微量白蛋白测定试剂盒(免疫比浊法)产品技术要求丹大
尿微量白蛋白(MAU)测定标准操作程序SOP文件

其它适合的质控品
贮存条件:置2-8℃冰箱至有效期。
准备:直接使用。
质控间隔时间及限制:应视不同地区及各自实验室情况而定。质控结果应在限定的范围之内,如果超出范围,实验室应根据情况采取措施。
ABCD医院
生化实验室
文件编号:
ABCD-SOP-04-13
10.5 MAU的浓度低于3000mg/l时不会有HOOK效应。
ABCD医院
生化实验室
文件编号:
ABCD-SOP-04-13
尿微量白蛋白(MAU)测定
版序:ABCD
页码:第3页,共3页
11临床意义
白蛋白是非糖类蛋白,分子量为66,000道而顿。其在肝实质细胞中合成,每天大约为14g/天。白蛋白是血浆、脑脊液、尿液中最重要的蛋白成份(约大于50%)。尿液中少量的但是非正常的白蛋白排泌称为尿微量白蛋白。
<37mg albumin /g crea
7.2 24-hour尿液:<20mg/l
<30mg/24h
8性能指标
本法线性范围为3-120mg/dl,不准确度允许范围 ±10%,不精密度血液:CV=3.3%,尿液:CV=4.3%灵敏度为1.5mg/dl。
9注意事项
9.1血清标本出现溶血、脂血、黄疸或抗坏血酸的干扰情况参见抗干扰能力。
微量白蛋白尿可以由小球、小管及肾后性等原因引起。白蛋白是不同的类型的蛋白尿的标记蛋白。
对于选择性的肾小球性蛋白尿,排泌量约为100-3000mg/g crea。对于非选择性的肾小球性蛋白尿,其特点为高分子量蛋白的分泌增加。肾前性蛋白尿通过总蛋白与白蛋白之间的不同来确认(白蛋白少于30%)。白蛋白及微量蛋白同事增加可见于球管性蛋白尿(肾小管的重吸收少于肾小球的滤过率)。肾小球、肾小管以及间质性肾炎、糖尿病或其它原因引起的肾衰。血浆中的蛋白质具有两种功能:维持渗透压以及参与体内物质的转运。对于低水溶性物质来说,白蛋白是最重要的转运蛋白。白蛋白水平的降低可以由高血压、肝细胞合成减少、血管内分泌异常、血管内外分布异常,白蛋白丢失及降解加快等原因引起。
总局关于发布医疗器械注册单元划分指导原则的通告(2017年第187号)2017.11.23

总局关于发布医疗器械注册单元划分指导原则的通告(2017年第187号)2017年11月23日发布为加强医疗器械产品注册工作的管理和指导,进一步规范医疗器械注册申报和技术审评工作,根据《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)和《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)有关要求,国家食品药品监督管理总局组织制定了《医疗器械注册单元划分指导原则》(见附件),现予发布。
特此通告。
附件:医疗器械注册单元划分指导原则食品药品监管总局2017年11月17日2017年第187号通告附件.docx—1—附件医疗器械注册单元划分指导原则本指导原则根据《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)和《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)有关要求制定。
注册单元划分着重考虑产品的技术原理、结构组成、性能指标、适用范围及体外诊断试剂的包装规格等因素。
本指导原则包括有源医疗器械、无源医疗器械及体外诊断试剂注册单元划分的指导原则,并列举了有关注册单元划分的实例,部分要求需结合相关的注册技术审查指导原则或标准进行综合判断。
本指导原则是基于现行医疗器械注册申报工作实际情况制定的,随着法规体系的不断完善、科学技术的不断发展以及认知水平的提升,本指导原则相关内容也将适时进行调整。
一、有源医疗器械注册单元划分指导原则(一)技术原理不同的有源医疗器械原则上划分为不同的注册单元。
(二)技术原理相同,但产品主要结构、组成的不同对安全有效性有影响的相同种类有源医疗器械原则上划分为不同注册单元。
(三)当产品性能指标差异导致适用范围或作用机理不同时,原则上划分为不同的注册单元。
—2—(四)技术原理和设计结构相同,但产品适用范围有实质不同的相同种类有源医疗器械,原则上划分为不同的注册单元。
(五)与有源医疗器械配合/组合使用的无源类耗材原则上与该有源医疗器械划分为不同的注册单元。
尿微量白蛋白测定卡(胶体金免疫层析法)产品技术要求yicheng

尿微量白蛋白测定卡(胶体金免疫层析法)适用范围:本产品用于体外定量检测人体尿液中的白蛋白水平。
1.1 产品型号1.2 产品规格1支/袋×1袋/盒;1支/袋×2袋/盒;1支/袋×5袋/盒;1支/袋×10袋/盒;1支/袋×20袋/盒。
1.3 产品组成每盒含有测定卡、1支校准卡、尿杯(选配)、滴管(选配)。
每个铝箔袋中包括1支尿微量白蛋白测定卡和1包干燥剂。
尿微量白蛋白测定卡由样品垫(玻璃纤维素膜)、金标垫(氯金酸约14μg、鼠抗人白蛋白单克隆抗体约0.6μg、无机盐约60%)、硝酸纤维素膜(T线包被有人血清白蛋白;C线包被有羊抗鼠IgG)、吸水纸、背板(PVC板)。
2.1 外观2.1.1 外观平整,材料附着牢固,内容齐全。
2.1.2 包装完整,标签清晰。
2.2 物理检查2.2.1 膜条宽度应不低于3.0mm。
2.2.2 液体移行速度应不低于10mm/min。
2.3 准确度应在85%~115%范围内。
2.4 重复性重复测试低、中、高浓度后样本,测试结果的变异系数CV≤20%。
2.5 线性在[10,30]mg/L的范围内,线性相关系数(r)应不小于0.990。
样品中白蛋白浓度为[10,20)mg/L时,测量平均值与理论值的绝对偏差不超过±3.0mg/L;样品中白蛋白浓度[20,30]mg/L时,测量平均值与理论值的相对偏差不超过±15%。
2.6 空白限空白限应不超过3mg/L。
2.7 批间差批间相对极差R≤15%。
2.8 特异性用下列浓度的干扰物加入后测定结果的相对偏差应不超过±15%。
2.9 稳定性4℃~30℃保存,有效期(15个月)到期后三个月内的留样,应符合2.3、2.4、2.5、2.6的要求。
2.10 校准信息溯源性校准卡中的校准曲线,按照GB/T 21415-2008 《体外诊断医疗器械生物样品中量的测量标准品和控制物质赋值的计量学溯源性》的要求溯源,溯源到怡成公司企业校准品,选用罗氏尿微量白蛋白测试系统对校准品进行比对赋值。
普利莱基因技术微量蛋白质定量试剂盒 (BCA 法) 使用说明说明书

微量蛋白质定量试剂盒(BCA法)使用说明P1513描述:Bicinchoninic acid(BCA)法是近来广为应用的蛋白定量方法。
其原理与Lowery法蛋白定量相似,即在碱性环境下蛋白质与Cu2+络合并将Cu2+还原成Cu1+。
BCA与Cu1+结合形成稳定的紫蓝色复合物,在562 nm处有高的光吸收值并与蛋白质浓度成正比,据此可测定蛋白质浓度。
与Lowery法相比,BCA蛋白测定方法灵敏度高,操作简单,试剂及其形成的颜色复合物稳定性俱佳,并且受干扰物质影响小。
与Bradford 法相比,BCA法的显著优点是不受去垢剂的影响。
组成与储存:(1)BCA Reagent100ml,室温保存;(2)Cu Reagent2.5ml,室温保存;(3)BSA standard4mg/ml1ml,−20ºC冻存。
1.5年有效。
可进行500次微板(microplate)测定或100次1ml比色杯测定。
所需设备:比色计、酶标仪、或微板比色仪,最佳工作波长562nm,可在540-590nm之间。
工作溶液(Working Reagent,WR)配制:将50体积BCA Reagent与1体积Cu Reagent混合即为WR工作试剂,呈嫩绿色,立即使用或者4℃保存不超过一天。
标准蛋白溶液配制:用双蒸水、0.9%生理盐水、PBS、或与待测蛋白样品匹配之缓冲液进行倍比稀释,得到BSA标准溶液80、40、20、10、5、2.5、1.25µg/ml。
蛋白测定微量蛋白质浓度线性检测范围为1-100µg/ml。
标准测定用1cm光程玻璃或塑料比色皿,反应终体积1.2ml,用比色计测定。
微板测定用96孔板,反应终体积240µl,用酶标仪、微板比色仪测定。
1.标准测定:将0.2ml标准品或待测样本与1ml WR工作溶液混合。
微板测定:将40µl标准品或待测样本与200µl WR工作溶液混合。
免疫球蛋白A测定试剂盒(免疫比浊法)产品技术要求xksm

免疫球蛋白A测定试剂盒(免疫比浊法)
组成:
试剂1(R1)主要成分:Tris缓冲液(pH 6.0~9.0)4mmol/L;
试剂2(R2)主要成分:抗IgA抗血清适量。
适用范围:用于体外定量检测人血清中免疫球蛋白A的浓度。
2.1 外观
外包装完整无破损,标签清晰;试剂1应为无色透明溶液;试剂2应为无色透明溶液。
2.2 净含量
应不低于试剂瓶标示装量。
2.3 试剂空白
在340nm处测定试剂空白吸光度,应≤0.20。
2.4 分析灵敏度
测定浓度为450mg/dl的样品,吸光度变化(ΔA)应不低于0.15。
2.5 线性
2.5.1在[18.0,621.0]mg/dl范围内,线性回归的相关系数应不低于0.990;
2.5.2测试浓度[50.0,621.0]mg/dl的样品,相对偏差应不超过±15%;测试浓度[18.0,50.0)mg/dl的样品,绝对偏差应不超过±7.5mg/dl。
2.6 重复性
2.6.1 批内重复性
变异系数(CV)应不超过5%。
2.6.2 批间差
对同一份样品进行重复测定,相对极差(R)应不超过10%。
2.7 准确度
回收率应在85%~115% 范围内。
2.8空白限
试剂空白限为18mg/dL。
2.9 稳定性
原包装试剂在2℃~8℃条件下有效期为18个月,取到效期后6个月内的试剂盒检测,应符合本技术要求2.1、2.3、2.4、2.5、2.6.1、2.7、2.8之规定。
白蛋白(ALB)测定试剂(盒)(溴甲酚绿法)产品技术要求新产业

白蛋白(ALB)测定试剂(盒)(溴甲酚绿法)
2.性能指标
2.1外观
试剂应为清澈透明的液体,无沉淀、悬浮物和絮状物。
2.2净含量
试剂装量的装量应按表 2,液体装量的最大允许负偏差应为 5%。
表 2 净含量
2.3试剂空白吸光度
试剂(盒)以纯化水为空白在 37 ℃±1℃ 、630 nm 波长、1 cm 光径条件下,试剂空白吸光度应≤0.40。
2.4分析灵敏度
试剂(盒)测试 43.7 g/L 的被测物时,吸光度变化(ΔA)应在 1.00~2.66 的范围内。
2.5线性范围
试剂(盒)在(0~60) g/L范围内的分析性能应符合如下要求:
a)线性相关系数r≥0.990;
b)(0~10) g/L 范围内,线性绝对偏差应在±1 g/L以内;(10~60) g/L 范围内,线性
相对偏差应在±10%以内。
2.6测量精密度
2.6.1重复性
用校准品重复测试所得结果的变异系数CV≤4%。
2.6.2批间差
试剂(盒)批间相对偏差R≤6%。
2.7准确度
校准品的相对偏差 B 在±10%以内。
2.8分析特异性
血红蛋白浓度在 400 mg/dL 内、甘油三酯浓度在 500 mg/dL 内、抗坏血酸浓度在 30 mg/dL 内、胆红素浓度在 40 mg/dL 内,对试剂检测结果的偏差影响应在±10%以内。
免疫球蛋白(IgG、IgM、IgA)检测试剂盒(免疫比浊法)产品技术要求

医疗器械产品技术要求编号:免疫球蛋白(IgG、IgA、IgM)检测试剂盒(免疫比浊法)1.产品型号/规格及其划分说明序号规格12×200Tests2R1:2×80ml、R2:2×20ml3R1:2×60ml、R2:2×15ml4R1:2×40ml、R2:2×10ml5R1:2×50ml、R2:1×25ml6校准品(选配):1×1ml2.性能指标2.1外观试剂R1溶液应无色、无颗粒、无杂质、无沉淀和悬浮物;试剂R2溶液应呈淡黄色、无颗粒、无杂质、无沉淀和悬浮物;校准品为米白色至浅黄色冻干粉末。
2.2净含量试剂盒各试剂装量应不小于标示值。
2.3试剂空白吸光度应为≤0.4,IgA、用蒸馏水作为样品加入试剂测试,IgG试剂空白吸光度A570nm应为≤0.4。
IgM试剂空白吸光度A340nm2.4分析灵敏度测试IgG:4.85g/L、IgA:0.94g/L、IgM:0.47g/L被测物时,吸光度差值(△A)IgG应不小于0.20、IgA应不小于0.20、IgM应不小于0.05。
2.5线性范围IgG:在(0~35.0)g/L范围内,其线性相关系数r≥0.990;浓度≥5.0g/L 时,相对偏差≤20%;浓度<5.0g/L时,绝对偏差≤1.0g/L。
IgA:在(0~8.0)g/L范围内,其线性相关系数r≥0.990;浓度≥1.3g/L 时,相对偏差≤20%;浓度<1.3g/L时,绝对偏差≤0.4g/L。
IgM:在(0~4.8)g/L范围内,其线性相关系数r≥0.990;浓度≥0.4g/L时,相对偏差≤20%;浓度<0.4g/L时,绝对偏差≤0.2g/L。
2.6测量精密度2.6.1重复性用控制血清重复测试所得结果的重复性(变异系数,CV)应≤6.0%。
2.6.2批间差批间差应≤10.0%。
2.7准确度用参考物质进行测试,其相对偏差应≤20.0%。
转铁蛋白检测试剂盒(免疫比浊法)说明书

转铁蛋白检测试剂盒(免疫比浊法)说明书转铁蛋白检测试剂盒(免疫比浊法)说明书【产品名称】通用名称:转铁蛋白检测试剂盒(免疫比浊法)英文名称:TRF Kit【包装规格】R1:R2 ,1×30ml;1×10ml1×60ml;1×20ml 2×60ml;2×20ml【预期用途】转铁蛋白检测试剂盒临床上用于定量测定人体血清中转铁蛋白的含量。
转铁蛋白(TRF)连接上铁离子之后可以防止铁中毒以及通过肾的流失。
其水平的升高常见于铁缺乏症、怀孕、雌性激素的控制以及类脂肪的肾病。
其水平的降低常见于睾丸激素的控制、感染、急性的炎症、某些类型的肾炎、血色素缺失、急性的疟疾以及营养不良。
【检验原理】人体中的转铁蛋白与试剂中抗人转铁蛋白抗体在缓冲液中快速形成抗原抗体复合物,使反应液出现浊度。
当反应液中保持抗体过剩时,形成的复合物随抗原量增加而增加,反应液的浊度亦随之增加,在340nm以终点法检测吸光度变化,与校准品对照,即可计算出未知蛋白的含量。
【主要组成成分】组成主要成分R1 NaH2PO4缓冲液R2 抗人转铁蛋白抗体注意不同批号的试剂盒的组分不能混用。
校准品:用户自行购买利德曼公司的多项高值免疫标准液,标准值见说明书;质控品:用户自行购买利德曼公司多项免疫质控血清,质控值见说明书;【储存条件及有效期】1.包装试剂均应在2?,8?避光储存,可稳定至标签所示失效日期;2( 试剂有效期为12个月;3( 开瓶有效期:10天(开瓶后在2?,8?保存);【适用仪器】包装规格适用机型1×30ml;1×10ml 日立7060、1×60ml;1×20ml 日立7170、东芝-40 2×60ml;2×20ml 日立7020、奥林巴斯AU640、贝克曼CX4 【样本要求】1、标本为离心或分离除去血液凝块的新鲜血清。
2、血清样本在2~8?储存不超过一周。
医疗器械注册单元划分指导原则

医疗器械注册单元划分指导原则本指导原则根据《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)和《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)有关要求制定。
注册单元划分着重考虑产品的技术原理、结构组成、性能指标、适用范围及体外诊断试剂的包装规格等因素。
本指导原则包括有源医疗器械、无源医疗器械及体外诊断试剂注册单元划分的指导原则,并列举了有关注册单元划分的实例,部分要求需结合相关的注册技术审查指导原则或标准进行综合判断。
本指导原则是基于现行医疗器械注册申报工作实际情况制定的,随着法规体系的不断完善、科学技术的不断发展以及认知水平的提升,本指导原则相关内容也将适时进行调整。
一、有源医疗器械注册单元划分指导原则(一)技术原理不同的有源医疗器械原则上划分为不同的注册单元。
(二)技术原理相同,但产品主要结构、组成的不同对安全有效性有影响的相同种类有源医疗器械原则上划分为不同注册单元。
(三)当产品性能指标差异导致适用范围或作用机理不同时,原则上划分为不同的注册单元。
(四)技术原理和设计结构相同,但产品适用范围有实质不同的相同种类有源医疗器械,原则上划分为不同的注册单元。
(五)与有源医疗器械配合/组合使用的无源类耗材原则上与该有源医疗器械划分为不同的注册单元。
(六)适用范围相同,需要配合使用但各自独立的有源医疗器械原则上划分为不同的注册单元。
体外诊断设备以系统申报的情况例外。
(七)有源医疗器械附件与连接使用的主机原则上作为同一个注册单元申报。
对于单独注册的作为医疗器械管理的附件,不同预期用途的附件原则上划分为不同的注册单元,有源和无源附件原则上划分为不同的注册单元。
如果有源和无源附件在同一个无菌包装内,原则上划分为同一注册单元。
(八)适用范围、产品性能和结构组成基本相同的不同型号医疗器械,原则上划分为同一注册单元。
但如果各型号间在适用范围、性能、结构方面差异较大,则划分为不同的注册单元。
尿微量白蛋白测定试剂盒(免疫比浊法)产品技术要求zhongshengbeikong

尿微量白蛋白测定试剂盒(免疫比浊法)适用范围:本试剂盒与ABBOTT ARCHITECT c4000/c8000/c16000全自动生化分析仪配套使用,用于体外定量测定人尿液中白蛋白的浓度。
1.1规格液体双剂型试剂1(R1):60mL×2,试剂2(R2): 6mL×2,校准品(4个浓度):1mL×4/套;试剂1(R1):65mL×2,试剂2(R2): 10mL×2,校准品(4个浓度):1mL×4/套。
1.2规格划分说明根据净含量划分规格。
1.3主要组成成分试剂盒由试剂1(R1)液体、试剂2(R2)液体及校准品液体组成。
1.3.1 试剂1(R1)液体主要组分:三羟甲基氨基甲烷(pH 7.5) 200mmol/L1.3.2 试剂2(R2)液体主要组分:羊抗人白蛋白抗体浓度根据效价而定1.3.3 校准品液体主要组分:PBS缓冲液基质(4个浓度)白蛋白①0 mg/L、②30mg/L~50mg/L、③70mg/L~100mg/L、④ 160mg/L~185mg/L。
(每批定值)2.1 外观试剂盒中各组件的外观应满足:a) 试剂1(R1)应为无色透明溶液,无杂质、无絮状物,外包装完整无破损;b) 试剂2(R2)应为浅黄色粘稠溶液,无杂质、无絮状物,外包装完整无破损;c) 校准品应为无色液体,无杂质、无絮状物,外包装完整无破损。
2.2 净含量液体试剂净含量应不少于标示值。
2.3 试剂空白吸光度在波长340nm处(光径1cm),试剂空白吸光度值(A)应≤0.2。
2.4 准确度用中生试剂和已上市同类试剂分别测定40个在测定范围内不同浓度的样本,在[20,180]mg/L检测范围内,比对两组数据的相关系数(r)及测值的偏差,要求r≥0.975,相对偏差应不超过±15%。
2.5 分析灵敏度对应于浓度为50mg/L的MALB所引起的吸光度变化差值(△A)的绝对值应在0.15~0.60的范围内。
cusabio 微量白蛋白尿(MAU ALB)检测试剂盒使用说明书

Human microalbunminuria(MAU/ALB) ELISA kit Catalog Number. CSB-E08970hFor the quantitative determination of human microalbunminuria(MAU/ALB) concentrations in serum, plasma, urine.This package insert must be read in its entirety before using this product.If You Have ProblemsTechnical Service Contact informationPhone: 86-27-87582341Fax: 86-27-87196150Email:****************Web: In order to obtain higher efficiency service, please ready to supply the lot numberof the kit to us (found on the outside of the box).1PRINCIPLE OF THE ASSAYThis assay employs the competitive inhibition enzyme immunoassay technique. Antibody specific for MAU has been pre-coated onto a microplate. Standards and samples are pipetted into the wells with biotin-conjugated MAU. A competitive inhibition reaction is launched between MAU (Standards or samples) and biotin-conjugated MAU with the pre-coated MAU antibody. After washing, avidin conjugated Horseradish Peroxidase (HRP) is added to the wells. Following a wash to remove any unbound reagent, a substrate solution is added to the wells and color develops in opposite to the amount of MAU bound in the initial step. The color development is stopped and the intensity of the color is measured.DETECTION RANGE0.078 µg/ml-5 µg/ml.SENSITIVITYThe minimum detectable dose of human MAU is typically less than 0.019 µg/ml. The sensitivity of this assay, or Lower Limit of Detection (LLD) was defined as the lowest human MAU concentration that could be differentiated from zero.SPECIFICITYThis assay has high sensitivity and excellent specificity for detection of human MAU. No significant cross-reactivity or interference between human MAU and analogues was observed.Note: Limited by current skills and knowledge, it is impossible for us to complete the cross-reactivity detection between human MAU and all the analogues, therefore, cross reaction may still exist.2PRECISIONIntra-assay Precision (Precision within an assay): CV%<8%Three samples of known concentration were tested twenty times on one plate to assess.Inter-assay Precision (Precision between assays):CV%<10%Three samples of known concentration were tested in twenty assays to assess.LIMITATIONS OF THE PROCEDUREFOR RESEARCH USE ONLY. NOT FOR USE IN DIAGNOSTIC PROCEDURES.The kit should not be used beyond the expiration date on the kit label.Do not mix or substitute reagents with those from other lots or sources.If samples generate values higher than the highest standard, dilute the samples with Sample Diluent and repeat the assay.Any variation in Sample Diluent, operator, pipetting technique, washing technique, incubation time or temperature, and kit age can cause variation in binding.This assay is designed to eliminate interference by soluble receptors, binding proteins, and other factors present in biological samples. Until all factors have been tested in the Immunoassay, the possibility of interference cannot be excluded.3MATERIALS PROVIDEDReagents QuantityAssay plate (12 x 8 coated Microwells) 1(96 wells) Standard (Freeze dried) 2Biotin-conjugate (100 x concentrate) 1 x 60 µlHRP-avidin (100 x concentrate) 1 x 120 µlBiotin-conjugate Diluent 1 x 10 mlHRP-avidin Diluent 1 x 20 ml Sample Diluent 2 x 20 mlWash Buffer (25 x concentrate) 1 x 20 mlTMB Substrate 1 x 10 mlStop Solution 1 x 10 ml Adhesive Strip (For 96 wells) 4Instruction manual 1STORAGEUnopenedkitStore at 2 - 8°C. Do not use the kit beyond the expiration date.Opened kitCoated assayplateMay be stored for up to 1 month at 2 - 8°C.Try to keep it in a sealed aluminum foil bag,and avoid the damp.Standard May be stored for up to 1 month at 2 - 8° C.If don’t make recent use, better keep it storeat -20°C.HRP-avidinBiotin-conjugateBiotin-conjugateDiluentMay be stored for up to 1 month at 2 - 8°C. HRP-avidinDiluentSample DiluentWash BufferTMB SubstrateStop Solution*Provided this is within the expiration date of the kit.4OTHER SUPPLIES REQUIREDMicroplate reader capable of measuring absorbance at 450 nm, with the correction wavelength set at 540 nm or 570 nm.An incubator which can provide stable incubation conditions up to 37°C±0.5°C.Squirt bottle, manifold dispenser, or automated microplate washer.Absorbent paper for blotting the microtiter plate.100ml and 500ml graduated cylinders.Deionized or distilled water.Pipettes and pipette tips.Test tubes for dilution.PRECAUTIONSThe Stop Solution provided with this kit is an acid solution. Wear eye, hand, face, and clothing protection when using this material.5SAMPLE COLLECTION AND STORAGESerum Use a serum separator tube (SST) and allow samples to clot for30 minutes before centrifugation for 15 minutes at 1000 x g, 2 - 8°C.Remove serum and assay immediately or aliquot and store samples at -20°C or -80°C. Avoid repeated freeze-thaw cycles. Centrifuge the sample again after thawing before the assay.Plasma Collect plasma using EDTA, or heparin as an anticoagulant.Centrifuge for 15 minutes at 1000 x g, 2 - 8°C within 30 minutes of collection. Assay immediately or aliquot and store samples at -20°C or -80°C. Avoid repeated freeze-thaw cycles. Centrifuge the sample again after thawing before the assay.Urine Use a sterile container to collect urine samples. Remove any particulates by centrifugation for 15 minutes at 1000xg, 2 - 8°C and assay immediately or aliquot and store samples at -20°C or -80°C. Avoid repeated freeze-thaw cycles. Centrifuge again before assaying to remove any additional precipitates that may appear after storage.SAMPLE PREPARATIONRecommend to dilute the serum or plasma samples 50000-fold before test.The suggested 50000-fold dilution can be achieved by adding 2µl sample to 398µl of normal saline. Complete the 50000-fold dilution by adding 2µl of this solution to 498µl of Sample Diluent. The recommended dilution factor is for reference only. The optimal dilution factor should be determined by users according to their particular experiments.Recommend to dilute the urine samples with Sample Diluent(1:40) before test. The suggested 40-fold dilution can be achieved by adding 6µl sample to 234µl of Sample Diluent. The recommended dilution factor is for reference only. The optimal dilution factor should be determined by users according to their particular experiments6Note:1. CUSABIO is only responsible for the kit itself, but not for the samplesconsumed during the assay. The user should calculate the possible amount of the samples used in the whole test. Please reserve sufficient samples in advance.2. Samples to be used within 5 days may be stored at 2-8°C, otherwisesamples must be stored at -20°C (≤1month) or -80°C (≤2month) to avoid loss of bioactivity and contamination.3. Grossly hemolyzed samples are not suitable for use in this assay.4. If the samples are not indicated in the manual, a preliminary experiment todetermine the validity of the kit is necessary.5. Please predict the concentration before assaying. If values for these arenot within the range of the standard curve, users must determine the optimal sample dilutions for their particular experiments.6. Tissue or cell extraction samples prepared by chemical lysis buffer maycause unexpected ELISA results due to the impacts of certain chemicals.7. Owing to the possibility of mismatching between antigen from otherresource and antibody used in our kits (e.g., antibody targets conformational epitope rather than linear epitope), some native or recombinant proteins from other manufacturers may not be recognized by our products.8. Influenced by the factors including cell viability, cell number and alsosampling time, samples from cell culture supernatant may not be detected by the kit.9. Fresh samples without long time storage are recommended for the test.Otherwise, protein degradation and denaturalization may occur in those samples and finally lead to wrong results.7REAGENT PREPARATIONNote:Kindly use graduated containers to prepare the reagent. Please don't prepare the reagent directly in the Diluent vials provided in the kit. Bring all reagents to room temperature (18-25°C) before use for 30min.Prepare fresh standard for each assay. Use within 4 hours and discard after use.Making serial dilution in the wells directly is not permitted.Please carefully reconstitute Standards according to the instruction, and avoid foaming and mix gently until the crystals have completely dissolved.To minimize imprecision caused by pipetting, use small volumes and ensure that pipettors are calibrated. It is recommended to suck more than 10µl for once pipetting.Distilled water is recommended to be used to make the preparation for reagents. Contaminated water or container for reagent preparation will influence the detection result.1. Biotin-conjugate (1x) - Centrifuge the vial before opening.Biotin-conjugate requires a 100-fold dilution. A suggested 100-fold dilution is 10 µl of Biotin-conjugate + 990 µl of Biotin-conjugate Diluent.2. HRP-avidin (1x) - Centrifuge the vial before opening.HRP-avidin requires a 100-fold dilution. A suggested 100-fold dilution is 10 µl of HRP-avidin + 990 µl of HRP-avidin Diluent.3. Wash Buffer(1x)- If crystals have formed in the concentrate, warm up toroom temperature and mix gently until the crystals have completely dissolved. Dilute 20 ml of Wash Buffer Concentrate (25 x) into deionized or distilled water to prepare 500 ml of Wash Buffer (1 x).894.StandardCentrifuge the standard vial at 6000-10000rpm for 30s.Reconstitute the Standard with 1.0 ml of Sample Diluent . Do not substitute other diluents. This reconstitution produces a stock solution of 5 µg/ml. Mix the standard to ensure complete reconstitution and allow the standard to sit for a minimum of 15 minutes with gentle agitation prior to making dilutions.Pipette 150 µl of Sample Diluent into each tube (S0-S6). Use the stock solution to produce a 2-fold dilution series (below). Mix each tube thoroughly before the next transfer. The undiluted Standard serves as the high standard (5 µg/ml). Sample Diluent serves as the zero standard (0 µg/ml).Tube S7 S6 S5S4 S3 S2 S1 S0 µg/ml52.51.250.6250.3120.1560.078ASSAY PROCEDUREBring all reagents and samples to room temperature before use. Centrifuge the sample again after thawing before the assay.It is recommended that all samples and standards be assayed in duplicate.1. Prepare all reagents, working standards, and samples as directed in theprevious sections.2. Refer to the Assay Layout Sheet to determine the number of wells to beused and put any remaining wells and the desiccant back into the pouch and seal the ziploc, store unused wells at 4°C.3. Set a Blank well without any solution.4. Add 50µl of standard and sample per well.5. Add 50µl of Biotin-conjugate(1x) to each well(not to Blank well). Coverwith a new adhesive strip. Incubate for 60 minutes at 37°C.(Biotin-conjugate(1x) may appear cloudy. Warm up to room temperature and mix gently until solution appears uniform.)6. Aspirate each well and wash, repeating the process two times for a total ofthree washes. Wash by filling each well with Wash Buffer (200µl) using a squirt bottle, multi-channel pipette, manifold dispenser, or autowasher, and let it stand for 2 minutes, complete removal of liquid at each step is essential to good performance. After the last wash, remove any remaining Wash Buffer by aspirating or decanting. Invert the plate and blot it against clean paper towels.7. Add 100µl of HRP-avidin(1x) to each well(not to Blank well). Cover themicrotiter plate with a new adhesive strip. Incubate for 60 minutes at 37°C.8. Repeat the aspiration/wash process for five times as in step 6.9. Add 90µl of TMB Substrate to each well. Incubate for 20 minutes at 37°C.Protect from light.10. Add 50µl of Stop Solution to each well, gently tap the plate to ensurethorough mixing.1011. Determine the optical density of each well within 5 minutes, using amicroplate reader set to 450 nm. If wavelength correction is available, set to 540 nm or 570 nm. Subtract readings at 540 nm or 570 nm from the readings at 450 nm. This subtraction will correct for optical imperfections in the plate. Readings made directly at 450 nm without correction may be higher and less accurate.*Samples may require dilution. Please refer to Sample Preparation section. Note:1. The final experimental results will be closely related to validity of theproducts, operation skills of the end users and the experimental environments.2. Samples or reagents addition: Please use the freshly prepared Standard.Please carefully add samples to wells and mix gently to avoid foaming. Do not touch the well wall as possible. For each step in the procedure, total dispensing time for addition of reagents or samples to the assay plate should not exceed 10 minutes. This will ensure equal elapsed time for each pipetting step, without interruption. Duplication of all standards and specimens, although not required, is recommended. To avoid cross-contamination, change pipette tips between additions of each standard level, between sample additions, and between reagent additions.Also, use separate reservoirs for each reagent.3. Incubation: To ensure accurate results, proper adhesion of plate sealersduring incubation steps is necessary. Do not allow wells to sit uncovered for extended periods between incubation steps. Once reagents have been added to the well strips, DO NOT let the strips DRY at any time during the assay. Incubation time and temperature must be observed.4. Washing: The wash procedure is critical. Complete removal of liquid ateach step is essential to good performance. After the last wash, remove any remaining Wash Solution by aspirating or decanting and remove any drop of water and fingerprint on the bottom of the plate. Insufficient washing will result in poor precision and falsely elevated absorbance reading. When using an automated plate washer, adding a 30 second soak period following the addition of wash buffer, and/or rotating the plate 180 degrees between wash steps may improve assay precision.115. Controlling of reaction time: Observe the change of color after adding TMBSubstrate (e.g. observation once every 10 minutes), TMB Substrate should change from colorless or light blue to gradations of blue. If the color is too deep, add Stop Solution in advance to avoid excessively strong reaction which will result in inaccurate absorbance reading.6. TMB Substrate is easily contaminated. TMB Substrate should remaincolorless or light blue until added to the plate. Please protect it from light.7. Stop Solution should be added to the plate in the same order as the TMBSubstrate. The color developed in the wells will turn from blue to yellow upon addition of the Stop Solution. Wells that are green in color indicate that the Stop Solution has not mixed thoroughly with the TMB Substrate.1213ASSAY PROCEDURE SUMMARY*Samples may require dilution. Please refer to Sample Preparation section.CALCULATION OF RESULTSUsing the professional soft "Curve Expert" to make a standard curve is recommended, which can be downloaded from our web.Average the duplicate readings for each standard and sample and subtract the average optical density of Blank.Create a standard curve by reducing the data using computer software capable of generating a four parameter logistic (4-PL) curve-fit. As an alternative, construct a standard curve by plotting the mean absorbance for each standard on the x-axis against the concentration on the y-axis and draw a best fit curve through the points on the graph. The data may be linearized by plotting the log of the MAU concentrations versus the log of the O.D. and the best fit line can be determined by regression analysis. This procedure will produce an adequate but less precise fit of the data.If samples have been diluted, the concentration read from the standard curve must be multiplied by the dilution factor.14人尿微量白蛋白(MAU/ALB)酶联免疫试剂盒使用说明书【产品编号】CSB-E08970h【预期应用】ELISA法定量测定人血清、血浆、尿液中MAU含量。
【指导原则】医疗器械注册单元划分指导原则(2017年第187号)

附件医疗器械注册单元划分指导原则本指导原则根据《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)和《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)有关要求制定。
注册单元划分着重考虑产品的技术原理、结构组成、性能指标、适用范围及体外诊断试剂的包装规格等因素。
本指导原则包括有源医疗器械、无源医疗器械及体外诊断试剂注册单元划分的指导原则,并列举了有关注册单元划分的实例,部分要求需结合相关的注册技术审查指导原则或标准进行综合判断。
本指导原则是基于现行医疗器械注册申报工作实际情况制定的,随着法规体系的不断完善、科学技术的不断发展以及认知水平的提升,本指导原则相关内容也将适时进行调整。
一、有源医疗器械注册单元划分指导原则(一)技术原理不同的有源医疗器械原则上划分为不同的注册单元。
(二)技术原理相同,但产品主要结构、组成的不同对安全有效性有影响的相同种类有源医疗器械原则上划分为不同注册单元。
(三)当产品性能指标差异导致适用范围或作用机理不同时,原则上划分为不同的注册单元。
—1—(四)技术原理和设计结构相同,但产品适用范围有实质不同的相同种类有源医疗器械,原则上划分为不同的注册单元。
(五)与有源医疗器械配合/组合使用的无源类耗材原则上与该有源医疗器械划分为不同的注册单元。
(六)适用范围相同,需要配合使用但各自独立的有源医疗器械原则上划分为不同的注册单元。
体外诊断设备以系统申报的情况例外。
(七)有源医疗器械附件与连接使用的主机原则上作为同一个注册单元申报。
对于单独注册的作为医疗器械管理的附件,不同预期用途的附件原则上划分为不同的注册单元,有源和无源附件原则上划分为不同的注册单元。
如果有源和无源附件在同一个无菌包装内,原则上划分为同一注册单元。
(八)适用范围、产品性能和结构组成基本相同的不同型号医疗器械,原则上划分为同一注册单元。
但如果各型号间在适用范围、性能、结构方面差异较大,则划分为不同的注册单元。
关于印发体外诊断试剂质量管理体系考核范围有效覆盖判定原则及认定程序的通知(国食药监械[2009]320号)
![关于印发体外诊断试剂质量管理体系考核范围有效覆盖判定原则及认定程序的通知(国食药监械[2009]320号)](https://img.taocdn.com/s3/m/b731d15d804d2b160b4ec019.png)
关于印发体外诊断试剂质量管理体系考核范围有效覆盖判定原则及认定程序的通知国食药监械[2009]320号2009年06月15日发布各省、自治区、直辖市食品药品监督管理局(药品监督管理局):为进一步明确《体外诊断试剂质量管理体系考核实施规定(试行)》(国食药监械〔2007〕239号)有关质量管理体系考核报告中考核范围有效覆盖问题,国家局组织制定了《体外诊断试剂质量管理体系考核范围有效覆盖判定原则及认定程序》,现予印发,自印发之日起施行。
国家食品药品监督管理局二○○九年六月十五日体外诊断试剂质量管理体系考核范围有效覆盖判定原则及认定程序一、体系覆盖原则(一)原理相同、生产工艺和控制过程相同的品种可以覆盖。
根据原理、生产工艺和控制过程相同原则将体外诊断试剂分为16个体系考核类型(见附表1)。
(二)原理、工艺和控制过程相同,类别不同应有条件覆盖:1.Ⅲ类通过,可以覆盖Ⅱ类,Ⅱ类通过不能覆盖Ⅲ类2.不同体系考核机构考核的品种不能相互覆盖(三)原理、工艺相同,品种性质和控制过程不同应区别对待:病原微生物、激素、毒品相关试剂之间原则上不能相互覆盖,与其他类试剂也不能相互覆盖。
当试剂组分中具有污染性、传染性及高风险生物活性物料的制备或操作采取了必要的防护措施(如至少在万级环境下进行,使用单独的空气净化系统,与相邻区域保持负压等),则其它部分可以覆盖。
(四)厂房、设施和设备应与体外诊断试剂生产的品种数量和/或规模相适应。
二、体系覆盖补充说明(一)在原理、生产工艺和控制过程相同的情况下,类别高的可以覆盖类级别低的,如:属于Ⅲ类的“C EA定量测定试剂盒(酶联免疫法)”质量管理体系考核范围可以覆盖属于Ⅱ类的“雌二醇(E2)检测试剂(微粒子化学发光法)”。
(二)考虑不同的机构管理权限不同,为了避免管理权限交叉可能引起的混乱,规定不同的机构考核的品种不能相互覆盖。
例如:属于Ⅲ类、由国家局认证管理中心负责考核的过敏原类试剂如“过敏原IgE 测定试剂盒(微粒子化学发光法)”,将不能覆盖同属Ⅲ类且原理、生产工艺和控制过程相同的由省局负责考核的“Beta2-微球蛋白酶免试剂盒”。
尿微量白蛋白质控品产品技术要求mairui

2性能指标
2.1外观和性状
2.1.1颜色性状
质控品应为液体,无沉淀、悬浮物和絮状物。
2.1.2包装
分装瓶应为白色硬质塑料瓶,盖有塑料外盖;盒贴、瓶贴、标签标识应完整、清晰。
塑料外盖应无明显划痕、崩缺。
2.2装量
液体质控品的含量应不少于标示值。
2.3均一性
2.3.1瓶内均一性
质控品瓶内均一性应不大于表 2 的要求(SD 和CV 值满足其一即可)。
表 2 质控品瓶内均一性要求
2.3.2瓶间均一性
质控品瓶间均一性应不大于表3 的要求(SD 和CV 值满足其一即可)。
表3 质控品瓶间均一性要求
1
2.4参考值及参考范围
每批质控品均应提供本批产品的参考值表。
经迈瑞校准品校准的BS 系列生化分析仪及配套试剂对质控品进行测定,测定结果应在给定的参考范围内。
2。
艾威德 AS-420 AS-660 AS-1200 微球免疫比浊法 β2 微球蛋白(β2-MG)测定

β2微球蛋白(β2-MG)测定试剂盒(胶乳免疫比浊法)说明书【产品名称】β2微球蛋白(β2-MG)测定试剂盒(胶乳免疫比浊法)【包装规格】a)试剂1:1×20mL试剂2:1×5mLb)试剂1:2×40mL试剂2:1×20mLc)试剂1:4×60mL试剂2:2×30mLd)试剂1:2×80mL试剂2:2×20mL【预期用途】用于体外定量测定人血清中β2-微球蛋白的含量。
血清β2-MG浓度受体内合成速度与尿液排泄率的影响。
一些导致β2-MG合成增加的疾病会使血清β2-MG浓度升高。
在急性肾炎、慢性肾炎及慢性肾功能不全等疾病时,因肾小球滤过率(GFR)及肾血流量降低,导致β2-MG 滤过减少,血清β2-MG浓度升高。
自身免疫疾病时,尤其是系统性红斑狼疮(SLE)活动期,血清β2-MG往往升高。
β2-微球蛋白主要来自于肾脏[1]。
临床上测定β2-微球蛋白常用于急性肾炎、慢性肾炎及慢性肾功能不全等疾病时的辅助诊断。
【检验原理】样本中的β2-MG与胶乳颗粒的抗β2-MG抗体特异性反应,形成免疫复合物,使反应液产生浑浊,在600nm波长处检测其吸光度的变化,其变化程度与样本中β2-MG含量成正相关。
【主要组成成分】试剂1主要组分三羟甲基氨基甲烷缓冲液20mmol/L 聚乙二醇6000 3.5%表面活性剂及稳定剂适量试剂2主要组分三羟甲基氨基甲烷缓冲液20mmol/L 包被有β2-MG抗体的胶乳颗粒适量表面活性剂及稳定剂适量注:不同批号试剂盒中各组分未经试验不可互换。
【储存条件及有效期】1.试剂原包装在2~8℃储存,有效期为12个月,生产日期、有效期见标签。
2.开口后的试剂在仪器仓中(2~8℃)可稳定30天。
【适用仪器】艾威德AS-420/AS-660/AS-1200;日立HITACHI7020型/7060型/7180型/7600型/LABOSPECT008AS型;贝克曼AU400/AU480/AU640/AU680/ AU2700/AU5400/AU5800/AU5811/AU5821;佳能TBA-FX8/TBA-120FR/ TBA-2000FR;罗氏cobas8000c702/cobas8000c701/cobas8000c502;西门子SIEMENS ADVIA1800/ADVIA2400;雅培ABBOTT ARCHITECT c8000/ARCHITECT c16000/ARCHITECT ci8200;西森美康SYSMEX BM6010/C;科华KHB卓越310/卓越330/卓越400/卓越450/ZY-1200/ZY-1280;迪瑞CS-240/CS-T300/CS-300B/CS-380/CS-400A/CS-400B/CS-600A/ CS-600B/CS-800A/CS-800B/CS-1200/CS-1200ISE/CS-1300B/CS-1400;迈瑞MINDRAY BS-220/BS-330/BS-350E/BS-380/BS-390/BS-400/BS-430/BS-600/ BS-800/BS-2000M;颐兰贝ES-200/ES-380/ES-480;赛诺迈德SUNMATIK-9050型;雷杜Chemray420;英诺华D280;特康TC6010L;锦瑞GS400;普康6066。
人尿微量白蛋白m-Alb检测试剂盒透射比浊法
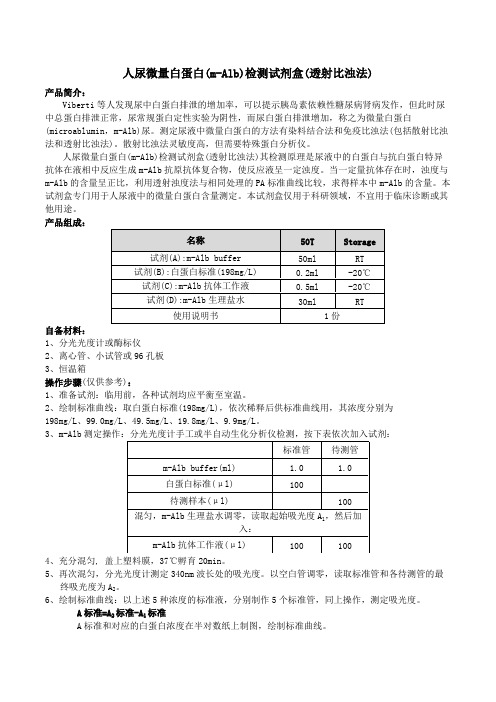
参考区间:
健康成年人尿液白蛋白:
24h尿:<30mg/ 24h。
定时尿:<20μg/min。
随意尿:<30μg/mg肌酐。
注意事项:
1、上述试剂避免反复冻融,以免失效或效率下降。
2、本法属于浊度反应,试剂中有可见混浊出现后,应弃用。
3、如果没有分光光度计,也可以使用酶标仪测定。
使用酶标仪测定蛋白浓度时,每个试剂盒可以测定的样品数量可能会显著增加。
4、待测样本如不能及时测定,应置于2~8℃保存,2天内稳定。
5、本法线性范围在4~200mg/L,如果样本白蛋白浓度大于500mg/L,结果可能呈假性降低。
应用m-Alb生理盐水稀释至4~200mg/L后重测,结果乘以稀释倍数。
有效期:12个月有效。
蛋白标准配制成溶液后应-20℃冻存。
免疫球蛋白G(IgG)测定试剂盒(免疫比浊法)产品技术要求sainuopu

免疫球蛋白G(IgG)测定试剂盒(免疫比浊法)适用范围:用于体外定量测定人体血清中免疫球蛋白G的含量。
1.1 试剂盒包装规格试剂1:1×25ml,试剂2:1×5ml;试剂1:2×60ml,试剂2:2×12ml;试剂1:3×40ml,试剂2:3×8ml;试剂1:4×60ml,试剂2:4×12ml;试剂1:2×400ml,试剂2:1×160ml;试剂1:1×10L,试剂2:1×2L;试剂1:2×40ml,试剂2:2×8ml。
校准品(选配):1×1ml,1×1.5ml,1×3ml。
1.2试剂盒主要组成成分2.1 外观液体双试剂:试剂1无色澄清液体;试剂2浅黄至微红色液体。
校准品:无色至浅黄色液体。
2.2 净含量液体试剂的净含量不得低于标示体积。
2.3 试剂空白吸光度在37℃、405nm波长、1cm光径条件下,试剂空白吸光度应不大于0.2。
2.4 分析灵敏度测定浓度为1g/L样本时,吸光度变化值(ΔA)应在(0.05,0.40)范围内。
2.5 线性范围在(0.5,20.00)g/L线性范围内,线性相关系数r不小于0.995。
在(6.00,20.00)g/L区间内线性相对偏差不大于±10%;(0.5,6.00]g/L区间内线性绝对偏差应不大于±0.60g/L。
2.6 重复性重复测试两份高低浓度的样本,所得结果的变异系数(CV%)应不大于5%。
2.7 批间差不同批号试剂测试同一份样本,测定结果的批间相对极差应不大于10%。
2.8 准确度相对偏差:相对偏差应不超过±10%。
2.9 校准品溯源性依据GB/T 21415-2008《体外诊断医疗器械生物样品中量的测量校准品和控制物质赋值的计量学溯源性》的要求,校准品溯源至IRMM生产的有证参考物质(ERM-DA470k)。
尿微量白蛋白(mALB)测定试剂盒(免疫比浊法)性能指标

尿微量白蛋白(mALB)测定试剂盒(免疫比浊法)性能指标1.性能指标1.1外观外观应符合以下要求:a)试剂盒各组分应齐全、完整,液体无渗漏;包装标签文字符号清晰。
b)R1:无色澄清液体。
c)R2:无色至淡黄色透明液体,目测均不得有任何沉淀及絮状悬浮物。
d)校准品、质控品:无色至淡黄色透明液体。
1.2装量液体试剂装量要求不低于标示量。
1.3空白限空白限不高于5 mg/L。
1.4分析灵敏度测定30 mg/L样本时,吸光度差值(△A)应≥0.02。
1.5线性范围1.5.1在[5,400]mg/L范围内,线性相关系数|r|≥0.990;1.5.2在[5,30]mg/L范围内,线性绝对偏差应不超过±3mg/L;在(30,400]mg/L范围内,线性相对偏差应不超过±10%。
1.6重复性变异系数(CV)应≤5%。
1.7批间差批间相对极差R≤10%。
1.8准确度回收率应在90%~110%。
1.9分析特异性当抗坏血酸≤100mg/dL,胆红素≤40mg/dL,肌酐≤100mg/dL,葡萄糖≤800mg/dL,尿素≤400mg/dL时,对试剂检测结果的偏差影响在±10%以内。
1.10量值溯源应明确分析物的量值溯源。
1.11校准品赋值结果及其不确定度的表示方式应使用规范的表示方式,主要表示方式可选择:a)赋值结果±扩展不确定度;b)赋值结果,扩展不确定度。
1.12校准品正确度量值传递的正确度应符合E≤1。
n1.13质控品赋值准确度在用校准品校准后的生化分析仪上测试定值质控品,结果应在制造商指定的赋值范围内。
1.14校准品均匀性应不大于10%。
1.14.1瓶内均匀性:CV瓶内应不大于10%。
1.14.2瓶间均匀性:CV瓶间1.15质控品均匀性应不大于10%。
1.15.1瓶内均匀性:CV瓶内1.15.2瓶间均匀性:CV应不大于10%。
瓶间。
尿微量白蛋白(MAL)测定试剂盒 (免疫比浊法)产品技术要求sainuopu

尿微量白蛋白(MAL)测定试剂盒 (免疫比浊法)适用范围:用于体外定量测定人体24小时尿液或随机尿液中微量白蛋白含量。
1.1 试剂盒包装规格试剂1:1×25ml,试剂2:1×5ml;试剂1:2×60ml,试剂2:2×12ml;试剂1:3×40ml,试剂2:3×8ml;试剂1:4×60ml,试剂2:4×12ml;试剂1:2×400ml,试剂2:1×160ml;试剂1:1×10L,试剂2:1×2L;试剂1:2×40ml,试剂2:2×8ml。
校准品(选配):1×1ml,1×1.5ml,1×3ml,1×5ml。
质控品(选配):1×1ml,1×1.5ml,1×3ml,1×5ml。
1.2试剂盒主要组成成分2.1 外观液体双试剂:试剂1无色澄清液体;试剂2浅橙色液体。
校准品:无色至浅黄色澄清液体。
质控品:无色至浅黄色澄清液体。
2.2 净含量液体试剂的净含量不得低于标示体积。
2.3 试剂空白吸光度在37℃、340nm波长、1cm光径条件下,试剂空白吸光度应不大于0.3。
2.4 分析灵敏度测定浓度为25mg/L样本时,吸光度变化值(ΔA)应在(0.005,0.12)范围内。
2.5 线性范围在(2,100)mg/L线性范围内,线性相关系数r不小于0.995。
在(30,100)mg/L区间内线性相对偏差应不大于±10%;(2,30]mg/L区间内线性绝对偏差应不大于±3.0mg/L。
2.6 重复性重复测试两份高低浓度的样本,所得结果的变异系数(CV%)应不大于10%。
2.7 批间差不同批号试剂测试同一份样本,测定结果的批间相对极差应不大于10%。
2.8 准确度相对偏差:相对偏差应不超过±10%。
尿微量白蛋白检测试剂的性能验证

0~37992mg/L;⑤ 钩状效应识别:当样本中白蛋白水 平 不 超 过 54610mg/L 时,仪 器 均 可 提 示 反 应 曲 线 错
误.结论 本研究评估的尿微量白蛋白免疫比浊法试剂性能 符 合 临 床 使 用 要 求,结 合 仪 器 反 应 曲 线 检 验 参 数,能 够 识 别
白蛋白水平不高于 54610mg/L 的样本产生的钩状效应.
on
c
en
t
r
a
t
i
oni
sl
e
s
s
yk
yt
qu
/
,
ehookede
f
f
e
c
t.
t
han54610mg L t
hek
i
tha
st
heab
i
l
i
t
or
e
c
ogn
i
z
yt
Ke
r
d
s:Mi
c
r
oa
l
bumi
n; Immuno
t
u
rb
i
d
ime
t
r
f
f
e
c
t
ywo
y; Hooke
尿微量 白 蛋 白 (mi
t
hei
n
s
t
r
umen
t.
Re
s
u
l
t
sThei
n
t
r
aba
t
ch,i
n
t
e
rba
t
chandt
o
t
a
lva
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
微量白蛋白测定试剂盒(免疫比浊法)
适用范围:本品用于体外定量测定人尿液中微量白蛋白的含量。
1.1规格
规格1: (试剂1:20mL;试剂2: 5mL);
规格2: (试剂1:40mL;试剂2:10mL);
规格3: (试剂1:80mL;试剂2:20mL);
校准品(冻干品):为选配:
规格1(0.3mL×1;1水平);规格2(0.5mL×1;1水平);
规格3(1.0mL×1;1水平);
质控品(冻干品):为选配
规格1(0.5mL×2;2水平);规格2(1.0mL×2;2水平)。
1.2组成
试剂盒组成见表1定试
表1 微量白蛋白测定试剂盒组成
2.1试剂
2.1.1外观
试剂盒外观应整洁,文字符号标识清晰;试剂1、试剂2均为无色透明液体,不得有沉淀和絮状物。
2.1.2装量
每瓶不少于标示值。
2.1.3试剂空白吸光度
用指定的空白样品测试试剂(盒),在光径1cm下,在A340nm处测定试剂空白吸光度A≤0.2。
2.1.4分析灵敏度
测定30 mg/L的样品,吸光度差值△A≥0.01。
2.1.5线性范围
2.1.5.1在[2, 400]mg/L内,相关系数R≥0.990。
2.1.5.2在[2, 30]mg/L内,线性绝对偏差不超过±
3.0mg/L;(30, 400]mg/L内,线性相对偏差不超过±10%。
2.1.6 重复性
重复测试(45±9)mg/L和(90±18)mg/L样本,所得结果的变异系数(CV%)应不大于5%。
2.1.7批间差
测定(45±9)mg/L和(90±18)mg/L样本,所得结果的批间相对极差(R)应不大于10%。
2.1.8准确度
)中加入一定体积高于400mg/L的白蛋在正常浓度范围的临床样本(C
白纯品(C
)或由纯品配制的标准溶液,回收率应在90%-110%范围内。
s
2.2校准品
2.2.1外观
校准品为冻干品。
2.2.2瓶间差
瓶间差CV≤10%
2.2.3准确度
与配套试剂组成测试系统,指标要求同2.1.8。
2.2.4 溯源性
根据GB/T21415-2008的要求,本产品校准品溯源至企业工作校准品,与已取得医疗器械产品注册证的企业产品对比赋值(德赛生物技术有限公司)。
2.3质控品
2.3.1外观
质控品为冻干品。
2.3.2 瓶间差
瓶间差CV≤10%。
2.3.3赋值有效性
试剂盒内的质控品,检测结果均在质控范围内。
2.4 效期稳定性
试剂(所有组份)有效期为12个月到效期后一个月内进行检测,测定结果应符合2.1.3-2.1.8(除2.1.7批间差)、2.2.3和2.3.3项要求。