良性肿瘤。囊内切除植骨术或边缘 精 整块切除术均是有效的治疗方法,化疗及放疗对本肿瘤无效。
【 1华 积德 肿瘤 讣稃 学【 】 北京 : 人民军 医出版 社 , 1 M 19 2 0 95 54
功 能、 甲功 T 、T 、T SH 均正 常 ,碱 性磷 酸酶 增 高 为 2 7 / f 7 U L 正常值 3 7 ̄2 8 L 。心脏 彩超示 动脉导 管赤 0 U/ ) 闭 ,二 尖 瓣 轻 度 关 闭 不 全。 X 线 :双 股骨 、胫 腓骨 增 粗,有骨 膜 反应 f 1 ,胫骨 弯 曲, 双恻 尺桡骨增 粗, 图 }
厚皮性骨膜病1例

镜下可见疥螨成虫及虫 卵 ( l0 x0 )
图 3 结 痂性 疥 疮 患者 皮屑 镜 下 所 见
图 2 结痂性疥疮患者皮损组织病理像 斑鳞 屑性 皮损 【 3 1 。本病 的临 床及 组织病理学改变 易与其他皮肤 病相混淆 , 从而造成误诊 和治疗 上的失误 。某些 患者可发生泛
发 性 淋 巴结 病 变 , 可 有外 周 血 嗜 酸 性 粒 细 胞 增 多 … 与普 通 疥 并 。
病; 后经皮 屑镜检查 , 见大 量疥螨 , 灭疥 治疗很快 痊愈 , 则确诊
Ari n G, Mo n la L ra MS,E ts SA,e a .Cic l tn g se t 1 r u a i g l E i p — n a
t ns w t riay n rse cbe 『] J i t i odn r e h ad cu t sais J. Me E t l d d no , mo 20 ,4 () 4 7 . 04 11 :7 — 7
其他病 因 .l 2 3 4染。 特别是可能存 在感
5
6
参 考 文 献
马 东 来 ,方 凯 ,刘 芳 挪 威疥【1 J'临床皮肤科 杂志,20 ,3 () 0 2 l4:
2 5- 0 . 0 2 6
疮相 比. 结痂性疥疮 可以引起更显著 的 IE升高[ g 4 1 。外周血 嗜酸 性粒细胞 的数量及血清 IE水平与皮肤 反应 的严重程度相关1 g 5 1 。 本例 患者在发病过程 中因系统使用糖 皮质激素 , 皮损一度
O i e hs o . Sa i 【1 N n l Me, 20 , 3 4 1) l lr C ol w cbe J v d s . E g J d 0 6 5 (6:
骨膜增生厚皮症介绍

骨膜增生症面部皮损组织病理像
二、临床表现
• (2)骨关节放射线显示全身骨膜骨赘形 成,特别是长骨远端及手足骨,可有骨 皮质增厚,关节肿胀,部分患者有关节 积液,偶有骨关节疼痛。 (见下图)
骨膜增生症患者四肢X线影像图
二、临床表现
• (3)其他表现:头发和阴毛减少、男性 乳房女性化。实验室检查:除骨放射线 典型表现外,多无阳性结果。
该病好发于男性,男女比例为9:1,发病年龄有两个高峰,分 别为1岁和15岁,多青春期起病,呈自限性,预后良好,有家族 聚集倾向。皮损处组织学显示:胶原纤维增生、增厚。
二、临床表现
• 临床表现以皮肤和骨关节为主,分别为: • (1)面部、头皮、手足皮肤增厚、粗糙,皮下脂肪过度增 生。头皮或前额可出现典型回纹状头皮,面部皮肤可呈“狮 面征”,下肢可有“象腿”样改变,面部皮肤油脂分泌旺盛, 可有痤疮样皮疹,呈忧郁面容,杵状指趾多见(89%)。
三、发病机制
发病机制尚不清楚,目前认为其与遗传性结缔组 织代谢紊乱有关 1、神经理论:迷走神经刺激导致血管扩张,增加血流量 导致骨膜增生厚皮症。
2、体液理论:生长因子或炎症介质的增加,导致成纤维 细胞增殖,致使骨膜增生厚皮症。
四、遗传规律
PDP是一种罕见的发育性遗传性疾病,属于多变的 常染色体显性遗传。主要见于男性,男女比例9:1, 约1/3患者有家族史。 • Martinez-Lavin等统计了英法德国132例PDP患者, 男性发病率明显高于女性,男女比例9:1,有家族史者 占38%,67%患者有明显的起病年龄,1岁和15岁是 发病的两个高峰段。Sinha报道过一个四代的家庭有 10例患PDP,其中4例为儿童;Lam等也报道过3例 PDP家族性发病,均有典型的杵状指(趾)、关节肿 胀、长骨周围有新骨形成。所有PDP患者在儿童及青 春期发病。
厚皮骨膜增生症1例

,
般情 况 好
,
。
系 统 体检
、
定 期 随访
2
未见异常
。
皮肤科情 况 :面 部 表情 僵 硬 沿 发 际 线 双 侧 鼻 唇
。
,
讨论
沟及 颏部 可 见 手 术疤 痕 头皮 增 厚 油 腻 呈 脑 回状 面 部 皮 肤
。
厚 皮 骨 膜增 生 症 即 原 发 性 ( 特 发 性 ) 肥 大 性 骨 关 节 炎
,
。
2007
一
年
10
增厚 见 图 4
,
。
月 再 次 出 现 四 肢关 节 疼 痛 双 膝关 节 肿胀 为 进
,
步治疗收
”
诊 断 :① 厚皮 骨 膜增 生 症 ;② 面 部 除 皱 术 后
。
入我院
。
既往体健
。
。
2006
年
7
月 于 外 院行 面 部 除 皱 术
“
。
治疗
7
m
:
予 行右膝关 节 腔抽液 术+ 复方倍 他米松 注射液
。
父 母 非近亲结婚
否 认 家 族 中 类 似 病 史 及 其 他 家族 性 遗 传
g
关 节 腔 内注 射
m
予 口 服 塞 来 昔布 胶 囊 0
,
.
2
次 g/ 来 氟 米
,
。
病史
。
特片 20
一
次 1 g/
. 。
(1 次/
外 用 青鹏 膏 剂 涂 双 膝控制关节症状
体格 检查 :发 育 正 常 智 力 正 常
To dd 。
.
生长激素
m
先天性厚甲症-1型基因突变分析

者进行基 因突变检测 , 为产前诊断和遗传 咨询 提供 依据 。方
采 用 巢 式 P R扩 增 K a基 因突 变 热 点 区 , 过 D A直 C 6 通 N 接 测 序 的方 法 , 先 天 性 厚 甲症 患 者 、 系 中 的正 常 对 照 和 对 家 10例 无 亲缘 关 系 的 正 常 人 进 行 K a突 变 检 测 。 结 果 在 0 6 K a基 因上 发 现 3个 错 义 突 变 ( 6 3 7 > K a19 T> 6 K a18 G C。 6 3 3
20 0 0 8— 3—1 收 7接 基 金 项 目: 国家 自然 科 学 基 金 ( 号 :00 4 0 编 3 50 4 ) 作 者单 位 : 徽 医 科 大学 第 一 附属 医 院皮 肤 性 病 科 , 肥 安 合 安 徽 医 科 大学 皮 肤病 研 究所 , 肥 合 2 0 3 30 2 2 02 30 2
病 率大 致相 等 , 临床 主要表 现 为指 ( ) 趾 甲的过 度 角 化增 厚 及 其 他 外 胚 叶缺 陷 的症 状 … 。 目前 临床 上 主要 分 为 1和 2两 型 , 中 1型 又称 Jds o—e 其 aas nL— h w no sy综合 征 ( C1 O M.620 , 现 为 厚 adwk P 一, MI 170 ) 表
一
先 天性 厚 甲症 ( a ho yhacn e i , C) P c yn ci o gnt P 是 a
河 北 医 科 大 学第 四医 院 皮肤 科 , 家 庄 石
种 罕见 的 常染 色 体 显性 遗 传 性 皮 肤病 。 由 Jd — aa
厚皮指症1例

1 临 床 资 料 患 者男 ,5岁 , 手 近端 指 关节 肿 胀 3 多 月 。3个 月前 l 双 个 无 明显 原 因 右手 中指近 端 指 间关 节 两 侧肿 胀 , 渐加 重 , 后 逐 随
右手食指 、 无名指 , 中 、 左手 指 无名指均出现类似症状 , 持笔受
影响而来就诊 。 患者平素健康 , 无慢性疾病史。 无家族史 , 父母 非近亲结婚。体格检查 : 患者发育正常 , 足关节及 四肢大关节
未 见 异常 。皮肤 科 检 查 : 双手 2 4指 近端 指 间 关节 粗 大 , 图 ~ 见 1 图 2 关 节 周 同 软 组 织 增 生 肿 胀 , 面 严 重 腹 侧 正 常 , 面 、 , 侧 表 皮肤正常, 中等 硬 度 , 压痛 . 节 活 动不 受 限 。 无 关 x线检 查 示 双
本病 主要应与淋巴瘤样丘疹病相 鉴别 。 后者 主要发生于 5 0岁左右男性 , 红斑 、 丘疹成批 出现 , 丘疹 中心可 出血坏死 ,
( 稿 1期 2 0 0 — 7) 收 3 叭 —5 1
厚皮指症 I 例
党林 , 杨海龙 , 玉珍 栗
( 哈尔 滨医科 大学 附0 6
本例患者为壮年男性 ,皮损 为暗红色或紫红色斑 丘疹 、 坏死 、 溃疡 及结痂 , 部分皮损 上覆 细薄鳞屑 , 皮损呈 多形性 , 临床及病理均较典 型。
参 考 文献 :
[ ]Mce H C ln E Ga s 1 keP , ao , rn R主 编 ; 学 骏 , 建 方 主 译 . 肤 朱 孙 皮
1例HPGD基因新缺失突变致儿童原发性肥大性骨关节病报道

1例HPGD基因新缺失突变致儿童原发性肥大性骨关节病报道刘霜;仇佳晶;孟岩;邱正庆【期刊名称】《基础医学与临床》【年(卷),期】2012(032)006【摘要】目的通过对1例临床疑似原发性肥大骨关节病的患者进行HPGD基因分析并确诊,提高对该疾病的认识.方法根据患儿症状,体征及骨骼系统放射学检查进行临床诊断,提取患儿及其父亲外周血DNA,PCR扩增HPGD基因编码氨基酸的7个外显子片段,测序检测突变.结果患儿女性,5岁,具有手指及足趾末端指节肥大,手足多汗,前额皮肤增厚等典型临床表现.PCR扩增片段直接测序示患儿HPGD外显子3发生c.308 _309delCT(p.Thr103Thrfs4X)纯合改变.结论原发性肥大骨关节病为一种少见的常染色体隐性遗传疾病,典型的临床及影像学表现有助于诊断,HPGD基因突变分析是确诊的主要方法.【总页数】4页(P656-659)【作者】刘霜;仇佳晶;孟岩;邱正庆【作者单位】中国医学科学院北京协和医学院北京协和医院儿科,北京100730;中国医学科学院北京协和医学院北京协和医院儿科,北京100730;中国医学科学院北京协和医学院北京协和医院儿科,北京100730;中国医学科学院北京协和医学院北京协和医院儿科,北京100730【正文语种】中文【中图分类】R729【相关文献】1.疑似假性肥大性肌营养不良症患儿Dystophin基因缺失突变分析 [J], 郭雅洁;王咏红;佟月娟;申晨2.HPGD突变导致罕见的以杵状指趾为主要表现的原发性肥厚性骨关节病家系研究[J], 吕芳;宋玉文;李路娇;王鸥;姜艳;夏维波;邢小平;李梅3.原发性肥大性骨关节病并SLCO2A1基因突变2例 [J], 金萍;张勤;何红晖;朱伟豪;龙晓丹;莫朝晖4.低分化鳞癌致肺性肥大性骨关节病1例报道 [J], 徐鸥;郝青林5.并发动脉导管未闭原发性肥大性骨关节病一例 [J], 崔冉;张悠扬;曲伸;盛辉因版权原因,仅展示原文概要,查看原文内容请购买。
厚皮性骨膜病1 例报道

YANG Zhi-yi, ZENG Rui, DONG Shu-qin, HAI Bing* (Department of Respiratory and critical Care, the second affiliated Hospital of Kunming Medical University, Kunming
0 引言
厚 皮 性 骨 膜 病 (Pachydermoperiostosis,PDP) 又 称 原 发 性 肥 厚 性 骨 关 节 病,是 一 种 罕 见 的 遗 传 性 疾 病,最 早 发 现 于 1868 年。以指节、软组织增生和骨膜增生为特征。其发病机 制尚不明确,诊断依据主要是靠临床症状和影像学资料。现 将我院于 2020 年 1 月 19 日收治的 1 例 PDP 报告如下。
Yunnan 650000)
ABSTRACT: In a 15-year-old male patient, the swelling of multiple joints in both hands lasted for more than 1 year and worsened for 3 months. X-ray examination showed that the space of proximal interphalangeal joint of 2-5 fingers of both hands was slightly narrow, and the soft tissue of proximal interphalangeal joint of 2-4 fingers of both hands showed symmetrical fusiform swelling. Joint ultrasound showed bone erosion of the proximal interphalangeal joint of the right hand. Diagnosis: pachydermic periosteal disease. KEY WORDS: pachydermoperiostosis; clinical manifestation; diagnosis; treatment
原发性厚皮性骨膜病的遗传学研究进展

原发性厚皮性骨膜病的遗传学研究进展杜然;范亮亮;黄皓;项荣【期刊名称】《中华医学遗传学杂志》【年(卷),期】2016(033)001【摘要】原发性厚皮性骨膜病是一种较为罕见的遗传疾病,其主要临床表现为杵状指、骨膜增生、回状头皮和皮肤肥厚,有的患者还伴随有肢端骨质溶解症、多汗症等症状.目前已定位的相关基因有HPGD和SLCO2A1,其表达产物均参与前列腺素E2的运输和代谢,二者的异常是导致原发性厚皮性骨膜病的主要原因.本文对原发性厚皮性骨膜病的遗传基础及其与临床表型的相关性进行综述,为原发性厚皮性骨膜病的基础研究和临床诊断提供参考.%Pachydermoperiostosis is a rare genetic disease characterized by finger clubbing,periostosis,cutis verticis gyrata and pachydermia accompanied by acroosteolysis and hyperhidrosis.Recently,two susceptibility genes,HPGD and SLCO2A1,have been identified,whose protein products are involved in the transportation of prostaglandin and metabolism underlying pachydermoperiostosis.Here the genetic basis of pachydermoperiostosis and its correlation with its clinical phenotype are reviewed,which may provide a reference for basic research and clinic diagnosis for the disease.【总页数】3页(P105-107)【作者】杜然;范亮亮;黄皓;项荣【作者单位】410013长沙,中南大学生命科学学院医学遗传学国家重点实验室;410013长沙,中南大学生命科学学院医学遗传学国家重点实验室;410013长沙,中南大学生命科学学院医学遗传学国家重点实验室;410013长沙,中南大学生命科学学院医学遗传学国家重点实验室【正文语种】中文【相关文献】1.中医药防治原发性肝癌的表观遗传学研究进展2.原发性低钾型周期性麻痹的分子遗传学研究进展3.原发性开角型青光眼的分子遗传学研究进展4.原发性开角型青光眼的分子遗传学研究进展5.原发性低钾型周期性麻痹分子遗传学及其治疗的研究进展因版权原因,仅展示原文概要,查看原文内容请购买。
厚皮性骨膜病

厚皮性骨膜病
金诗怡;郑捷
【期刊名称】《中国麻风皮肤病杂志》
【年(卷),期】2000(016)003
【摘要】@@ 厚皮性骨膜病(pachydermoperiostosis)也称为原发性肥厚性骨关节病,是一种罕见的常染色体显性遗传疾病,以皮肤肥厚、骨膜增生及杵状指、回状头皮为主要表现[1,2].1868年,Friedreich首次报道了本病,但直到1935年才由Touraine、Solents和Gole把它确立为一个独立的疾病.中国人的首例病例在1976年被发现和报道[3].现将该病介绍如下.
【总页数】2页(P193-194)
【作者】金诗怡;郑捷
【作者单位】上海第二医科大学附属瑞金医院皮肤科,200025;上海第二医科大学附属瑞金医院皮肤科,200025
【正文语种】中文
【中图分类】R75
【相关文献】
1.厚皮性骨膜病1例并文献复习 [J], 谢宇萍;罗佐杰
2.厚皮性骨膜病1例 [J], 马红艳;康晓静;屈圆圆
3.厚皮性骨膜病误诊为幼年类风湿关节炎1例 [J], 王涛;范晓云
4.浅析厚皮性骨膜病综合征的临床特征及治疗 [J], 李爽
5.1例厚皮性骨膜病注射A型肉毒杆菌毒素的护理 [J], 杨木香;陈吉辉;陈思奇;邹琴
因版权原因,仅展示原文概要,查看原文内容请购买。
先天性厚甲综合征一例

先天性厚甲综合征一例
陶晓苹;伍友成;万慧颖;杨为
【期刊名称】《国际皮肤性病学杂志》
【年(卷),期】2007(33)2
【摘要】先天性厚甲综合征,又称厚甲,甲肥大症,先天性厚甲,先天性厚甲症。
为一少见的外胚叶缺陷病,系常染色体显性遗传,但也有常染色体隐性遗传的报告,本病除厚甲外,尚可见掌趾角化,毛囊角化,黏膜白色角化,掌跖多汗,大疱及毛发异常等多种损害。
【总页数】2页(P69-70)
【作者】陶晓苹;伍友成;万慧颖;杨为
【作者单位】610031,成都,四川省医学科学院、四川省人民医院皮肤病性病研究所;610031,成都,四川省医学科学院、四川省人民医院皮肤病性病研究所;610031,
成都,四川省医学科学院、四川省人民医院皮肤病性病研究所;610031,成都,四川省
医学科学院、四川省人民医院皮肤病性病研究所
【正文语种】中文
【中图分类】R75
【相关文献】
1.伴发溃疡的I型先天性厚甲症一例 [J], 王燕飞;柳君如;刘卫兵
2.Ⅰ型先天性厚甲症一例 [J], 徐小容;熊琦;曹碧兰
3.先天性厚甲综合征1例 [J], 李鑫;王哲;陈淑明
4.先天性厚甲综合征一例 [J], 赵志贤;张伟力
5.Ⅰ型先天性厚甲症一例 [J], 刘忠艳;王启华;周桂芝
因版权原因,仅展示原文概要,查看原文内容请购买。
一例1q41q44重复及3p26.2p26.1缺失患儿的表型与遗传学分析

一例1q41q44重复及3p26.2p26.1缺失患儿的表型与遗传学分析刘慧;杨成青;易致;宋振凤;李菲;薛姣;刘凯璇;张颖【期刊名称】《临床医学进展》【年(卷),期】2024(14)3【摘要】目的:探讨1例发育落后患儿染色体拷贝数变异的性质及来源,分析基因型与疾病表型的相关性。
方法:应用常规G显带分析患儿及其父母的外周血染色体核型,并对患儿进行二代测序(next generation sequencing, NGS)检测。
结果:染色体G显带核型分析显示患儿存在染色体结构异常,核型描述为46,XX,−3, der (3),t (1;3) (q41;p26.1),其父亲染色体核型为46,XY,t (1;3) (q32;p25),其母亲核型未见异常。
NGS检测显示患儿染色体1q41q44区存在约29.093 Mb的重复,3p26.2p26.1区存在约1.717 Mb的缺失。
结论:患儿染色体异常是来自其父亲的平衡易位。
1q41q44重复及3p26.2p26.1缺失是导致患儿表型异常的遗传学病因。
【总页数】7页(P1717-1723)【作者】刘慧;杨成青;易致;宋振凤;李菲;薛姣;刘凯璇;张颖【作者单位】青岛大学附属医院儿科青岛【正文语种】中文【中图分类】R72【相关文献】1.一例染色体微缺失和微重复患儿的细胞遗传学及分子生物学分析2.新发8p重复伴缺失综合征患儿细胞分子遗传学研究3.17p13.3微缺失/微重复与胎儿临床表型差异的遗传学分析——附五例报告4.1q21.1微缺失/微重复胎儿的临床表型和遗传学分析5.1例2p11.1-q21.1片段重复导致智力障碍患儿的遗传学及表型分析因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
【摘要】
目的通过基因测序,在分子水平确诊厚皮性骨膜病l例。方法收集1例26岁男性厚皮性骨膜
病患者及其父母外周血DNA,PCR扩增HPGD及SLC02A1基因外显子片段,通过基因测序查找有无突变。根据测 序结果进行蛋白质空间结构的同源性分析。结果基因测序结果显示,患者HPGD基因第3外显子存在移码突 变c.310 31ldelCT(p.L104AfsX3),为纯合子,其母为该突变的杂合子携带者,其父正常。蛋白空间结构预测显示,上 述基因突变可使编码蛋白缩短60%。结论厚皮性骨膜病的典型临床表现及影像学表现有助于诊断,HPGD、 SLC02A1基因突变测定则是确诊的主要方法。
l J J.
[11]Komatsu N,Saijoh
Based
Complement
Ahernat
Med.2013.2013:907016.
expression in the stratum
comeum and serum of atopic dermatitis
DOI:10.1155/2013/907016.
patients[J].Exp Dermat01.2007,16(6):513—519.DOI:10.1111/j.
atopic
16j
Kim BE,Leung DY.Epidermal barrier in Allergy Asthma
dermatitisl J J. 【12
1 600.0625.2007.00562.x. Sevilla LM,Nachat R,Groot KR,et a1.Mice deficient in involucrin, envoplakin.and periplakin )
STAT一6[J J.
(本文编辑:吴晓初)
HPGD基因缺失突变致厚皮性骨膜病一例
丁晨召 任蕾
乐昊李静赵天雪秦贵军
450052郑州大学第一附属医院内分泌科河南省内分泌及代谢病诊疗中心
通信作者:秦贵军,Email:hyqin舀@zzu.edu.ca
DOI:10.3760/cma.j.issn.0012-4030.2016.01.013
gene
prediction of spacial structure of proteins revealed that the above gene mutation could shorten the length of the encoded Typical clinical manifestations and imaging findings peptide by about 60%.Conclusion of diagnosis pachydermoperiostosis,while mutation analysis of HPGD and SLC02A1 genes is diagnosis.
are
a
helpful for the primary main approach to its final
【Key words】Osteoarthropathy,primary
hypertrophic;Mutation;HPGD gene;SLC02A 1 gene
万方数据
生堡廛腿抖盘查2Q!§生!旦筮塑鲞筮!翅£h也』堕婴熊!!,』g!H§Ⅱ2Q!§,y丛:!皇,盟Q:!
厚皮性骨膜病(pachydermo.periostosis)又称原发性肥大 性骨关节病(primary
hypertrophic
台、股骨下端边缘见骨质增生,关节间隙略变窄;骨膜增厚、 毛糙。彩超:左侧阴囊结石,双侧附睾头多发囊肿。额部正中 近发际线处皮肤组织病理检查:皮肤真皮层及皮下组织、结 缔组织、皮脂腺、汗腺、真皮内毛细血管等结构增生,毛囊间 真皮网状层存在大量嗜碱性粘液样物质。骨密度测定:腰椎 部呈轻度骨质疏松,股骨颈部骨量正常,大转子部、Wards 部轻度骨量减少。临床诊断:厚皮性骨膜病。 二、方法 1.DNA提取:患者及家属均签署知情同意书后,留取患 者及其父母晨起空腹外周静脉血2 IIll(肝素抗凝),采用 QIAamp
122(3):560—568.DOI:10.1016/j.jaci.2008.05.050. [9]Cork MJ,Danby SG,Vasilopoulos Y,et a1.Epidermal barrier dysfunction in atopic dermatitis lJ J.J Invest Dermatol,2009,129 (8):1892一1908.DOI:10.1038,jid.2009.133. 1
K,Kuk C,et a1.Human tissue kallikrein
XY,Jiang
in
W,et a1.Anti-inflammatory effect of
atopic dermatitis・like murine model
2007.11.006.
qingpeng Evid
ointment
生堡麈丛科塞盍2Q!§生!旦筮塑鲞筮!期£丛n』旦!四巫Q!,』塑女§型垫!§,yQ!:垒!,盟!!!
47
10.39696.issn.1009.1 157.2013.03.006. 15j
Li YZ,Lu
Clin
Immun01.2008.126(3):332—337.DOI:10.1016/j.clim.
Tianxue,Qin Guijun
Department of Endocrinology,First Affiliated Hospital ofZhengzhou University,Henan Clinical Center for Endocrine and
Metabolic Diseases,Zhengzhou 450052,China
HPGD(NM_000860)为本病的致病基因。Zhang等…认为本病
的发生亦与SLC02A1基因(NM_005630.2)突变有关,该基因 与前列腺素E2(PGE2)在细胞内外的转运密切相关。本研究 报道l例基因诊断明确的PHO患者。 一、病历资料 患者男,26岁。6岁时出现手指、足趾增粗,16岁时出现 皮肤粗糙增厚,近5年出现长距离行走后足跟疼痛。父母及 一姐体健,父母非近亲结婚。体检:皮肤粗厚,油腻潮湿,毛孔 粗大,头面部皮肤过度增生,头部皮肤形成深褶,呈脑回状 (图1)。手指、足趾末端增粗,呈杵状指(图2),不能做精细动 作,手指、足趾、双膝关节粗大,双足跟压痛。未见其他阳性体 征。实验室检查:血碱性磷酸酶135 u,L(0~40 U/L),肝肾功 能及血常规、血钙、血磷等生化指标均正常,血卵泡刺激素、 促黄体生成素、泌乳素、睾酮、雌二醇、生长激素、胰岛素样生 长因子1均正常,甲状腺激素及肾上腺激素正常。影像学检 查:垂体MRI正常。手足部正侧位x线片:双足第1脚趾远 端趾骨缺失,双踝关节及跗、跖关节模糊,间隙显示不清;双 手远端指间关节屈曲,关节面边缘骨质轻度增生,间隙存在; 双腕关节骨质密度稍减低,局部关节面边缘较模糊。尺桡骨 及胫骨正侧位x线片:双侧尺桡骨骨干端肥大增生,关节间 隙变窄;骨膜增厚、毛糙;左右膝关节形态结构完好,胫骨平
200706187.
[1 3 1
Hansson L,Backman A,Ny A,et a1.Epidermal overexpression of stratum corneum chymotryptic enzyme in mice:a model for chronic itchy
aggregate
Peripheral blood samples were obtained from
a
sequencing.Methods
26-year-old male
patient with pachydermoperiostosis and his parents,and DNA was extracted from these blood samples.Polymerase chain reaction(PCR)was performed to amplify all the exons of HPGD and SLC02A1 genes,and gene sequencing to identify Gene mutations.According to sequencing results,the spatial structure of relevant proteins was predicted.Results showed a exon 3 oftheHPGD sequencing homozygousframe-shiftingmutation c.310—31ldelCT(p.L104AfsX3)in genein the patient.His mother was a heterozygous carrier of the mutation,but no mutation was identified in his father.The
to
(KLK7).and
filaggrin(FLG)
DOI:10.1046/j.0022—202x.2001.01684.x.
risk[J]J
Allergy Clin Immunol,2008,
[14]Buraczewska
with
I,Berne B,Lindberg M,et affects the mRNA
【关键词】骨关节病,原发肥大性;突变;HPGD基因;SLC02A1基因
A of paehydermoperiostosis caused by
a
case
deletion mutation in the HPGD gene
Ding Chenzhao,Ren Lei,Yue
Hao,Li Jing,2hao