构建系统发育树的方法 PPT
系统发育树构建教程(PHYLIP)

系统发育树构建教程(PHYLIP)PHYLIP网址:/phylip.html(一)序列的前期准备1.用ENTREZ或SRS搜索同源DNA/蛋白质序列(same sequence in different organisms) 2.用CLUSTALX进行多条序列比对,在output format option选定PHY格式,构建进化树需要这个phy文件。
Figure 4.1 用clustalx进行多条序列比对3.解压缩phylip-3.68.exe,得到三个文件夹,doc文件夹里是关于所有PHYLIP子程序的使用说明,exe文件夹里是直接可以使用的各个子程序,src文件夹里是所有程序的源文件。
4.打开exe文件夹,双击SEQBOOTt子程序(SEQBOOT是一个利用bootstrap方法产生伪样本的程序),输入刚刚生成的phy文件的路径,点击enter。
5.所有PHYLIP程序默认的输入文件名为infile, 输出文件名为outfile。
如果在exe文件夹里找不到默认的输入文件,会提示can’t find input file “infile”。
Figure 4.2 seqboot程序起始界面6.进入程序参数选择页面(Figure 4.3)。
第一列中的D、J、%、B、R、W、C、S等代表可选的参数。
想改变哪个参数,就键入此参数对应的字母,并点击回车键,对应参数将会发生改变。
当我们设置好所有参数后,(这里我们可以不做任何修改),键入Y,按回车。
此时程序询问“random numbe r seed? <must be odd>”,这是询问生成随机数的种子是多少,输入一个4N+1的数,点击回车程序开始运行,输出结果到文件outfile,保存在当前文件夹里。
.Figure 4.3 seqboot程序参数选择页面主要参数解释:D: 数据类型,有Molecular sequence、discrete morphology、restriction sites和gene frequencies4个选项。
系统发育树构建PPT(共10张PPT)

系统发育树构建软件 窗Mo口de上lt面es的t&下Mr拉M菜od单elt可es让t(碱你基选替择换传模统型多筛重选比软对件和)轮廓比对需要的所有选项。
• BEAST 名CLUS称T:ALX-Un是cuCltLuUreSdTAbaLc多te重riu序m列cl比on对e 程YU序2的01WH1in0dows版本。
• 序列长度:353 • 相 似 比: 99%
• 核酸序列 • 分类地位
打开软件clustalx
• CLUSTALX-是CLUSTAL多重序列比对程序的Windows 版本。Clustal X为进行多重序列和轮廓比对和分析结果提 供一个整体的环境。 序列将显示屏幕的窗口中。采用多色彩的模式可以在比 对中加亮保守区的特征。窗口上面的下拉菜单可让你选 择传统多重比对和轮廓比对需要的所有选项。
D并is用ta系nc统e-进ba化se树d来m概eth括od生s物基间于的距这离种的亲方缘法关系。
• Figtree (树形显示软件) C窗lu口st上al面x比的对下结拉果菜是单构可建让系你统选发择育传树统的多前重提比对和轮廓比对需要的所有选项。
名Maxim称u:m paUrnscimulotunrye(dMbPa)最cte大riu简m约c法lone YU201H10
3
系统进化树
结点:表示一个分类单元。
进ቤተ መጻሕፍቲ ባይዱ支:两种以上生物(DNA序列 )及其祖先组成的树枝。
进化分支长度:用数值表示的进化枝 的变化程度(遗传距离) 距离标尺:生物体或序列之间差异的的 数字尺度。
根:所有分类的共同祖先。
外群:一个或多个无可争议的同源物 种,与分析序列相关且具有适当的亲缘 根 关系
系统发育树构建的三种方法(一)

系统发育树构建的三种方法(一)三种系统发育树构建方法在生物学中,系统发育树是指生物种类之间的亲缘关系。
通过构建系统发育树,我们可以了解不同生物种类之间的演化历程和联系。
下面我们来介绍三种系统发育树构建的方法。
距离法距离法是一种常用的构建系统发育树的方法。
它的基本思想是根据不同生物种类之间的距离进行分类。
距离可以是基于相似性的度量,也可以是基于差异性的度量。
常见的距离度量包括曼哈顿距离、欧几里德距离、切比雪夫距离等。
距离矩阵是距离法的重要组成部分,它是一个方阵,包含了所有生物种类之间的距离值。
构建系统发育树的过程就是通过对距离矩阵进行聚类(clustering)实现的,聚类的目标是将所有生物种类分为几个亚类,使得每个亚类内部的距离较小,不同亚类之间的距离较大。
距离法的优点是计算简单,易于理解和实现,但它也存在一些缺点,如可能会受到距离度量方法的影响,对于复杂的数据集不够精确等问题。
最大简约法最大简约法(Maximum Parsimony)是另一种常用的构建系统发育树的方法。
它的基本思想是在所有可能的构建树中,选择最优的一棵树使得总的分支长度最短。
在最大简约法中,每个生物种类都被认为有一个共同的祖先,并且所有的演化事件都发生在该祖先的分支上。
最大简约法的优点是具有高度的准确性和可靠性,但计算复杂度较高,需要考虑多个参数的优化问题。
最大似然法最大似然法(Maximum Likelihood)是一种统计学方法,它的基本思想是估计不同系统发育树的概率,并选择具有最高概率的一棵树。
最大似然法的关键在于建立一个模型,该模型需要考虑生物种类之间的演化速率、基因突变率、特征亲缘关系等因素。
最大似然法计算量较大,但是它的结果在重复实验中更加可靠和稳定。
三种系统发育树构建方法各有优缺点,在实际应用中,我们需要结合具体的研究问题和数据特点来选择最适合的方法。
通过不断的探索和实践,才能更好地理解和探索生命的奥秘。
总结系统发育树的构建是生物学和生物信息学中一个重要的研究领域。
系统发育进化树构建

系统发育进化树构建1. 什么是系统发育进化树?系统发育进化树(Phylogenetic Tree),也称为系统树或进化树,是生物学中常用的一种图形表示方法,用于展示不同物种之间的亲缘关系以及它们的进化历史。
系统发育进化树可以帮助我们理解生物多样性的起源、演化以及物种之间的关系。
2. 构建系统发育进化树的方法2.1 形态学特征比较法形态学特征比较法是构建系统发育进化树最早也是最常用的方法之一。
通过比较不同物种的形态特征,如体型、颜色、器官结构等,来推断它们之间的亲缘关系。
这种方法适用于无法进行分子遗传学研究的古生物学领域。
2.2 分子遗传学方法分子遗传学方法是目前构建系统发育进化树的主要手段之一。
它利用DNA、RNA、蛋白质等分子的序列信息来推断不同物种之间的亲缘关系。
常用的方法包括序列比对、构建进化模型、计算进化距离等。
2.3 组织化石记录法组织化石记录法是通过研究化石中的细胞结构、细胞组织等信息,来推断不同物种之间的亲缘关系。
这种方法适用于无法获取分子遗传学信息的古生物学领域。
3. 构建系统发育进化树的步骤3.1 收集相关数据构建系统发育进化树的第一步是收集相关的数据,包括形态学特征数据、分子序列数据或化石记录数据。
数据的准确性和全面性对于构建准确的进化树非常重要。
3.2 数据处理与分析在收集到数据后,需要对数据进行处理和分析。
对于形态学特征数据,可以通过比较不同物种的特征值来计算相似性矩阵;对于分子序列数据,可以进行序列比对和计算进化距离等操作。
3.3 构建进化模型在数据处理与分析的基础上,需要选择合适的进化模型来描述不同物种之间的进化关系。
常用的进化模型包括NJ(Neighbor-Joining)方法、ML(Maximum Likelihood)方法和Bayesian方法等。
3.4 构建进化树在选择了合适的进化模型后,可以利用计算机软件或在线工具来构建进化树。
常用的软件包括MEGA、PAUP*和MrBayes等。
系统发育树的构建

1.Sequence analysis of the complete mitochondrial DNA molecule of the hedgehog, Erinaceus europaeus, and the phylogenetic position of the Lipotyphla ,1995. 2.Murphy, W.J., et al., Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science, 2001. 294(5550): p. 2348-51.
贝叶斯法(Bayesin)
基本思想:
1.先验概率; 2.后验概率;
贝叶斯法(Bayesin)
每棵树的后验概率是无法直接计算的,通常采用MCMC法近似估计后验 概率的密度分布和相对比例。
比较项目 原理 序列信息利用 进化模型 模型参数 目标函数 函数计算 树搜索 结果
可靠性评估 系统误差 运算速度 混合性状分析 适用范围
2.长枝吸引(Long-branch Attraction,LBA) 克服长枝吸引的方法:
1.排除法
去除序列中受选择压力较少的位点
去除分类群中进化速率较快的长枝分类元
2.打断长枝法 增加与长枝分类元关系较近的分类元进行系统发育分析, 以打断 长枝。多数情况下, 这种方法能够避免形成长枝吸引。
3.使用多种建树方法 NJ 和MP容易造成长枝吸引,改ML或bayesin 可改善。
构建进化树的方法
UPGMA法 (Unweighted Pair Group Method using Arithmetic average) 1.距离法
MEGA软件——系统发育树构建方法
MEGA软件——系统发育树构建方法(图
文讲解)
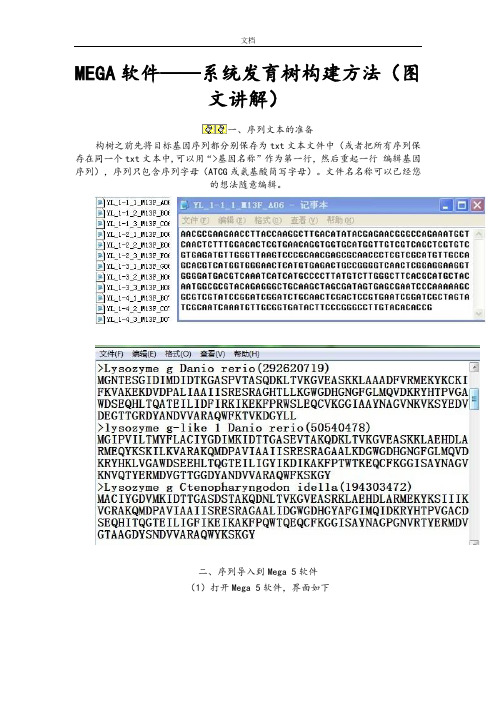
一、序列文本的准备
构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行编辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您
的想法随意编辑。
二、序列导入到Mega 5软件
(1)打开Mega 5软件,界面如下
(2)导入需要构建系统发育树的目的序列
OK
选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击Prot
ein)
出现新的对话框,创建新的数据文件
选择序列类型
导入序列
导入序列成功。
(3)序列比对分析
点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对齐
碱基
(4)系统发育分析
关闭窗口,选择保存文件路径,自定义文件名称
三、系统发育树构建
根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例
Bootstrap 选择1000,点击Compute,开始计算
计算完毕后,生成系统发育树。
文档
根据不同目的,导出分析结果,进行简单的修饰,保存。
系统发育树构建的三种方法

系统发育树构建的三种方法
1. 距离法(Distance Method):该方法将各个物种之间的差异转化为距离值,并根据这些距离值构建系统发育树。
距离可以基于基因序列或形态特征等进行计算。
该方法不考虑进化模式和序列的进化过程,仅提供基于相似性的分支结构。
2. 最大简约法(Maximum Parsimony):该方法基于最小进化原则,即最可能的树是具有最少次数的进化事件的树。
它寻求在进化树上使得进化事件(如插入、缺失、突变)的次数最少的树。
该方法是需要较多计算的方法,但树的建立结果更加准确。
3. 最大似然法(Maximum Likelihood):该方法也是基于最小进化原则,但它考虑进化模式和序列的进化过程,并将最可能的进化树视为产生的序列数据的最大概率估计。
该方法需要更复杂的计算,但对于数据信息的准确推断较好。
用PhyML构建系统发育树 PPT课件

PhyML简介
• PhyML是采用最大似然法估计核苷酸或氨基 酸序列系统发生分析的软件。
PhyML 3.0: new algorithms, methods and utilities
• http://www.atgc-montpellier.fr/phyml/
PhyML可以在线使用了
四个碱基比例键入后,E选项变为user defined
键入K,设置进化模型,为自己定制的模型起名字
根据modeltest结果依次键入碱基替代模型
自定义碱基替代模型成功
这一步设置可变位点的比例。
这一步设置碱基替代的类别数,默认为4,最佳一般为6
*这一步为设置是否优化替代参数,自己订置一定要选为no,否 则最终替代参数会与设置的有差异。
invariable sites (fixed/estimated)默 认为0. The gamma shape
parameter can be
fixed by the user or
estimated via
maximum-likelihood.
这一步设置进化模型。 phyML3.0可直接提供数种模型,每一个 模型均代表一种碱基替代类型。除GTR外,其它模型均可设置 其中的具体参数。同时还可以自已订置替代模型。其中自己订 置替代模型一般为通过modeltest3.7获得的最佳模型。
此选项为序列信息:包括DNA或蛋白序列以有序列为交互或 连续(?)。不用设置,输入tution model界面
Ts/tv ratio (fixed/estimated) 只在K80, HKY85 or TN93 models中选择 才有意义。
Proportion of
构建系统发育树的方法课件PPT

2021/3/10
12
构建发育树
再点击phylogency----construct/test neighborjioning tree-----然后选择上述保存的文件.mas 然后参数选择Bootstrap method 1000 其他默 认,点击compute
2021/3/10
13
得到系统发育树
系统发育树构建
赵慧
2021/3/10
1
16SrRNA片段获取
细菌培养液 DNA提取
基因组DNA
PCR扩增 16SrRNA
2021/3/10
测序
2
序列比对
1.使用DNAstar中的SeqMan对未知菌测序得到 的两条序列进行组装连接 SeqMan使用方法:1.打开软件如图
2021/3/10
3
2.点击Add Sequence 然后选择侧得的序列 (带有图的)
6.Blast DAN
2021/3/10
8
7.输入组装的DNA序列
2021/3/10
9
8.选择相似的菌种
2021/3/10
10
9.下载FASTA格式
2021/3/10
11
10.MEGA7比对
1.打开软件点击Align-Edit/Build Alignment 然后 OK-DNA 再然后是Edit ---insert sequence from file----选择序列(相似菌和目的菌,fasta 格式)----选中序列---点击Alignment ---Align by clustalW----保存(mas格式)
打开软件点击aligneditbuildalignment然后okdna再然后是editinsertsequencefromfile选择序列相似菌和目的菌fasta格式选中序列点击alignmentalign构建发育树再点击phylogencyconstructtestneighborjioningtree然后选择上述保存的文件
【精编版】MEGA构建系统发育树PPT文档共15页

谢谢!
【精编版】MEGA构建系统发育树
46、法律有权打破平静。——马·格林 47、在一千磅法律里,没有一盎司仁 爱。— —英国
48、法律一多,公正就少。——托·富 勒 49、犯罪总是以惩罚相补偿;只有处 罚才能 使犯罪 得到偿 还。— —达雷 尔
50、弱者比强者更能得到法律的保护 。—— 威·厄尔
61、奢侈是舒适的,否则就不是奢侈 。——CocoCha nel 62、少而好学,如日出之阳;壮而好学 ,如日 中之光 ;志而 好学, 如炳烛 之光。 ——刘 向 63、三军可夺帅也,匹不可夺志也。 ——孔 丘 64、人生就是学校。在那里,与其说好 的教师 是幸福 ,不如 说好的 教师是 不幸。 ——海 贝尔 65、接受挑战,就可以享受胜利的喜悦 。——杰纳勒 尔·乔治·S·巴顿
系统发育树的构建
分子钟
➢ 进化论的发展 ➢ 分子进化学说 ➢ 进化模型 ➢ 生命树概述 ➢ 进化树构建常用方法 ➢ 进化树检验 ➢ 进化树构建中的问题 ➢ 最近的工作
DNA序列进化模型
AG
A
AT
AC
GA
G
GT
GC
CT
C
CA
CG
TC
T
TA
TG
JC69 K80
HKY85
➢ 进化论的发展 ➢ 分子进化学说 ➢ 进化模型 ➢ 生命树概述 ➢ 进化树构建常用方法 ➢ 进化树检验 ➢ 进化树构建中的问题 ➢ 最近的工作
➢ 进化论的发展 ➢ 分子进化学说 ➢ 进化模型 ➢ 生命树概述 ➢ 进化树构建常用方法 ➢ 进化树检验 ➢ 进化树构建中的问题 ➢ 最近的工作
分子进化的中性学说
分子进化的中性学说
1.分子水平上的突变大多是中性的,不影响蛋白和核酸的功能; 2.中性突变不影响其对生活环境的生存适合度; 3.分子进化的速率是由中性突变的速率来决定的,即对每个蛋白或基 因来说,aa/nt的每个位点每年的替代率对所有生物是恒定的,称为分 子钟(molecular evolutionary clock)。
基于性状
距离法 UPGMA法
前提条件: 在进化过程中,每一世代发生趋异的次数相同,即碱基或氨基酸 的替换速率是均等且恒等的。
根据求得的距离系数,所有比较的分类单元的成对距离构成一个t×t方阵,即建立一 个距离矩阵M。
对于一个给定的距离矩阵,寻求最小距离值Dpq。
定义类群p和q之间的分支深度Lpq=Dpq/2。
1.达尔文的自然性”
2.新拉马克主义(neo-Lamarckism) 强调用进废退,强调功能决定结构
系统发育树 ppt

度和1/2节点与2之间的分支
长度相等,则表明物种进化
是同一速率的。
-
6
二.系统发育树的构建方法及原理
步骤:
1.选择 一个相 关序列
2.得到 多个序 列比对
3.是否具有显 是 著的序列相似 性?
最大简约法
三
集
否
大
4.是否可清晰 分辨序列相似
是 距离法
方
性? 否
法
最大似然法
-
7
1.相关序列:可以是DNA或蛋白质序列:每一类 型 有不同的程序选项,作为进化相关性指标。
优点:对多重序列排列的每一列进行分析,将 考虑所有可能的树,对其序列变化数进行分析,变 化数越多则树越不像,类似最大简约法。正是如此, 其可以通过不同谱系的突变率差异来评价树,可以 用于探索远源序列的关系,因而强于最大简约法。
缺点:计算过于复杂!
-
16
PHYLIP软件包中包括2个最大似然分析程序: 1.DNAML:用于对核苷酸序列估计系统发育关系。 2.DNAMLK:与DNAML不同在于假设存在分子钟(分 支上进化速率恒定)。
-
17
三.系统发育树的构建软件
1.PHYLIP 是一个包含了大约30个程序的软件,基本囊括了系统 发育分析的所有方面,而且是免费软件,如上面提到 的DNADIST和PROTDIST。 其处理DNA序列的软件和处理蛋白质序列的软件不同: 用最大节约法构建进化树时,DNA序列采用DNADIST 软件,蛋白质采用PROTPARS软件;用距离法构建树 时,DNA采用DNADIST软件,蛋白质采用PROTDIST 软件;用最大似然法构建树时,DNA采用DNAML、 DNAMLK,蛋白质采用PROTML或PROTMLK软件。
系统发育树的详细构建方法

cgaatagggt ctcgacttcc gtccaatggg tgcacaatga gtgggcaagc ggcgagctca 16 tgggggcttt cccggaaagg cgagctgaac gtacacaatt cgtgcggcgg tcgcaagcag 1 200 MIY sisneusn ag allegnaiJ 170136YA> …… c aggcggtga gcagtcgttt cttcgttcga agaaggacaa tggcaagctg 16
明说为行一第即(式格 ATSAF 以列序把 。列序的索搜要需贴粘于用 �框本文的大个一有都们 它。异差所有序程的用所但�多不差上面界在务服 TSALB 的供提站网些这。JBDD 的本日 和 IBE 的洲欧、IBCN 的国美、IBC 的内国如比�站网用常的务服 TSALB 供提到录登先首
。段片列序似相的长能可尽出找以 �伸延端两向核内为作并 �段片的高最度程性似相间之列序 标目和列序测检出找先首是路思本基的法算 TSALB。器务服 TSALB 的 beW 于基供提都心
)群外义定(KO → puorgtuo >> puorgni → …puo rgtuo enifeD -eerT )值数示显()eulav partstooB( slebal egde lanretni wohS -eerT
�bhpbjn.*件文树化进成生�中程过此在。关有置配机算计及以量数、度长列序与间时待等 ……gnitiaw → KO
。看查开打 weiVeerT 用以可
seerT J-N warD-seerT )6(
bhpbjn.a-cj\pmet\:C :SA EERT PILYHP EVAS bjn.a-cj\pmet\:C :SA EERT LATSULC EVAS
系统发育分析 PPT

treefile
系统发育分析
• 将treefile更名results3后,双击打开CONSENSE.EXE 工具
打开CONSENSE工具 把treefile更名为results3
系统发育分析
• 输入results3 • 修改O选项,输入18 • 默认R选项,构建无根树 • 其他设置默认,输入Y,回车 • 计算生成out文件
简约法相关工具
似然法相关工具
距离法相关工具 系统树统计学检验工具
• TreeView:
Nigerian
South Amerind
Australian
Papuan
North Amerind
Southern Chinese Korean
0.02
Nigerian Pygmy
Bantu
Japanese Finn
系统发育分析
• 输入results1 • 选择修改M选项,输入100 • 其他设置默认,输入Y,回车 • 计算生成新的outfile文件
输入results1
距离模型 是否处理多样本数 据集,默认为否 选择M,要处理多样本数据集 输入多样本数据集的样本集数目100, 与Seqboot中的设置要一致
其他设置默认,输入Y
• 计算生成out文 件
输入results2 选用的距离法 选择外类群 是否处理多样本数据集,默认为否 输入O,要设置外类群 输入18,表示是第18条序列作为外类群
选择M,要处理多样本数据集, 输入多样本数据集的样本集数目, 与前面步骤中的设置要一致
其他设置默认,输入Y
系统发育分析
outfile
构建的系统 发育树,每 个样本对应 一个系统树
系统发育分析
系统发育树构建方法及其应用

系统发育树构建方法及其应用简介:系统发育树(Phylogenetic tree)是生物学中常用的工具,用于表示不同物种之间的进化关系。
构建一个准确的系统发育树对于研究生物进化历史、分类和演化过程有着重要的意义。
本文将介绍系统发育树的构建方法以及其在生物学研究中的应用。
一、系统发育树构建方法1. 分子系统发育树构建方法分子系统发育树是通过比较不同物种基因或蛋白质序列的差异性来构建的。
常用的分子系统发育树构建方法包括:(1) 距离法(Distance-based methods):通过计算不同物种之间的序列相似性距离来构建系统发育树。
这种方法基于假设,认为进化关系越近,序列之间的相似性越高。
(2) 个体基因树法(Gene tree methods):通过基因序列的比对和进化关系的推断来构建系统发育树。
这种方法通常被用于研究基因家族在不同物种之间的进化关系。
(3) 群体基因树法(Coalescent-based methods):通过比较人口遗传学和种族学数据来构建系统发育树。
这种方法可以帮助我们理解不同群体之间的种群历史和迁移模式。
2. 形态系统发育树构建方法形态系统发育树是通过比较不同物种形态特征的异同来构建的。
常用的形态系统发育树构建方法包括:(1) 分离法(Cladistic methods):通过对比物种形态特征的共性和差异性来构建系统发育树。
这种方法基于假设,认为进化趋势是分支与分化的结果。
(2) 综合法(Integrated methods):结合形态特征和分子遗传学数据,综合分析不同物种间的形态和分子演化关系。
二、系统发育树的应用1. 生物分类学系统发育树为生物分类学提供了关键的工具。
通过构建系统发育树,我们可以清晰地了解不同物种之间的亲缘关系,进而对它们进行分类和命名。
2. 进化历史研究系统发育树可以帮助研究者重建物种的进化历史,并揭示不同物种之间的共同祖先及其衍生物的关系。
这有助于我们理解生物进化的模式和过程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1
16SrRNA片段获取
细菌培养液 DNA提取
基因组DNA
PCR扩增 16SrRNA
测序
2
序列比对
1.使用DNAstar中的SeqMan对未知菌测序得到 的两条序列进行组装连接 SeqMan使用方法:1.打开软件如图
3
2.点击Add Sequence 然后选择侧得的序列 (带有图的)
4
3.点击 Assemble 得到如下图,然后双击 Contig1
5
4.得到下图,减掉两端不好的序列,点击 Contig,再点击save consensus ,保存。
6
NCBI比对
5.将得到的序列输入到NCBI上进行比对
7
6.Blast DAN
8
7.输入组装的DNA序列
9
大家有疑问的ylogency----construct/test neighborjioning tree-----然后选择上述保存的文件.mas 然后参数选择Bootstrap method 1000 其他默 认,点击compute
14
得到系统发育树
谢谢
15
可以互相讨论下,但要小声点
10
8.选择相似的菌种
11
9.下载FASTA格式
12
10.MEGA7比对
1.打开软件点击Align-Edit/Build Alignment 然后 OK-DNA 再然后是Edit ---insert sequence from 选择序列(相似菌和目的菌,fasta格式) ----选中序列---点击Alignment ---Align by clustalW----保存(mas格式)