重氮化反应 氨基变磺酰氯 m-TRIFLUOROMETHYLBENZENESULFONYL CHLORIDE
重氮化反应 氨基变肼 PHENYLHYDRAZINE

Organic Syntheses, Coll. Vol. 1, p.442 (1941); Vol. 2, p.71 (1922).PHENYLHYDRAZINE[Hydrazine, phenyl-]Submitted by G. H. ColemanChecked by J. B. Conant and H. R. Thompson.1. ProcedureIn a 12-l. round-bottomed flask, fitted with a mechanical stirrer, is placed 1045 cc. of concentrated commercial hydrochloric acid (sp. gr. 1.138). The flask is surrounded with a freezing mixture of ice and salt, and, when the contents are at 0°, stirring is started and 500 g. of cracked ice is added, or more ice can be added and the external cooling dispensed with; then 372 g. (364 cc., 4 moles) of aniline, also cooled to 0°, is run in during five minutes. The mixture is treated with 500 g. more of cracked ice, and a cold solution (0°) of 290 g. (4 moles) of technical sodium nitrite dissolved in 600 cc. of water is allowed to run in slowly (twenty to thirty minutes) from a separatory funnel, the end of which is drawn to a small tip and reaches nearly to the bottom of the flask. During this addition, the stirrer is operated rather vigorously, and the temperature is kept as near 0° as possible by the frequent addition of cracked ice (about 1 kg.).In the meantime, a sodium sulfite solution is prepared by dissolving 890 g. (20 moles) of sodium hydroxide, of about 90 per cent purity, in about 1 l. of water and then diluting to 6 l. A few drops of phenolphthalein solution are added and sulfur dioxide passed in, first until an acid reaction is indicated and then for two or three minutes longer. During the addition of the sulfur dioxide, the solution is cooled with running water. On account of the strong alkaline solution, the original color produced by the phenolphthalein is very faint, but this slowly increases until it becomes deep just before the acid point is reached. It is best to remove a small sample of the liquid from time to time, dilute with three or four volumes of water, and add a drop more of phenolphthalein(Note 1).The sodium sulfite solution is placed in a 12-l. flask and cooled to about 5°. Approximately 500 g. of cracked ice is added, and then, with mechanical stirring, the diazonium salt solution is run in as rapidly as possible (Note 2). The mixture becomes a bright orangered. The flask is now warmed to about 20° on a steam bath, until the solid sodium sulfite, which has separated while cooling, redissolves. The total amount of liquid is now about 10 l. One-half of this is poured into another 12-l. flask, and both halves are warmed on the steam bath to 60–70°, until the color becomes quite dark (thirty to sixty minutes). Sufficient hydrochloric acid (300–400 cc.) is now added (Note 3) to each flask to make the solutions acid to litmus. The heating is continued and the color gradually becomes lighter until, after four to six hours, the solutions have become nearly colorless; they may be heated overnight, if desired.To the hot solutions is now added about one-third of their volume of concentrated hydrochloric acid(2 l. to each portion), and the mixtures are cooled, first in running water, then in a freezing mixture, to 0° (Note 4). The phenylhydrazine hydrochloride precipitates in the form of slightly yellowish or pinkish crystals which may be filtered off and dried (Note 5).The free base is liberated by adding to the phenylhydrazine hydrochloride 1 l. of a 25 per cent solution of sodium hydroxide. The phenylhydrazine separates and is taken up with benzene (two 300-cc. portions). The combined extractions are well dried with 200 g. of solid sodium hydroxide(Note 6), poured off, and distilled. Most of the benzene may be distilled under ordinary pressure, and the remainder, and any low-boiling impurities, under diminished pressure. The pure phenylhydrazine boils at 137–138°/18 mm., and is obtained as a pale yellow liquid (Note 7). It can be crystallized on cooling in an ice bath; the crystals melt at 23°. The crude phenylhydrazine from two lots of aniline (744 g.) is best distilled at one time and gives 695–725 g. of pure product (80–84 per cent of the theoretical amount).2. Notes1. If the sodium sulfite solution contains an excess of alkali, a black tar tends to form when the solution is warmed, and very little phenylhydrazine is obtained. Great care must be taken in determining the end point in the neutralization of the sodium hydroxide by the sulfur dioxide.It is best to use freshly prepared sodium sulfite for the reduction, since the commercial quality is poor and gives a lower yield of phenylhydrazine. A cylinder of liquid sulfur dioxide should, of course, be available.2. The rapid addition of the diazonium salt solution to the sodium sulfite seems to be advantageous.3. If the sodium sulfite-diazonium salt mixture is acidified before warming or before becoming dark, the red color of the solution does not disappear on heating, and the precipitated phenylhydrazine hydrochloride obtained is colored red.Most published directions for the preparation of phenylhydrazine specify the use of zinc dust and acetic acid following the reduction with sodium sulfite. No improvement in the quality or quantity of the product was obtained by using zinc and acetic acid.4. In order to obtain the maximum yield, it is necessary to cool the hydrochloric acid solution of the phenylhydrazine hydrochloride from 20° to 0°, before filtration. From 5 to 10 per cent of product separates between these two temperatures. When this is done, no more phenylhydrazine hydrochloride is obtained by concentration of the mother liquor. An increase in the amount of hydrochloric acid above 2 l. for the precipitation of the hydrochloride produces no increase in yield of product.5. The phenylhydrazine hydrochloride may be purified by crystallizing from water. A 600-cc. portion of water is used for 100 g. of crude hydrochloride, and the solution boiled a short time with a few grams of animal charcoal. After filtering, 200 cc. of concentrated hydrochloric acid is added, and the mixture cooled to 0°. Pure white crystals in a yield of 85–90 g. are obtained.6. The benzene solution of phenylhydrazine should be well dried before distilling, since the presence of moisture causes an increased amount of foaming to take place just after the benzene has distilled off. When the distillation is carried out carefully, practically no phenylhydrazine distills with the benzene or other low-boiling impurities.Care should be taken that the free base contains no trace of hydrochloride, for this compound catalyzes the decomposition of phenylhydrazine above 100°.7. Pure phenylhydrazine dissolves in dilute acetic acid to yield a perfectly clear solution.Care should be taken when working with large quantities of phenylhydrazine, since the product may cause serious injury to the skin. The vapors of phenylhydrazine should not be inhaled.3. DiscussionPhenylhydrazine can be prepared by the reduction of benzenediazonium salts by means of a miscellany of reducing agents, of which sodium sulfite1 is preferred. Although this method is given in several laboratory manuals, the results were not found entirely satisfactory. The present directions provide for a lengthy but essential heating of the diazonium-sulfite mixture, omit the useless zinc dust reduction, and supply exact details for preparation on a fairly large laboratory scale. The electrolytic reduction of benzenediazonium salts has been reported to furnish a quantitative yield of phenylhydrazine.2References and Notes1.Fischer, Ann. 190, 79 (1878); Reychler, Ber. 20, 2463 (1887). Thompson, J. Soc. DyersColourists 37, 7 (1921) [C. A. 15, 1513 (1921)].2.Takayanagi, J. Chem. Soc. Japan 53, 427 (1932) [C. A. 27, 233 (1933)].AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sodium sulfite-diazonium salthydrochloric acid (7647-01-0)acetic acid (64-19-7)Benzene (71-43-2)aniline (62-53-3)sodium sulfite (7757-83-7)sodium hydroxide (1310-73-2)sulfur dioxide (7446-09-5)sodium nitrite (7632-00-0)Phenylhydrazine,Hydrazine, phenyl- (100-63-0)zinc (7440-66-6)phenolphthalein (77-09-8)phenylhydrazine hydrochloride (59-88-1)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化以及重氮基的转化反应

总结: 上述各种质点的活泼性次序依次是: ON+ > ON—Br > ON—Cl > ON—NO2 > ON—OH
表 不同无机酸中重氮化亲电质点
无机酸 亲电质点
活性
浓H2SO4 NO+
HBr NOBr 大
HCl NOCl
小
稀H2SO4 N2O3
重氮化反映历程是N—亚硝化—脱水反映,可简单表示 如下:
偶氮基进入羟基的
邻对位。
NH2OH
HO3S
NH2
OH NH2
HO3S
SO3H
H酸
OH
J酸
HO3S
γ酸
(4)含有活泼亚甲基的化合物
O
O
CH3 C CH2 C NH
R
CH2 C CH3
O
N N
CH3 CONH2
HO N O
乙酰乙酰芳胺
吡唑酮衍生物
吡啶酮衍生物
(一)偶合反应机理
偶合反应是一个亲电芳环取代反应, 发生作用的是 重氮盐阳离子和游离胺、酚或活泼亚甲基化合物的阴 离子。在反应过程中, 第一步是重氮盐阳离子和偶合 组分结合形成一个中间产物;第二步是这个中间产物 释放质子给质子接受体, 而生成偶氮化合物。
反应所需卤化铜催化剂通常需要新鲜制备, 用量一般为重 氮盐的1/5~1/10(化学计算量)。也可用铜粉与卤化氢代 替卤化亚铜, 这种改良反应称为盖特曼(Gatterman)反 应反。应温然度后一配般合要物求经40电~子8转0℃移,生有成些芳反自应由也基可A在r•室;温下进行。 反应中常加入适量无机卤化物, 使卤离子浓度增加, 但需保 持较高酸性, 以加速卤置换反应, 提高收率, 减少偶氮、联 芳烃及氢化副产物。
重氮化反应 氨基变卤素

Organic Syntheses, Coll. Vol. 2, p.351 (1943); Vol. 19, p.55 (1939).IODOBENZENE[Benzene, iodo-]Submitted by H. J. Lucas and E. R. Kennedy.Checked by John R. Johnson and P. L. Barrick.1. ProcedureIn a 3- or 5-gallon stoneware crock are placed 950 cc. (1130 g., 11.7 moles) of concentrated hydrochloric acid (sp. gr. 1.19), 950 cc. of water, 200 g. (196 cc., 2.15 moles) of aniline, and 2 kg. of ice (Note 1). The mixture is agitated by a mechanical stirrer, and, as soon as the temperature drops below 5°, a chilled solution of 156 g. (2.26 moles) of sodium nitrite in a measured volume (700–1000 cc.) of water is introduced fairly rapidly from a separatory funnel, the stem of which projects below the surface of the reaction mixture. The addition should not be fast enough to cause the temperature to rise to 10° or to cause evolution of oxides of nitrogen. The last 5 per cent of the nitrite solution is added more slowly, and the reaction mixture is tested with starch-iodide paper at intervals until an excess of nitrous acid is indicated.Stirring is continued for ten minutes, and if necessary the solution is filtered rapidly through a loose cotton plug in a large funnel. An aqueous solution of 358 g. (2.16 moles) of potassium iodide is added and the reaction mixture allowed to stand overnight. The mixture is transferred to a large flask (or two smaller flasks) and heated on a steam bath, using an air-cooled reflux condenser, until no more gas is evolved, then allowed to cool and stand undisturbed until the heavy organic layer has settled thoroughly.A large part of the upper aqueous layer is siphoned off, and discarded (Note 2). The residual aqueous and organic layers are made alkaline by the cautious addition of strong sodium hydroxide solution (100–125 g. of solid technical sodium hydroxide is usually required) and steam-distilled at once. The last one-third of the steam distillate is collected separately and combined with the aqueous layer separated from the earlier portions of the distillate. This mixture is acidified with 5–10 cc. of concentrated sulfuric acid and steam-distilled again. The iodobenzene from this operation is combined with the main portion and dried with 10–15 g. of calcium chloride(Note 3) and (Note 4). Distillation under reduced pressure gives 327–335 g. (74–76 per cent of the theoretical amount) of iodobenzene, b.p. 77–78°/20 mm. or 63–64°/8 mm. (Note 5).2. Notes1. If more ice is used a portion remains unmelted after the diazotization is completed.2. If a good separation has been made not more than 1–2 g. of iodobenzene is lost with the upper layer.3. An appreciable amount of iodobenzene is retained by the solid calcium chloride. By treating the spent drying agent with water 8–12 g. of iodobenzene can be recovered.4. The crude iodobenzene weighs 350–355 g. (80 per cent of the theoretical amount) and is pure enough for many purposes without redistillation.5. If the distillation is carried too far, the distillate will be colored.3. DiscussionThe preparation of iodobenzene by iodination of benzene, with iodine and nitric acid, and a survey of preparative methods have been given in an earlier volume.1 The present procedure, based upon the method of Gattermann,2 gives a purer product.This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 660z Org. Syn. Coll. Vol. 5, 665References and Notes. Syn. Coll. Vol. I, 1941, 323.2.Gattermann-Wieland, "Laboratory Methods of Organic Chemistry," p. 283. Translated from thetwenty-fourth German edition by W. McCartney, The Macmillan Company, New York, 1937.AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)oxides of nitrogencalcium chloride (10043-52-4)sulfuric acid (7664-93-9)hydrochloric acid (7647-01-0)Benzene (71-43-2)aniline (62-53-3)sodium hydroxide (1310-73-2)nitric acid (7697-37-2)potassium iodide (7681-11-0)sodium nitrite (7632-00-0)nitrous acid (7782-77-6)iodine (7553-56-2)Iodobenzene,Benzene, iodo-(591-50-4)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应

重氮化反应1,历史回顾:1858 年,Peter Griess首次发现了芳香重氮化合物。
1884 年,德国化学家T.Sandmeyer在用乙炔铜和苯胺的重氮盐(PhN2Cl)合成苯乙炔时,得到的主产物却是氯苯,经过仔细研究,发现原来是由于反应中产生的CuCl催化使重氮基被氯取代。
随后,Sandmeyer发现用CuBr和CuCN也能得到相应的溴苯和苯甲腈,因此我们把这一类反应称为Sandmeyer反应。
1890 年,L.Gatterman发现直接用铜粉和盐酸或氢溴酸也能从苯胺得到相应的氯苯或溴苯,这种类型的反应称为Gatterman反应。
1927 年,同样是德国的化学家G.Balz和G.Schiemann发现直接加热苯胺的硼氟酸重氮盐能得到氟苯,这就是Balz-Schiemann反应。
1935 年,F.B.Dains和 F.Eberly用KI去处理重氮盐,成功合成了碘代苯。
随后重氮化羟基取代和重氮化去胺反应也相继被发现,加上偶氮反应,形成了比较完善的芳香重氮化合物反应体系。
2,定义:是指一级胺与亚硝酸在低温下作用生成重氮盐(dizaonium salt)的反应(diazotization)。
脂肪族伯胺与亚硝酸反应,可以订量释放出来氮气,这是测量脂肪伯胺非常好的方法,可以形成碳正离子,副反应比较多,在合成上很少使用。
芳香族伯胺和亚硝酸作用生成的重氮盐,由于氮正离子与苯环的共轭,稳定性大大提高,这样一个中间体,在酸性介质中,在0-5oC可以稳定存在。
一旦遇到光照和加热,会马上分解。
所以这样的一个重氮盐在合成中要现制现用。
3,反应机理:芳胺的重氮化反应需经2步,首先是亲电试剂进攻芳胺氮原子生成不稳定的中间产物,然后不稳定中间产物迅速分解,整个反应受第一步控制。
无机酸不同,参与重氮化反应的亲电试剂也不同。
稀硫酸中参与反应的是N2O3(一为不稳定结构ONONO,一为稳定结构ONNO2),盐酸中参与反应的是亚硝酰氯NOCl;在浓硫酸中则是亚硝基正离子N+O。
重氮化反应 氨基变肼 PHENYLHYDRAZINE
Organic Syntheses, Coll. Vol. 1, p.442 (1941); Vol. 2, p.71 (1922).PHENYLHYDRAZINE[Hydrazine, phenyl-]Submitted by G. H. ColemanChecked by J. B. Conant and H. R. Thompson.1. ProcedureIn a 12-l. round-bottomed flask, fitted with a mechanical stirrer, is placed 1045 cc. of concentrated commercial hydrochloric acid (sp. gr. 1.138). The flask is surrounded with a freezing mixture of ice and salt, and, when the contents are at 0°, stirring is started and 500 g. of cracked ice is added, or more ice can be added and the external cooling dispensed with; then 372 g. (364 cc., 4 moles) of aniline, also cooled to 0°, is run in during five minutes. The mixture is treated with 500 g. more of cracked ice, and a cold solution (0°) of 290 g. (4 moles) of technical sodium nitrite dissolved in 600 cc. of water is allowed to run in slowly (twenty to thirty minutes) from a separatory funnel, the end of which is drawn to a small tip and reaches nearly to the bottom of the flask. During this addition, the stirrer is operated rather vigorously, and the temperature is kept as near 0° as possible by the frequent addition of cracked ice (about 1 kg.).In the meantime, a sodium sulfite solution is prepared by dissolving 890 g. (20 moles) of sodium hydroxide, of about 90 per cent purity, in about 1 l. of water and then diluting to 6 l. A few drops of phenolphthalein solution are added and sulfur dioxide passed in, first until an acid reaction is indicated and then for two or three minutes longer. During the addition of the sulfur dioxide, the solution is cooled with running water. On account of the strong alkaline solution, the original color produced by the phenolphthalein is very faint, but this slowly increases until it becomes deep just before the acid point is reached. It is best to remove a small sample of the liquid from time to time, dilute with three or four volumes of water, and add a drop more of phenolphthalein(Note 1).The sodium sulfite solution is placed in a 12-l. flask and cooled to about 5°. Approximately 500 g. of cracked ice is added, and then, with mechanical stirring, the diazonium salt solution is run in as rapidly as possible (Note 2). The mixture becomes a bright orangered. The flask is now warmed to about 20° on a steam bath, until the solid sodium sulfite, which has separated while cooling, redissolves. The total amount of liquid is now about 10 l. One-half of this is poured into another 12-l. flask, and both halves are warmed on the steam bath to 60–70°, until the color becomes quite dark (thirty to sixty minutes). Sufficient hydrochloric acid (300–400 cc.) is now added (Note 3) to each flask to make the solutions acid to litmus. The heating is continued and the color gradually becomes lighter until, after four to six hours, the solutions have become nearly colorless; they may be heated overnight, if desired.To the hot solutions is now added about one-third of their volume of concentrated hydrochloric acid(2 l. to each portion), and the mixtures are cooled, first in running water, then in a freezing mixture, to 0° (Note 4). The phenylhydrazine hydrochloride precipitates in the form of slightly yellowish or pinkish crystals which may be filtered off and dried (Note 5).The free base is liberated by adding to the phenylhydrazine hydrochloride 1 l. of a 25 per cent solution of sodium hydroxide. The phenylhydrazine separates and is taken up with benzene (two 300-cc. portions). The combined extractions are well dried with 200 g. of solid sodium hydroxide(Note 6), poured off, and distilled. Most of the benzene may be distilled under ordinary pressure, and the remainder, and any low-boiling impurities, under diminished pressure. The pure phenylhydrazine boils at 137–138°/18 mm., and is obtained as a pale yellow liquid (Note 7). It can be crystallized on cooling in an ice bath; the crystals melt at 23°. The crude phenylhydrazine from two lots of aniline (744 g.) is best distilled at one time and gives 695–725 g. of pure product (80–84 per cent of the theoretical amount).2. Notes1. If the sodium sulfite solution contains an excess of alkali, a black tar tends to form when the solution is warmed, and very little phenylhydrazine is obtained. Great care must be taken in determining the end point in the neutralization of the sodium hydroxide by the sulfur dioxide.It is best to use freshly prepared sodium sulfite for the reduction, since the commercial quality is poor and gives a lower yield of phenylhydrazine. A cylinder of liquid sulfur dioxide should, of course, be available.2. The rapid addition of the diazonium salt solution to the sodium sulfite seems to be advantageous.3. If the sodium sulfite-diazonium salt mixture is acidified before warming or before becoming dark, the red color of the solution does not disappear on heating, and the precipitated phenylhydrazine hydrochloride obtained is colored red.Most published directions for the preparation of phenylhydrazine specify the use of zinc dust and acetic acid following the reduction with sodium sulfite. No improvement in the quality or quantity of the product was obtained by using zinc and acetic acid.4. In order to obtain the maximum yield, it is necessary to cool the hydrochloric acid solution of the phenylhydrazine hydrochloride from 20° to 0°, before filtration. From 5 to 10 per cent of product separates between these two temperatures. When this is done, no more phenylhydrazine hydrochloride is obtained by concentration of the mother liquor. An increase in the amount of hydrochloric acid above 2 l. for the precipitation of the hydrochloride produces no increase in yield of product.5. The phenylhydrazine hydrochloride may be purified by crystallizing from water. A 600-cc. portion of water is used for 100 g. of crude hydrochloride, and the solution boiled a short time with a few grams of animal charcoal. After filtering, 200 cc. of concentrated hydrochloric acid is added, and the mixture cooled to 0°. Pure white crystals in a yield of 85–90 g. are obtained.6. The benzene solution of phenylhydrazine should be well dried before distilling, since the presence of moisture causes an increased amount of foaming to take place just after the benzene has distilled off. When the distillation is carried out carefully, practically no phenylhydrazine distills with the benzene or other low-boiling impurities.Care should be taken that the free base contains no trace of hydrochloride, for this compound catalyzes the decomposition of phenylhydrazine above 100°.7. Pure phenylhydrazine dissolves in dilute acetic acid to yield a perfectly clear solution.Care should be taken when working with large quantities of phenylhydrazine, since the product may cause serious injury to the skin. The vapors of phenylhydrazine should not be inhaled.3. DiscussionPhenylhydrazine can be prepared by the reduction of benzenediazonium salts by means of a miscellany of reducing agents, of which sodium sulfite1 is preferred. Although this method is given in several laboratory manuals, the results were not found entirely satisfactory. The present directions provide for a lengthy but essential heating of the diazonium-sulfite mixture, omit the useless zinc dust reduction, and supply exact details for preparation on a fairly large laboratory scale. The electrolytic reduction of benzenediazonium salts has been reported to furnish a quantitative yield of phenylhydrazine.2References and Notes1.Fischer, Ann. 190, 79 (1878); Reychler, Ber. 20, 2463 (1887). Thompson, J. Soc. DyersColourists 37, 7 (1921) [C. A. 15, 1513 (1921)].2.Takayanagi, J. Chem. Soc. Japan 53, 427 (1932) [C. A. 27, 233 (1933)].AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sodium sulfite-diazonium salthydrochloric acid (7647-01-0)acetic acid (64-19-7)Benzene (71-43-2)aniline (62-53-3)sodium sulfite (7757-83-7)sodium hydroxide (1310-73-2)sulfur dioxide (7446-09-5)sodium nitrite (7632-00-0)Phenylhydrazine,Hydrazine, phenyl- (100-63-0)zinc (7440-66-6)phenolphthalein (77-09-8)phenylhydrazine hydrochloride (59-88-1)Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变羟基 m-METHOXYBENZALDEHYDE

Organic Syntheses, Coll. Vol. 3, p.564 (1955); Vol. 29, p.63 (1949).m-METHOXYBENZALDEHYDE[Benzaldehyde, m-methoxy-]Submitted by Roland N. Icke, C. Ernst Redemann, Burnett B. Wisegarver, and Gordon A. Alles.Checked by H. R. Snyder and Frank X. Werber.1. ProcedureA. m-Hydroxybenzaldehyde. In a 2-l. three-necked flask, equipped with a mechanical stirrer, a thermometer, and a 250-ml. dropping funnel, 575 ml. of 6 N sulfuric acid is cooled to 0° by means of a salt-ice bath. The acid is stirred and maintained at 0° or below while 167 g. (1 mole) of m-aminobenzaldehyde dimethylacetal(p. 59) is added dropwise. The solution becomes deep orange or red. When the addition of the amino compound is complete, a solution of 71 g. (1 mole) of 97% sodium nitrite in about 175 ml. of water is introduced slowly while the temperature of the acid solution is maintained at 5°. Stirring at 5° is continued for 1 hour to complete the reaction.In each of two 4-l. beakers are placed 450 ml. of water and 50 ml. of concentrated sulfuric acid, and the solutions are heated to boiling with large burners. The cold diazonium solution is divided into two approximately equal portions which are placed in 500-ml. separatory funnels suspended above the beakers containing the boiling acid. The two portions of the diazonium solution are run dropwise into the strongly heated acid at such a rate that boiling continues. The solutions are boiled for 5 minutes after the additions are complete. They are then allowed to cool to room temperature and are finally stored overnight in a refrigerator. The crude product separates as a dark oil which crystallizes (Note 1) and becomes lighter in color upon standing. It is collected on a Büchner funnel and used in part B without purification (Note 2).Methyl sulfate is quite toxic. Caution! The methylation should be carried out in a good hood.B. m-Methoxybenzaldehyde. The crude m-hydroxybenzaldehyde is dissolved in about 550 ml. of 2 N sodium hydroxide in a 2-l. three-necked flask equipped with a mechanical stirrer, a thermometer, and a 125-ml. dropping funnel. The dark-colored solution is stirred while 126 g. (95 ml., 1 mole) of methyl sulfate(Note 3) is added dropwise and the temperature is maintained at 40–45°. When the addition is complete the mixture is stirred for 5 minutes. A 275-ml. portion of 2 N sodium hydroxide(Note 4) is added in one lot, and then 63 g. (47.5 ml.) of methyl sulfate is added as before, except that the temperature is allowed to rise to 50°. Stirring at 50° is continued for 30 minutes, the mixture is cooled, and the organic layer is extracted with ether(Note 5). The ether solution is dried over anhydrous sodium sulfate for 8 hours, then filtered and concentrated by distillation. The residue is distilled under reducedpressure. The yield of m-methoxybenzaldehyde, a pale yellow liquid boiling at 88–90° /3 mm., is 86–98 g. (63–72%) (Note 6).2. Notes1. Seeding the mixture helps to initiate crystallization.2. If m-hydroxybenzaldehyde is desired, the crude product may be purified as described elsewhere (p. 453).3. A good technical grade of methyl sulfate was used.4. The optimum amount of sodium hydroxide solution apparently varies according to the amount of acid remaining in the crude, wet hydroxybenzaldehyde employed in the methylation. The checkers found it advisable to increase the amount added at this point to 345 ml. It is wise to test the reaction mixture with litmus paper occasionally during the final heating period and to add alkali as necessary to keep the solution from becoming acid.5. If the methylation is not complete, some m-hydroxybenzaldehyde will remain dissolved in the aqueous phase. This may be recovered by acidifying the alkaline solution and collecting any crystalline solid which separates.6. As with other aromatic aldehydes, m-methoxybenzaldehyde is susceptible to air oxidation and should be stored in a bottle which will just hold the product, so that air space above the liquid is minimized.3. Discussionm-Methoxybenzaldehyde has been prepared by the reduction of m-methoxybenzoic acid,1 by the reaction of diazotized m-aminobenzaldehyde with methanol,2 by an acid hydrolysis of the phenylhydrazone which was obtained by oxidation of the hydrazine analog,3 and by the methylation of m-hydroxybenzaldehyde, with methyl iodide,4,5,6,7 and with methyl sulfate.2,7,8,9References and Notes1.Asano and Huziwara, J. Pharm. Soc. Japan, 50, 141 (1939).2.Noelting, Ann. chim., (8) 19, 541 (1910).3.Grammaticakis, Compt. rend., 210, 303 (1940).4.Tiemann and Ludwig, Ber., 15, 2043 (1882).5.Pschorr and Jaeckel, Ber., 33, 1826 (1900).6.Staudinger and Kon, Ann., 384, 90 (1911).7.Späth, Monatsh., 34, 1998 (1913).8.Posner, J. prakt. Chem., (2) 82, 431 (1910).9.Livshits, Bazilevskaya, Bainova, Dobrovinskaya, and Preobrazhenskii, J. Gen. Chem. U.S.S.R.,17, 1671 (1947) [C. A., 42, 2606 (1948)].AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)phenylhydrazonesulfuric acid (7664-93-9)methanol (67-56-1)ether(60-29-7)sodium hydroxide (1310-73-2)sodium sulfate (7757-82-6)sodium nitrite (7632-00-0)hydroxybenzaldehyde (90-02-8)Methyl iodide (74-88-4)methyl sulfate (75-93-4)m-Hydroxybenzaldehyde (100-83-4)m-aminobenzaldehyde (1709-44-0)m-Aminobenzaldehyde dimethylacetal (53663-37-9)m-Methoxybenzaldehyde,Benzaldehyde, m-methoxy- (591-31-1)m-methoxybenzoic acid (586-38-9) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应氨基变羟基m-METHOXYBENZALDEHYDE

重氮化反应氨基变羟基m-METHOXYBENZALDEHYDE Organic Syntheses, Coll. Vol. 3, p.564 (1955); Vol. 29, p.63 (1949).m-METHOXYBENZALDEHYDE[Benzaldehyde, m-methoxy-]Submitted by Roland N. Icke, C. Ernst Redemann, Burnett B. Wisegarver, and Gordon A. Alles.Checked by H. R. Snyder and Frank X. Werber.1. ProcedureA. m-Hydroxybenzaldehyde. In a 2-l. three-necked flask, equipped with a mechanical stirrer, a thermometer, and a 250-ml. dropping funnel, 575 ml. of 6 N sulfuric acid is cooled to 0° by means of a salt-ice bath. The acid is stirred and maintained at 0° or below while 167 g. (1 mole) of m-aminobenzaldehyde dimethylacetal(p. 59) is added dropwise. The solution becomes deep orange or red. When the addition of the amino compound is complete, a solution of 71 g. (1 mole) of 97% sodium nitrite in about 175 ml. of water is introduced slowly while the temperature of the acid solution is maintained at 5°. Stirring at 5° is continued for 1 hour to complete the reaction.In each of two 4-l. beakers are placed 450 ml. of water and 50 ml. of concentrated sulfuric acid, and the solutions are heated to boiling with large burners. The cold diazonium solution is divided into two approximately equal portions which are placed in 500-ml. separatory funnels suspended above the beakers containing the boiling acid. The two portions of the diazonium solution are run dropwise into the strongly heated acid at such a rate that boiling continues. The solutions are boiled for 5 minutes after the additions are complete. They are then allowed to cool to room temperature and are finally stored overnight in a refrigerator. The crude product separates as a dark oil which crystallizes (Note 1) and becomes lighter in color upon standing. It is collected on a Büchner funnel and used in part B without purification (Note 2).Methyl sulfate is quite toxic. Caution! The methylation should be carried out in a good hood.B. m-Methoxybenzaldehyde. The crude m-hydroxybenzaldehyde is dissolved in about 550 ml. of 2 N sodium hydroxide in a 2-l. three-necked flask equipped with a mechanical stirrer, a thermometer, and a 125-ml. dropping funnel. The dark-colored solution is stirred while 126 g. (95 ml., 1 mole) of methyl sulfate(Note 3) is added dropwise and the temperature is maintained at 40–45°. When the addition is complete the mixture is stirred for 5 minutes. A 275-ml. portion of 2 N sodium hydroxide(Note 4) is added in one lot, and then 63 g. (47.5 ml.) of methyl sulfate is added as before, except that the temperature is allowed to rise to 50°. Stirring at 50° is continued for 30 minutes, the mixture is cooled, and the organic layer is extracted withether(Note 5). The ether solution is dried over anhydrous sodium sulfate for 8 hours, then filtered and concentrated by distillation. The residue is distilled under reducedpressure. The yield of m-methoxybenzaldehyde, a pale yellow liquid boiling at 88–90° /3 mm., is 86–98 g. (63–72%) (Note 6).2. Notes1. Seeding the mixture helps to initiate crystallization.2. If m-hydroxybenzaldehyde is desired, the crude product may be purified as described elsewhere (p. 453).3. A good technical grade of methyl sulfate was used.4. The optimum amount of sodium hydroxide solution apparently varies according to the amount of acid remaining in the crude, wet hydroxybenzaldehyde employed in the methylation. The checkers found it advisable to increase the amount added at this point to 345 ml. It is wise to test the reaction mixture with litmus paper occasionally during the final heating period and to add alkali as necessary to keep the solution from becoming acid.5. If the methylation is not complete, some m-hydroxybenzaldehyde will remain dissolved in the aqueous phase. This may be recovered by acidifying the alkaline solution and collecting any crystalline solid which separates.6. As with other aromatic aldehydes, m-methoxybenzaldehyde is susceptible to air oxidation and should be stored in a bottle which will just hold the product, so that air space above the liquid is minimized.3. Discussionm-Methoxybenzaldehyde has been prepared by the reduction of m-methoxybenzoic acid,1 by the reaction of diazotized m-aminobenzaldehyde with methanol,2 by an acid hydrolysis of the phenylhydrazone which was obtained by oxidation of the hydrazine analog,3 and by the methylation of m-hydroxybenzaldehyde, with methyl iodide,4,5,6,7 and with methyl sulfate.2,7,8,9References and Notes1.Asano and Huziwara, J. Pharm. Soc. Japan, 50, 141 (1939).2.Noelting, Ann. chim., (8) 19, 541 (1910).3.Grammaticakis, Compt. rend., 210, 303 (1940).4.Tiemann and Ludwig, Ber., 15, 2043 (1882).5.Pschorr and Jaeckel, Ber., 33, 1826 (1900).6.Staudinger and Kon, Ann., 384, 90 (1911).7.Sp?th, Monatsh., 34, 1998 (1913).8.Posner, J. prakt. Chem., (2) 82, 431 (1910).9.Livshits, Bazilevskaya, Bainova, Dobrovinskaya, and Preobrazhenskii, J. Gen. Chem. U.S.S.R.,17, 1671 (1947) [C. A., 42, 2606 (1948)].AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)phenylhydrazonesulfuric acid (7664-93-9)methanol (67-56-1)ether(60-29-7)sodium hydroxide (1310-73-2)sodium sulfate (7757-82-6)sodium nitrite (7632-00-0)hydroxybenzaldehyde (90-02-8)Methyl iodide (74-88-4)methyl sulfate (75-93-4)m-Hydroxybenzaldehyde (100-83-4)m-aminobenzaldehyde (1709-44-0)m-Aminobenzaldehyde dimethylacetal (53663-37-9)m-Methoxybenzaldehyde,Benzaldehyde, m-methoxy- (591-31-1)m-methoxybenzoic acid (586-38-9) Copyright ? 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变烃基 1-(p-NITROPHENYL)-1,3-BUTADIEN

Organic Syntheses, Coll. Vol. 4, p.727 (1963); Vol. 31, p.80 (1951).1-(p-NITROPHENYL)-1,3-BUTADIENE[1,3-Butadiene, 1-(p-nitrophenyl)-]Submitted by Gus A. Ropp and Eugene C. Coyner1.Checked by Arthur C. Cope and David J. Marshall.1. ProcedureA. 1-(p-Nitrophenyl)-4-chloro-2-butene. p-Nitroaniline hydrochloride is prepared by heating 138 g.(1.0 mole) of p-nitroaniline(Note 1) with 240 ml. of concentrated hydrochloric acid and 100 ml. of water on a steam bath for 15 minutes with occasional stirring. The mixture is cooled in an ice-salt bath and stirred rapidly in order to precipitate the hydrochloride as fine crystals. Cracked ice (100 g.) is added, and a solution of 70 g. of sodium nitrite is added dropwise with rapid mechanical stirring during a 1-hour period while the temperature of the reaction mixture is held between −4° and +4.5° by cooling with the ice-salt bath. The mixture is stirred for an additional period of 20 minutes and then is filtered through a chilled funnel into an ice-cooled filter flask. The filtrate is kept below 4° (Note 2) and is added through a dropping funnel during 90 minutes to a cold, vigorously stirred mixture composed of 1 l. of acetone, a solution of 80 g. of sodium acetate trihydrate in 100 ml. of water, a solution of 30 g. of cupric chloride in 50 ml. of water, and 130 ml. of liquid butadiene(Note 3). The reaction mixture is kept at −3° to +2° by means of an ice-salt bath while the diazonium salt solution is added. After the addition is completed the cooling bath is removed and the mixture is stirred for 16 hours. One liter of ether is added, and after several minutes' stirring the ethereal layer is separated, washed with four 1-l. portions of water, and dried over 20 g. of anhydrous magnesium sulfate. The solvent is removed by distillation at 15 mm. by heating on a steam bath, leaving a dark brown oily residue (187–199 g.) of crude 1-(p-nitrophenyl)-4-chloro-2-butene(Note 4).B. 1-(p-Nitrophenyl)-1,3-butadiene. The crude 1-(p-nitrophenyl)-4-chloro-2-butene obtained in Part A is dissolved in a mixture of 500 ml. of ligroin, b.p. 90–100°, and 500 ml. of benzene; 5 g. of decolorizing carbon is added, and the mixture is heated under reflux for 2 hours. After filtration to separate the decolorizing carbon the solvents are removed by distillation from a steam bath under reduced pressure, and the residual clear oil is dissolved in 400 ml. of methanol. A solution of 112 g. of potassium hydroxide in 600 ml. of methanol is added from a dropping funnel during 30 minutes while the mixture is stirred mechanically and kept at 15–30° by cooling with a bath of cold water. After being stirred for an additional period of 5 minutes the mixture, which contains some precipitated product, ispoured into 1.2 l. of cold water. The crude product is collected on a filter, washed well with cold water, and air-dried. It is dissolved in 700 ml. of hot ligroin, b.p. 90–100°, and the solution is treated with 5 g. of decolorizing carbon, and filtered. On cooling, 1-(p-nitrophenyl)-1,3-butadiene separates as a yellow crystalline solid which is collected on a filter and dried in a desiccator. The yield of pure product, m.p. 77–79° (Note 5), is 100–108 g. (57–62% based on p-nitroaniline).2. Notes1. Either a pure grade of p-nitroaniline obtained from the Eastman Kodak Company or a technical grade purified by one recrystallization from ethanol was used, m.p. 147.5–148°.2. The filtrate is kept in an ice-salt bath and transferred to the dropping funnel in small amounts in order to keep the temperature below 4°.3. Butadiene from a commercial cylinder is passed through an 8-mm. glass tube leading to the bottom ofa graduated cylinder cooled with Dry Ice and acetone, where it condenses and is measured as a liquid.4. The submitters report that small samples of the crude product can be distilled in order to obtain pure 1-(p-nitrophenyl)-4-chloro-2-butene, b.p. 160–165°/1 mm.5. Two recrystallizations from ligroin raise the melting point of the 1-(p-nitrophenyl)-1,3-butadiene to a constant value of 78.6–79.4°. The product can be kept for several weeks in a dark bottle at room temperature without evidence of decomposition.3. Discussion1-(p-Nitrophenyl)-1,3-butadiene has been prepared only by the method described,2 which is an example of the Meerwein reaction (addition of diazonium salts to a carbon-carbon double bond with the elimination of nitrogen).3This preparation is referenced from:z Org. Syn. Coll. Vol. 6, 21References and Notes1.University of Tennessee, Knoxville, Tennessee.2.Coyner and Ropp, J. Am. Chem. Soc., 70, 2283 (1948); Dombrovskii, Doklady Akad. NaukS.S.S.R., 111, 827 (1956) [C. A., 51, 9507 (1957)].3.Müller, Angew. Chem., 61, 179 (1949).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)ligroinethanol (64-17-5)hydrochloric acid (7647-01-0)Benzene (71-43-2)methanol(67-56-1)ether (60-29-7)nitrogen (7727-37-9)sodium nitrite (7632-00-0)acetone (67-64-1)decolorizing carbon (7782-42-5)potassium hydroxide (1310-58-3)cupric chloride (7758-89-6)magnesium sulfate (7487-88-9)butadiene (106-99-0)sodium acetate trihydrate (6131-90-4)1-(p-Nitrophenyl)-1,3-butadiene,1,3-Butadiene, 1-(p-nitrophenyl)- (20264-89-5)p-nitroaniline (100-01-6)p-nitroaniline hydrochloride (15873-51-5)1-(p-Nitrophenyl)-4-chloro-2-butene (98011-65-5) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变磺酰氯 m-TRIFLUOROMETHYLBENZENESULFONYL CHLORIDE

Organic Syntheses, Coll. Vol. 7, p.508 (1990); Vol. 60, p.121 (1981).m-TRIFLUOROMETHYLBENZENESULFONYL CHLORIDE[Benzenesulfonyl chloride, m-(trifluoromethyl)-]Submitted by R. V. Hoffman1Checked by G. Saucy, G. P. Roth, and J. W. Scott.1. ProcedureCaution! All operations should be carried out in a hood! m-Trifluoromethylbenzenesulfonyl chloride is a lachrymator. Spills should be treated with saturated sodium carbonate.α,α,α-Trifluoro-m-toluidine(m-aminobenzotrifluoride) (96.7 g, 0.6 mol) (Note 1) is added in one portion to a mixture of concentrated hydrochloric acid (200 mL) and glacial acetic acid (60 mL) in a 1000-mL beaker arranged for efficient mechanical stirring (Note 2). The white hydrochloride salt precipitates (Note 3). The beaker is placed in a dry ice-ethanol bath and, when the temperature of the stirred mixture has reached −10°C, a solution of sodium nitrite (44.8 g, 0.65 mol) in water (65 mL) is added dropwise at such a rate that the temperature does not exceed −5°C (Note 4). After all the sodium nitrite solution has been added, the mixture is stirred for 45 min while the temperature is maintained between −10°C and −5°C (Note 5).While the diazotization is being completed, glacial acetic acid (600 mL) is placed in a 4000-mL beaker and stirred magnetically. Sulfur dioxide is introduced by a bubbler tube with a fritted end immersed below the surface of the acetic acid until saturation is evident (Note 6). Cuprous chloride (15 g) (Note 7) is added to the solution. The introduction of sulfur dioxide is continued until the yellow–green suspension becomes blue-green. Most of the solids dissolve during this time (20–30 min). The mixture is then placed in an ice bath and cooled with stirring. When the temperature approaches 10°C, the diazotization reaction mixture (Note 8) is added in portions over a 30-min period to the sulfur dioxide solution. Considerable foaming occurs after each addition, and this can be disrupted with a few drops of ether. The temperature rises during the addition, but it should not exceed 30°C. After all the diazonium salt mixture has been added, the mixture is poured into ice water (1 : 1, 2000 mL), stirred magnetically until the ice has melted, and added to a 4000-mL separatory funnel. The product separates as a yellow oil that is drawn off. The reaction mixture is extracted with 200-mL portions of ether until the ether washings are colorless (Note 9), and these washings are added to the initial product. The combined organic fraction is washed with saturated aqueous sodium bicarbonate until neutral (Note 10), then with water, and is then dried with magnesium sulfate. The solvent is removed with a rotary evaporator, and the residue is distilled (bp 54–55°C, 0.1 mm) through a 10-cm vacuum-jacketed Vigreux column to give m-trifluoromethylbenzenesulfonyl chloride (100–115 g, 68–79%) as a colorless or slightly yellow, clear liquid (Note 11), (Note 12).2. Notes1. α,α,α-Trifluoro-m -toluidine was obtained from Aldrich Chemical Company, Inc. The checkers distilled this material prior to use (bp 187–189°C).2. A chain beaker clamp is very satisfactory for supporting the beaker, as it can later be used as a handle to pour the diazonium solution. For efficient stirring the blade of the stirrer was made by trimming the ends of a large Teflon stirring paddle to the diameter of the beaker. The paddle was inverted (straight edge on bottom) and should rotate 1–1.5 cm from the bottom of the beaker.3. If solid amines are used, they should be thoroughly crushed in a mortar and pestle before adding to the acid mixture.4. Temperature control during the sodium nitrite addition is essential to the success of the preparation. The temperature can go as low as −15°C but must not exceed −5°C. The addition takes ca. 1 hr. At temperatures greater than −5°C, dark-red by-products form which lower the yield.5. Temperature control is conveniently accomplished by raising and lowering the dry-ice bath. It does not seem to matter if longer reaction times are employed, but the temperature should be lowered to −10°C or below after 45 min.6. Saturation, which requires 15–30 min, is conveniently noted by observing that most sulfur dioxide bubbles reach the surface of the acetic acid .7. The original literature 2 suggests that copper(II) chloride dihydrate can be used as a catalyst, since it is reduced by the sulfur dioxide to copper(I). It has been noted on several occasions that catalytically inactive mixtures result. If copper(II) chloride dihydrate is used, it is expedient to add copper(I) chloride (1 g) to ensure efficient catalysis in the early stages of reaction.8. This mixture should be a pale tan suspension, and it should be cooled between additions.9. The first portion of ether may be larger (400 mL) since much dissolves in the aqueous mixture. A total of 1000 mL of ether is usually sufficient. 10. A considerable amount of acid is present in the ether extracts, so vigorous gas evolution occurs during the sodium bicarbonate extraction. Caution must be exercised at this point. 11. The product is sufficiently pure for most purposes. A second distillation affords a colorless product (lit.3 bp 88–90°C, 6 mm). 12. With many anilines used as precursors in this reaction, the sulfonyl chloride product is a solid and an alternate workup procedure is used. After the reaction is quenched with ice water, the solid product is filtered with suction and washed copiously with cold water. The crude product tends to occlude water and copper salts, which may be detrimental in later reactions. A good washing protocol involves rinsing the solid on the filter with water (200 mL), then suspending the solid in cold water (1000 mL), stirring briskly, and filtering with suction. The latter process should be repeated three times. The final water wash should be only very slightly yellow. After air drying the product can be recrystallized from an appropriate solvent.3. Discussionm -Trifluoromethylbenzenesulfonyl chloride has been prepared by treatment of m -trifluoromethylbenzenediazonium chloride with sulfur dioxide and hydrochloric acid 4 and by conversion of benzotrifluoride to m -trifluoromethylbenzenesulfonic acid with oleum, followed by chlorination with phosphorus pentachloride .3 Derivatives of this compound, such as esters and amides, are quite useful in that they display reactivities similar to p - and m -nitrobenzenesulfonyl compounds but have greatly improved solubilities.The described procedure essentially follows that described by Meerwein et al.2 as modified slightly by Yale and Sowinski.4 This same method can be used for a great variety of substituted anilines with good results. As evident in Table I, good yields are obtained in most cases, and the reaction works better for anilines with electron-withdrawing substituents. The identical procedure has been used to prepare many other examples, such as m -F, o -F, 3,5-di-CF 3.5 This method readily provides many unavailable arylsulfonyl chlorides; it is experimentally straightforward, and the products are isolated without complications.There are two general routes to arylsulfonyl chlorides. The first involves the conversion of analready sulfur-substituted aromatic compound to the sulfonyl chloride. Thus arylsulfonic acids or their alkali metal salts yield sulfonyl chlorides by treatment with a variety of chlorinating agents such as phosphorus pentachloride , thionyl chloride , phosgene , and chlorosulfonic acid . Alternatively, substituted thiophenols or aryl disulfides can be oxidized by chlorine-water to the sulfonyl chloride.6The second route utilizes the introduction of the chlorosulfonyl substituent directly onto the aromatic nucleus. The reaction of substituted benzenes with chlorosulfonic acid gives good yields of arylsulfonyl chlorides; however, the aryl substituent dictates the position of attachment of the chlorosulfonyl function in this electrophilic aromatic substitution.7 The method described herein allows replacement of a diazotized amine function by the chlorosulfonyl group. The ready availability of substituted anilines makes this the method of choice for the preparation of arylsulfonyl chlorides.Arylsulfonyl chlorides are pivotal precursors for the preparation of many diverse functional types including sulfonate esters,8 amides,4 sulfones,9 sulfinic acids,10 and others.11 Furthermore, sulfonyl fluorides are best prepared from sulfonyl chlorides.12 The sulfonyl fluorides have many uses, among which is their utilization as active site probes of chymotrypsin and other esterases.13 The trifluoromethyl group also plays valuable roles in medicinal chemistry.14References and Notes1.Department of Chemistry, Box 3C, New Mexico State University, Las Cruces, NM 88003.2.Meerwein, H.; Dittmar, G.; Gollner, R.; Hafner, K.; Mensch, F.; Steinfort, O. Chem. Ber . 1957,90, 841–852.3.Yagupolskii, L. M.; Troitskaya, V. I. Zh. Obsch. Khim . 1959, 29, 552–556; Chem. Abstr . 1960,54, 356f.4.Yale, H. L.; Sowinski, F. J. Org. Chem . 1960, 25, 1824–1826.5.Gerig. J. T.; Roe, D. C. J. Am. Chem. Soc . 1974, 96, 233–238.6.Muth. F., In Houben-Weyl, "Methoden der Organischen Chemie," Müller, E., Ed.; Georg ThiemeVerlag: Stuttgart, Germany, 1955; Vol. 9 pp. 563–585.7.Gilbert, E. E. "Sulfonation and Related Reactions"; Interscience: New York, 1965; pp. 84–87.8.See: Coates, R. M.; Chen, J. P. Tetrahedron Lett . 1969, 2705–2708 and references cited therein. 9.Truce, W. E. In "Organic Chemistry of Sulfur," Oae, S., Ed.; Plenum Press: New York, 1977; pp.532–536.10.Truce, W. E.; Murphy, A. M. Chem. Rev . 1951, 48, 68–124.11.Trost, B. M. Acc. Chem. Res . 1978, 11, 453–461.12.Davis W.; Dick, J. H. J. Chem. Soc . 1932, 483–484; DeCat, A., Van Poucke, R.; Verbrugghe, M.J. Org. Chem . 1965, 30, 1498–1502.TABLE IC ONVERSION OF ARYLAMINES TOARYLSULFONYL CHLORIDESAmine, XC 6H 4NH 2, X =Yield (%) of XC 6H 4SO 2Clm -CF 372 p -NO 268 m -NO 286 p -Cl90p -CO 2CH 390 3,5-di-NO 281 m -CH 371 H53p -OCH 3 2713.See 5 for a good introduction.14.Yale, H. L. J. Med. Pharm. Chem. 1959, 1, 121.AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sulfonyl chlorideaminep- and m-nitrobenzenesulfonylhydrochloric acid (7647-01-0)acetic acid (64-19-7)ether (60-29-7)chlorosulfonic acid (7790-94-5)phosphorus pentachloride (10026-13-8)thionyl chloride (7719-09-7)sodium bicarbonate (144-55-8)sodium carbonate (497-19-8)sulfur dioxide (7446-09-5)sodium nitrite (7632-00-0)phosgene (75-44-5)cuprous chloride,copper(I) chloride (7758-89-6)magnesium sulfate (7487-88-9)copper(I)benzotrifluoride (98-08-8)copper(II) chloride dihydrate (10125-13-0)m-aminobenzotrifluoride,α,α,α-Trifluoro-m-toluidine(98-16-8)m-Trifluoromethylbenzenesulfonyl chloride,Benzenesulfonyl chloride, m-(trifluoromethyl)- (777-44-6)m-trifluoromethylbenzenediazonium chloridem-trifluoromethylbenzenesulfonic acidCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
第13章重氮化反应

13.2 重氮化反应
13.2.1 重氮化反应的一般条件
• 芳伯胺的重氮化是亲电反应,反应进行的难易与多种因素 有关。
(1)芳胺的影响
• 当芳伯胺的芳环上连有供电子基团时,芳伯胺碱性增强, 反应速度加快;当芳伯胺的芳环上连有吸电子基团时,芳 伯胺碱性减弱,反应速度变慢。
衢州学院化学与材料工程学院
(2)无机酸性质和浓度的影响 • 使用不同性质的无机酸时,在重氮化反应中向芳胺进攻 的亲电质点也不同。
ON+ >ON-Br>ON-Cl>ON-ON2>ON-OH
无机酸 亲电质点 活性 浓H2SO4 NO+ 大
衢州学院化学与材料工程学院
HBr NOBr
HCl NOCl 小
稀H2SO4 N 2 O3
重氮化反应历程是N-亚硝化-脱水反应,可简要表示如下:
衢州学院化学与材料工程学院
• 无机酸的作用与用量
衢州学院化学与材料工程学院来自• 重氮化合物兼有酸和碱的特性,它既可以与酸生成盐,又 可以与碱生成盐。在水介质中,重氮盐的结构转变如下图 所示:
其中亚硝酸和亚硝胺盐比较稳定,而重氮盐、重氮酸和重氮酸盐则比 较活泼,所以重氮盐的反应一般是在强酸性到弱碱性介质中进行的。
衢州学院化学与材料工程学院
• 重氮盐可发生两类反应:一类是重氮基转化为偶氮基(偶 合)或肼基(还原),非脱落氮原子的反应;另一类是重 氮基被其他取代基所置换,同时脱落两个氮原子放出氮气 的反应。 • 重氮盐性质活泼,本身价值不高,但通过上述两类重氮盐 的反应,可制得一系列重要的有机中间体。
• 为了避免生成的酚与重氮盐发生偶合反应生成羟基偶氮染 料,转化反应最好在强酸性溶液(40-50%)中进行,通 常是将冷的重氮盐水溶液滴加到沸腾的稀硫酸中,使重氮 盐迅速水解。
重氮化反应 氨基变巯基

Organic Syntheses, Coll. Vol. 2, p.580 (1943); Vol. 12, p.76 (1932).THIOSALICYLIC ACID[Benzoic acid, o -mercapto-]Submitted by C. F. H. Allen and D. D. MacKay.Checked by Roger Adams and A. E. Knauf. 1. ProcedureCaution! Recently it was reported to us that workers, following the procedure in Coll. Vol. II, pg 580 (diazotization of anthranilic acid and its reaction with sodium disulfide) but substituting 2,3-dimethylaniline for anthranilic acid, experienced a serious explosion upon addition of the diazonium salt solution to the disulfide solution. We urge that extreme caution should always be exercised in the handling of diazonium salts even when they are in solution.In a 4-l. beaker, 290 cc. of water is heated to boiling, and 260 g. (1.1 moles) of crystallized sodium sulfide (Na 2S·9H 2O) and 34 g. of powdered sulfur are dissolved by heating and stirring. A solution of 40 g. of sodium hydroxide in 100 cc. of water is then added and the mixture cooled, first in cold water, and finally by a freezing mixture of ice and salt.In a 2-l. beaker, set in a freezing mixture and provided with a stirrer and a thermometer for reading temperatures to 0°, are placed 500 cc. of water, 137 g. (1 mole) of anthranilic acid , and 200 cc. of concentrated hydrochloric acid ; the stirrer is started and the mixture cooled to about 6°. Meanwhile 69 g. (1 mole) of sodium nitrite is dissolved in 280 cc. of hot water and the solution cooled in ice; portions are then placed in a separatory funnel of convenient size, supported in such a way that the lower end of the stem extends beneath the surface of the anthranilic acid solution. When the temperature has fallen to 5°, the nitrite solution is run in; about 500 g. of cracked ice is added at such a rate as to keep the temperature below 5°. This takes about ten minutes (Note 1). A drop of the solution should give an immediate blue color with starch-iodide paper.The stirrer and thermometer are now transferred to the alkaline sulfide solution, the temperature of which must be below 5°. The diazo solution is added over a period of twenty to thirty minutes along with 950 g. of ice to prevent the temperature from rising above 5°. When addition is complete, the waterbath is removed and the mixture allowed to warm up to room temperature; after two hours the evolution of nitrogen ceases (Note 2). About 180 cc. of concentrated hydrochloric acid is added until the solution is acid to Congo red paper, and the precipitate of dithiosalicylic acid is filtered and washed with water.To remove the excess sulfur, the precipitate is dissolved by boiling with a solution of 60 g. of anhydrous sodium carbonate (soda ash) in 2 l. of water, and the mixture is filtered while hot. It is divided into five equal parts (Note 3), and the dithiosalicylic acid is reprecipitated as before with concentrated hydrochloric acid. The solid is filtered, the cake being sucked as dry as possible.The moist cake is mixed with 27 g. of zinc dust and 300 cc. of glacial acetic acid in a 1-l. round-bottomed flask, and the mixture is refluxed vigorously for about four hours (Note 4). When the reduction is complete, the mixture is cooled and filtered with suction. The filter cake is washed once with water and then transferred to a 1-l. beaker. The cake is suspended in 200 cc. of water, and the suspension is heated to boiling. The hot solution is made strongly alkaline by the addition of about 40 cc. of 33 per cent aqueous sodium hydroxide solution. The alkaline solution is boiled for about twenty minutes to ensure complete extraction of the product from the filter cake, filtered from the insoluble material (Note 5), and the thiosalicylic acid is then precipitated by the addition of sufficient concentrated hydrochloric acid to make the solution acid to Congo red paper. The product is filtered with suction, washed once with water, and dried in an oven at 100–110°. The yield of a product which melts at 162–163° is 110–130 g. (71–84 per cent of the theoretical amount based on the anthranilic acid).This product is sufficiently pure for most purposes (Note 6).For recrystallization 5 g. of this material is dissolved in 20 cc. of hot 95 per cent alcohol, and 40 cc. of water is added. The solution is boiled with a little decolorizing carbon, filtered hot, and then allowed to cool. The product crystallizes in yellow flakes. The yield of recrystallized material is 4.7 g.; the melting point of the material is 163–164°.2. Notes1. This method is much more rapid than when external cooling alone is used (Org. Syn. Coll. Vol. I, 1941, 374). The total volume of the solution is not important since the insoluble dithiosalicylic acid is readily filtered.2. Foaming sometimes becomes very during the evolution of nitrogen. The addition of a few cubic centimeters of ether from time to time helps to keep this foaming under control.3. The dithiosalicylic acid may be precipitated all at once if desired and the entire amount reduced in one operation. If this is done, the reduction must be carried out in a 5-l. flask fitted with a good stirrer. The mixture needs to be refluxed about ten hours over a ring burner. In the laboratory, this is much less convenient than it is to divide the material and reduce in smaller amounts. The yield is not materially lowered by making the reduction in one portion.4. The reduction does not always run smoothly. If the zinc lumps and becomes inactive more must be added. To determine whether reduction is complete, a sample is removed, cooled, and filtered. The precipitate is boiled with strong sodium hydroxide solution, filtered, and then acidified with hydrochloric acid. If the reduction is complete, the precipitated material will melt at 164° or lower. If the reduction is not complete, the precipitated material will melt above 164°. If the reduction is not complete, the refluxing of the main portion must be continued (and perhaps more zinc must be added) until a test portion shows that the reaction is complete.In determining the melting point of the material, the capillary tube containing the test sample should be inserted in a bath previously heated to 163–164°.5. When the reduction is carried out in five portions, one extraction with sodium hydroxide is usually sufficient for each portion. If the reduction is carried out in one operation, several extractions are usually required. When the material is to be extracted more than once, it is best to boil the residue from the first alkaline treatment with hydrochloric acid, filter, and then treat again with the alkali.6. Thiosalicylic acid is used for the preparation of oxythionaphthene and many thioindigoid dyes.3. DiscussionOf the several methods described for the production of thiosalicylic acid, only the following are of preparative interest: heating o-halogenated benzoic acids with an alkaline hydrosulfide at 150–200° in the presence of copper or copper salts,1, 2 or with sodium sulfide at 200°;3 and reduction of dithiosalicylic acid with glucose,4 or metals5, 2 in alkaline solution. The dithiosalicylic acid is prepared by treating diazotized anthranilic acid with sodium disulfide in alkaline solution.5This preparation is referenced from:z Org. Syn. Coll. Vol. 3, 809References and Notes1.(a) Cassella and Company, Ger. pat. 189,200 [C. A. 2, 607 (1908)]; (b) Cain, "IntermediateProducts for Dyes," p. 151.2.Chem. Age 21, Dyestuffs Suppl. p. 11 (1929).3.Cassella and Company, Ger. pat. 193,290 [C. A. 2, 1514 (1908)]; Ref. 1(b).4.Claasz, Ber. 45, 2427 (1912).5.Kalle and Company, Ger. pat. 204,450 [C. A. 3, 1695 (1909)]; Ref. 1(b).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sodium sulfide (Na2S·9H2O)dithiosalicylic acid sodium carbonate (soda ash) copper or copper saltso-halogenated benzoic acidsalcohol (64-17-5) hydrochloric acid (7647-01-0) acetic acid (64-19-7)ether (60-29-7) sodium hydroxide (1310-73-2) nitrogen (7727-37-9) sodium nitrite(7632-00-0)sulfur (7704-34-9)decolorizing carbon (7782-42-5)zinc (7440-66-6)sodium sulfide (1313-82-2)sodium disulfideAnthranilic Acid (118-92-3)glucose (492-62-6)hydrosulfideThiosalicylic acid,Benzoic acid, o-mercapto- (147-93-3)oxythionaphtheneCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变卤素 氯 m-CHLOROBENZALDEHYDE

Organic Syntheses, Coll. Vol. 2, p.130 (1943); Vol. 13, p.28 (1933).m -CHLOROBENZALDEHYDE[Benzaldehyde, m -chloro-]Submitted by Johannes S. Buck and Walter S. Ide.Checked by John R. Johnson and Paul W. Vittum.1. ProcedureA solution of 450 g. (2 moles) of stannous chloride crystals (Note 1) in 600 cc. of concentrated hydrochloric acid is placed in a 3-l. beaker provided with an efficient mechanical stirrer and cooled in an ice bath. When the temperature of the solution has fallen to +5°, 100 g. (0.66 mole) of m -nitrobenzaldehyde (Note 2) is added in one portion. The temperature rises slowly at first, reaching 25–30° in about five minutes, then rises very rapidly to about 100°. Stirring must be vigorous or the reaction mixture may be forced out of the beaker (Note 3). During the reaction the nitrobenzaldehyde dissolves, and an almost clear, red solution is obtained. The solution is cooled in an ice-salt mixture until the temperature has fallen to about +2°. During the cooling, orange-red crystals separate and a pasty suspension results.A 250-cc. separatory funnel is fixed so that its stem extends below the surface of the pasty suspension. A solution of 46 g. (0.67 mole) of sodium nitrite in 150 cc. of water is placed in the funnel and is slowly added to the well-stirred mixture until it shows a positive starch-iodide test for nitrous acid . The temperature of the mixture is maintained between 0° and +5° (Note 4) throughout the addition of the nitrite solution, which requires about ninety minutes. Usually, all but 5–8 cc. of the nitrite solution must be added before a positive test for nitrous acid appears.During the latter part of the diazotization of the aminobenzaldehyde , a hot solution of cuprous chloride is prepared. In a 5-l. round-bottomed flask, 189 g. (0.75 mole) of powdered copper sulfate crystals and 161 g. of sodium chloride are dissolved in 600 cc. of hot water, and to this solution is added a solution of 41 g. (0.22 mole) of sodium metabisulfite (Na 2S 2O 5)and 27 g. (0.67 mole) of sodiumhydroxide in 300 cc. of water. The final temperature of the resulting cuprous chloride solution should be about 75°.The diazonium solution is added to the hot cuprous chloride solution while the latter is shaken by hand but is not cooled. After the solutions are thoroughly mixed, 840 cc. of concentrated hydrochloric acid is added and the mixture is allowed to stand overnight. The reaction mixture is steam-distilled to separate the m-chlorobenzaldehyde, which is collected practically completely in the first 1.5 l. of distillate. The m-chlorobenzaldehyde is removed from the aqueous distillate by extraction with two 150-cc. portions of ether, and the ethereal solution is dried with 10–15 g. of anhydrous calcium chloride. After being decanted from the drying agent, the ether is distilled, and the residual liquid is distilled under diminished pressure. The m-chlorobenzaldehyde boils at 84–86°/8 mm., 107–109°/26 mm. (Note 5). The yield is 70–74 g. (75–79 per cent of the theoretical amount) (Note 6).2. Notes1. A chemically pure grade of stannous chloride crystals (SnCl2·2H2O) was used. Lower yields wereobtained when technical stannous chloride was used.2. A practical grade of m-nitrobenzaldehyde was used; m.p. 52–55°.3. During the vigorous reaction it is advisable to keep the cooling bath and the reaction mixture well stirred. Less satisfactory yields were obtained when the reaction was moderated by adding the nitrobenzaldehyde in several portions.4. At temperatures below 0° the speed of diazotization is markedly decreased. Above +5° some decomposition of the diazonium salt takes place.5. Since m-chlorobenzaldehyde is oxidized easily by atmospheric oxygen, it should be stored in a tightly corked or sealed container.6. According to the submitters m-bromobenzaldehyde can be prepared by the same general procedure using, in place of cuprous chloride, a solution of cuprous bromide prepared from 189 g. of copper sulfate, 91 g. of sodium bromide, 41 g. of sodium metabisulfite, and 27 g. of sodium hydroxide. Instead of 840 cc. of concentrated hydrochloric acid, 200 cc. of 48 per cent hydrobromic acid is added after the diazonium solution has been mixed with the cuprous bromide. The m-bromobenzaldehyde boils at 93–98°/8 mm. The yield is 80 g. or 65 per cent of the theoretical amount.It is reported, however, that m-bromobenzaldehyde prepared in this way may contain as much as 20 per cent of m-chlorobenzaldehyde. This contamination can be avoided by using stannous bromide as the reducing agent.A solution of stannous bromide is prepared by heating 119 g. (1 gram atom) of mossy tin with 705 g. (4 moles) of 46 per cent hydrobromic acid for two hours on a steam bath, with mechanical stirring. The solution is cooled to 40°, and 50 g. (0.33 mole) of m-nitrobenzaldehyde is added in one portion, with continued stirring. The temperature rises from the heat of reaction and finally reaches about 105°. After heating for one-half hour longer on a steam bath, the reaction mixture is cooled to 0° and the aminobenzaldehyde diazotized by the gradual addition of 23 g. (0.33 mole) of sodium nitrite in 75 cc. of water. The diazonium solution is poured into a hot suspension of cuprous bromide, 100 cc. of 46 per cent hydrobromic acid is added, with stirring, and the mixture is allowed to stand overnight. The mixture is steam-distilled and the m-bromobenzaldehyde isolated by ether extraction and vacuum distillation; b.p. 90–92°/4 mm. The yield is 41 g. (67 per cent of the theoretical amount). (F. T. Tyson, private communication.)3. Discussionm-Chlorobenzaldehyde has been prepared by the chlorination of benzaldehyde1 and by the oxidation of m-chlorobenzyl alcohol2 and of m-chlorotoluene.3 It is most conveniently prepared from m-nitrobenzaldehyde through m-aminobenzaldehyde and the diazonium reaction.4 The procedure given above is essentially that described in the patent literature.4This preparation is referenced from:z Org. Syn. Coll. Vol. 2, 583z Org. Syn. Coll. Vol. 3, 453References and Notes1.Müller, Ger. pat. 30,329; 33,064 [Frdl. 1, 143, 146 (1877–87)].2.Mettler, Ber. 38, 2812 (1905).w and Perkin, J. Chem. Soc. 93, 1636 (1908).4.Meister, Lucius, and Brüning, Ger. pat. 31,842 [Frdl. 1, 144 (1877–87)]; Erdmann andSchwechten, Ann. 260, 59 (1890); Eichengrün and Einhorn, ibid. 262, 135 (1891).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sodium metabisulfitecalcium chloride (10043-52-4)hydrochloric acid (7647-01-0)ether (60-29-7)sodium hydroxide (1310-73-2)sodium chloride (7647-14-5)HYDROBROMIC ACID (10035-10-6)sodium bromide (7647-15-6)oxygen (7782-44-7)copper sulfate (7758-98-7)sodium nitrite (7632-00-0)nitrous acid (7782-77-6)tin (7440-31-5)stannous chloridebenzaldehyde (100-52-7)cuprous bromide (7787-70-4)cuprous chloride (7758-89-6)nitrobenzaldehyde(552-89-6)aminobenzaldehyde (529-23-7)stannous bromide (10031-24-0)m-Bromobenzaldehyde (3132-99-8)m-Chlorobenzaldehyde,Benzaldehyde, m-chloro- (587-04-2)m-Nitrobenzaldehyde (99-61-6)m-chlorobenzyl alcohol (873-63-2)m-chlorotoluene (108-41-8)m-aminobenzaldehyde (1709-44-0) Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变巯基m-THIOCRESOL

Organic Syntheses, Coll. Vol. 3, p.809 (1955); Vol. 27, p.81 (1947).m-THIOCRESOL[m-Toluenethiol; m-tolyl mercaptan]Submitted by D. S. Tarbell and D. K. Fukushima.Checked by C. F. H. Allen and John R. Byers, Jr..1. ProcedureAll the steps in this preparation, including sealing the product in bottles or ampoules, should be carried out under a good hood. Care should be exercised to avoid contact with m-thiocresol or its solutions since it is a skin irritant.In a 1-l. flask, equipped with a mechanical stirrer and thermometer for reading low temperatures, and immersed in an ice bath, are placed 150 ml. of concentrated hydrochloric acid (sp. gr. 1.18) and 150 g. of crushed ice. The stirrer is started, and 80 g. (0.75 mole) of m-toluidine (b.p. 92–93°/15 mm.) is slowly added. The mixture is cooled to 0°, and a cold solution of 55 g. (0.8 mole) of sodium nitrite in 125 ml. of water is slowly added, the temperature being kept below 4°.In a 2-l. flask equipped with a thermometer, dropping funnel, and stirrer is placed a solution of 140 g. of potassium ethyl xanthate(Note 1) in 180 ml. of water. This mixture is warmed to 40–45° and kept in that range during the slow addition of the cold diazonium solution (Note 2); about 2 hours is required (Note 3). After an additional 30 minutes at this temperature to ensure complete decomposition of the intermediate compound, the red, oily m-tolyl ethyl xanthate is separated and the aqueous layer is extracted twice, using 100-ml. portions of ether. The combined oil and extracts are washed once with 100 ml. of 10% sodium hydroxide solution (Note 4) and then with several portions of water until the washings are neutral to litmus. The ether solution is dried over 25 g. of anhydrous calcium chloride, and the ether is removed by distillation. The crude residual m-tolyl ethyl xanthate is dissolved in 500 ml. of 95% ethanol, the solution brought to boiling, and the source of heat removed. To this hot solution is added slowly 175 g. of potassium hydroxide pellets so that the solution keeps boiling, and the mixture is refluxed until a sample is completely soluble in water (about 8 hours). Approximately 400 ml. of ethanol is then removed by distillation on a steam bath, and the residue is taken up in the minimum of water (about 500 ml.). The aqueous solution is extracted with three 100-ml. portions of ether, the extract being discarded. The aqueous solution is now made strongly acid to Congo red paper, using 6 N sulfuric acid(Note 5) (625–650 ml.). The acidified solution is placed in a 3-l. flask, 2 g. of zinc dust is added, and the m-thiocresol is distilled with steam. The lower layer of the m-thiocresol is separated; the aqueous layer is extracted with three 100-ml. portions of ether, the extracts being added to the oil. Afterdrying with 50 g. of Drierite, the ether is removed by distillation, and the oily residue is distilled under reduced pressure. The yield of colorless m -thiocresol , b.p. 90–93°/25 mm., is 59–69 g. (63–75%) (Note 6) and (Note 7). It is best preserved in sealed glass bottles because of its disagreeable odor.2. Notes1. Eastman Kodak Company technical potassium ethyl xanthate was used.2. The diazonium solution is left in the ice bath, and only 10- to 15-ml. portions are placed in the dropping funnel at one time.3. Many diazonium solutions have been reported to react explosively with solutions of metallic polysulfides even at low temperatures.1,2 A violent reaction with xanthates is mentioned only in one report.3 Neither the authors nor the checkers observed any unusual reactivity during this preparation or with the procedure given for dithiosalicylic acid.4 On a large scale (100 moles of m -toluidine ) flashes of light have been occasionally observed (private communication, L. J. Roll).4. This wash serves to remove any m -cresol present.5. This acidification liberates carbon oxysulfide, which has a very disagreeable odor.6. The refractive index is n D 1.568–1.571.7. Other boiling points are 195°/760 mm.; 120°/100 mm.; 107°/50 mm.3. DiscussionThe only practical laboratory procedure for preparing m -thiocresol is by the alkaline hydrolysis of m -tolyl ethyl xanthate , obtained from m -toluenediazonium chloride and potassium ethyl xanthate .3,5 The procedure described is essentially that of Bourgeois.5This preparation is referenced from:z Org. Syn. Coll. Vol. 5, 1050References and Notes1.Nawiasky, Ebersole, and Werner, Chem. Eng. News , 23, 1247 (1945). 2.Hodgson, Chemistry & Industry , 1945, 362. 3.Leuckart, J. prakt. Chem., [2] 41, 189 (1890). 4.Org. Syntheses Coll. Vol. 2, 580 (1943). 5.Bourgeois, Rec. trav. chim., 18, 447 (1899).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)carbon oxysulfideDrieriteethanol (64-17-5)calcium chloride (10043-52-4)sulfuric acid (7664-93-9)25hydrochloric acid (7647-01-0)ether (60-29-7)sodium hydroxide (1310-73-2)sodium nitrite (7632-00-0)potassium hydroxide (1310-58-3)zinc (7440-66-6)potassium ethyl xanthate (140-89-6)m-Thiocresol,m-Toluenethiol,m-tolyl mercaptan (108-40-7)m-toluidine (108-44-1)m-tolyl ethyl xanthatem-cresol (108-39-4)m-toluenediazonium chloride Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变巯基
Organic Syntheses, Coll. Vol. 2, p.580 (1943); Vol. 12, p.76 (1932).THIOSALICYLIC ACID[Benzoic acid, o -mercapto-]Submitted by C. F. H. Allen and D. D. MacKay.Checked by Roger Adams and A. E. Knauf. 1. ProcedureCaution! Recently it was reported to us that workers, following the procedure in Coll. Vol. II, pg 580 (diazotization of anthranilic acid and its reaction with sodium disulfide) but substituting 2,3-dimethylaniline for anthranilic acid, experienced a serious explosion upon addition of the diazonium salt solution to the disulfide solution. We urge that extreme caution should always be exercised in the handling of diazonium salts even when they are in solution.In a 4-l. beaker, 290 cc. of water is heated to boiling, and 260 g. (1.1 moles) of crystallized sodium sulfide (Na 2S·9H 2O) and 34 g. of powdered sulfur are dissolved by heating and stirring. A solution of 40 g. of sodium hydroxide in 100 cc. of water is then added and the mixture cooled, first in cold water, and finally by a freezing mixture of ice and salt.In a 2-l. beaker, set in a freezing mixture and provided with a stirrer and a thermometer for reading temperatures to 0°, are placed 500 cc. of water, 137 g. (1 mole) of anthranilic acid , and 200 cc. of concentrated hydrochloric acid ; the stirrer is started and the mixture cooled to about 6°. Meanwhile 69 g. (1 mole) of sodium nitrite is dissolved in 280 cc. of hot water and the solution cooled in ice; portions are then placed in a separatory funnel of convenient size, supported in such a way that the lower end of the stem extends beneath the surface of the anthranilic acid solution. When the temperature has fallen to 5°, the nitrite solution is run in; about 500 g. of cracked ice is added at such a rate as to keep the temperature below 5°. This takes about ten minutes (Note 1). A drop of the solution should give an immediate blue color with starch-iodide paper.The stirrer and thermometer are now transferred to the alkaline sulfide solution, the temperature of which must be below 5°. The diazo solution is added over a period of twenty to thirty minutes along with 950 g. of ice to prevent the temperature from rising above 5°. When addition is complete, the waterbath is removed and the mixture allowed to warm up to room temperature; after two hours the evolution of nitrogen ceases (Note 2). About 180 cc. of concentrated hydrochloric acid is added until the solution is acid to Congo red paper, and the precipitate of dithiosalicylic acid is filtered and washed with water.To remove the excess sulfur, the precipitate is dissolved by boiling with a solution of 60 g. of anhydrous sodium carbonate (soda ash) in 2 l. of water, and the mixture is filtered while hot. It is divided into five equal parts (Note 3), and the dithiosalicylic acid is reprecipitated as before with concentrated hydrochloric acid. The solid is filtered, the cake being sucked as dry as possible.The moist cake is mixed with 27 g. of zinc dust and 300 cc. of glacial acetic acid in a 1-l. round-bottomed flask, and the mixture is refluxed vigorously for about four hours (Note 4). When the reduction is complete, the mixture is cooled and filtered with suction. The filter cake is washed once with water and then transferred to a 1-l. beaker. The cake is suspended in 200 cc. of water, and the suspension is heated to boiling. The hot solution is made strongly alkaline by the addition of about 40 cc. of 33 per cent aqueous sodium hydroxide solution. The alkaline solution is boiled for about twenty minutes to ensure complete extraction of the product from the filter cake, filtered from the insoluble material (Note 5), and the thiosalicylic acid is then precipitated by the addition of sufficient concentrated hydrochloric acid to make the solution acid to Congo red paper. The product is filtered with suction, washed once with water, and dried in an oven at 100–110°. The yield of a product which melts at 162–163° is 110–130 g. (71–84 per cent of the theoretical amount based on the anthranilic acid).This product is sufficiently pure for most purposes (Note 6).For recrystallization 5 g. of this material is dissolved in 20 cc. of hot 95 per cent alcohol, and 40 cc. of water is added. The solution is boiled with a little decolorizing carbon, filtered hot, and then allowed to cool. The product crystallizes in yellow flakes. The yield of recrystallized material is 4.7 g.; the melting point of the material is 163–164°.2. Notes1. This method is much more rapid than when external cooling alone is used (Org. Syn. Coll. Vol. I, 1941, 374). The total volume of the solution is not important since the insoluble dithiosalicylic acid is readily filtered.2. Foaming sometimes becomes very during the evolution of nitrogen. The addition of a few cubic centimeters of ether from time to time helps to keep this foaming under control.3. The dithiosalicylic acid may be precipitated all at once if desired and the entire amount reduced in one operation. If this is done, the reduction must be carried out in a 5-l. flask fitted with a good stirrer. The mixture needs to be refluxed about ten hours over a ring burner. In the laboratory, this is much less convenient than it is to divide the material and reduce in smaller amounts. The yield is not materially lowered by making the reduction in one portion.4. The reduction does not always run smoothly. If the zinc lumps and becomes inactive more must be added. To determine whether reduction is complete, a sample is removed, cooled, and filtered. The precipitate is boiled with strong sodium hydroxide solution, filtered, and then acidified with hydrochloric acid. If the reduction is complete, the precipitated material will melt at 164° or lower. If the reduction is not complete, the precipitated material will melt above 164°. If the reduction is not complete, the refluxing of the main portion must be continued (and perhaps more zinc must be added) until a test portion shows that the reaction is complete.In determining the melting point of the material, the capillary tube containing the test sample should be inserted in a bath previously heated to 163–164°.5. When the reduction is carried out in five portions, one extraction with sodium hydroxide is usually sufficient for each portion. If the reduction is carried out in one operation, several extractions are usually required. When the material is to be extracted more than once, it is best to boil the residue from the first alkaline treatment with hydrochloric acid, filter, and then treat again with the alkali.6. Thiosalicylic acid is used for the preparation of oxythionaphthene and many thioindigoid dyes.3. DiscussionOf the several methods described for the production of thiosalicylic acid, only the following are of preparative interest: heating o-halogenated benzoic acids with an alkaline hydrosulfide at 150–200° in the presence of copper or copper salts,1, 2 or with sodium sulfide at 200°;3 and reduction of dithiosalicylic acid with glucose,4 or metals5, 2 in alkaline solution. The dithiosalicylic acid is prepared by treating diazotized anthranilic acid with sodium disulfide in alkaline solution.5This preparation is referenced from:z Org. Syn. Coll. Vol. 3, 809References and Notes1.(a) Cassella and Company, Ger. pat. 189,200 [C. A. 2, 607 (1908)]; (b) Cain, "IntermediateProducts for Dyes," p. 151.2.Chem. Age 21, Dyestuffs Suppl. p. 11 (1929).3.Cassella and Company, Ger. pat. 193,290 [C. A. 2, 1514 (1908)]; Ref. 1(b).4.Claasz, Ber. 45, 2427 (1912).5.Kalle and Company, Ger. pat. 204,450 [C. A. 3, 1695 (1909)]; Ref. 1(b).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sodium sulfide (Na2S·9H2O)dithiosalicylic acid sodium carbonate (soda ash) copper or copper saltso-halogenated benzoic acidsalcohol (64-17-5) hydrochloric acid (7647-01-0) acetic acid (64-19-7)ether (60-29-7) sodium hydroxide (1310-73-2) nitrogen (7727-37-9) sodium nitrite(7632-00-0)sulfur (7704-34-9)decolorizing carbon (7782-42-5)zinc (7440-66-6)sodium sulfide (1313-82-2)sodium disulfideAnthranilic Acid (118-92-3)glucose (492-62-6)hydrosulfideThiosalicylic acid,Benzoic acid, o-mercapto- (147-93-3)oxythionaphtheneCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变卤素 o-BROMOTOLUENE

Organic Syntheses, Coll. Vol. 1, p.135 (1941); Vol. 9, p.22 (1929).o-BROMOTOLUENE[Toluene, o-bromo-]Submitted by L. A. BigelowChecked by C. S. Marvel and S. V. Puntambeker.1. ProcedureA solution of 162 g. (1.5 moles) of commercial o-toluidine in 880 cc. (6 moles) of 40 per cent commercial hydrobromic acid(Note 1) in a 3-l. flask is cooled to 10° and diazotized with 116 g. (1.7 moles) of coarsely powdered commercial sodium nitrite, added about 10 g. at a time. After each addition the flask is stoppered and shaken until all the red fumes are absorbed. The temperature must be kept below 10°. When diazotization is complete, 5 g. of copper powder(Note 2) is added, the flask is attached to a reflux condenser and heated very cautiously. As soon as the first sign of reaction is observed, the flask is cooled with ice. Nitrogen is evolved vigorously. When the reaction subsides, the mixture is heated one-half hour on the steam bath.Then 1 l. of water is added and the mixture is distilled with steam until about 1.5 l. has passed over. The distillate is made alkaline with about 10 g. of powdered sodium hydroxide and the lower red layer of crude o-bromotoluene separated (Note 3). This weighs about 140 g. It is washed with concentrated sulfuric acid, which removes almost all the color, and then twice with water. It is dried over a little calcium chloride, filtered and distilled twice from a modified Claisen flask (p. 130). The yield of pure product boiling at 178–181° is 110–120 g. (43–47 per cent of the theoretical amount).2. Notes1. If 48 per cent (constant boiling) hydrobromic acid is used the diazotization is very difficult to control. The reaction becomes very vigorous and forces out the stopper.2. Either reduced copper or fine copper filings may be used.3. This gives as good results as when the o-bromotoluene is extracted from the alkaline mixture with ether.3. Discussiono-Bromotoluene can be prepared by the bromination of toluene;1 by the bromination of potassium p-toluenesulfonate and subsequent hydrolysis;2 and by the diazotization of o-toluidine under different conditions.3References and Notes1.Jannasch and Hübner, Ann. 170, 117 (1873); Varma and Narayan, Quart. J. Indian Chem. Soc. 4,283 (1927) [Chem. Zentr. I, 489 (1928)].ler, J. Chem. Soc. 61, 1027 (1892).3.Wroblevsky, Ann. 168, 171 (1873); Körner, Gazz. chim. ital. 4, 305 (1874); Jackson, Am. Chem.J. 1, 100 (1879); Feitler, Z. physik. Chem. 4, 72 (1889); Neogi and Mitra, J. Chem. Soc. 1332 (1928).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)calcium chloride (10043-52-4)sulfuric acid (7664-93-9)ether (60-29-7)sodium hydroxide (1310-73-2)HYDROBROMIC ACID (10035-10-6)nitrogen (7727-37-9)sodium nitrite (7632-00-0)copper,copper powder,copper filings (7440-50-8)toluene (108-88-3)o-Bromotoluene,Toluene, o-bromo- (95-46-5)o-toluidine (95-53-4)potassium p-toluenesulfonateCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变卤素 p-FLUOROBENZOIC ACID
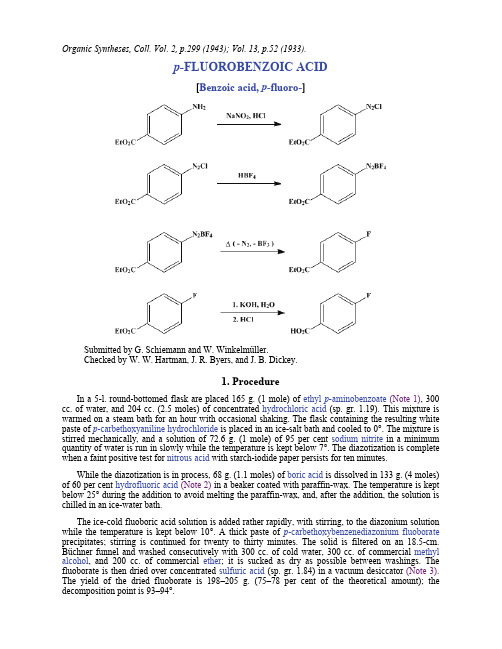
Organic Syntheses, Coll. Vol. 2, p.299 (1943); Vol. 13, p.52 (1933).p-FLUOROBENZOIC ACID[Benzoic acid, p-fluoro-]Submitted by G. Schiemann and W. Winkelmüller.Checked by W. W. Hartman, J. R. Byers, and J. B. Dickey.1. ProcedureIn a 5-l. round-bottomed flask are placed 165 g. (1 mole) of ethyl p-aminobenzoate(Note 1), 300 cc. of water, and 204 cc. (2.5 moles) of concentrated hydrochloric acid (sp. gr. 1.19). This mixture is warmed on a steam bath for an hour with occasional shaking. The flask containing the resulting white paste of p-carbethoxyaniline hydrochloride is placed in an ice-salt bath and cooled to 0°. The mixture is stirred mechanically, and a solution of 72.6 g. (1 mole) of 95 per cent sodium nitrite in a minimum quantity of water is run in slowly while the temperature is kept below 7°. The diazotization is complete when a faint positive test for nitrous acid with starch-iodide paper persists for ten minutes.While the diazotization is in process, 68 g. (1.1 moles) of boric acid is dissolved in 133 g. (4 moles) of 60 per cent hydrofluoric acid(Note 2) in a beaker coated with paraffin-wax. The temperature is kept below 25° during the addition to avoid melting the paraffin-wax, and, after the addition, the solution is chilled in an ice-water bath.The ice-cold fluoboric acid solution is added rather rapidly, with stirring, to the diazonium solution while the temperature is kept below 10°. A thick paste of p-carbethoxybenzenediazonium fluoborate precipitates; stirring is continued for twenty to thirty minutes. The solid is filtered on an 18.5-cm. Büchner funnel and washed consecutively with 300 cc. of cold water, 300 cc. of commercial methyl alcohol, and 200 cc. of commercial ether; it is sucked as dry as possible between washings. The fluoborate is then dried over concentrated sulfuric acid (sp. gr. 1.84) in a vacuum desiccator (Note 3). The yield of the dried fluoborate is 198–205 g. (75–78 per cent of the theoretical amount); the decomposition point is 93–94°.The thermal decomposition may be conveniently carried out in a 2-l. distilling flask. A second distilling flask of 1-l. capacity is connected directly to the side arm of the first to serve as a receiver. Attached to the side arm of the receiver is a rubber tube arranged to lead the escaping gases over 2 l. of water in a 5-l. round-bottomed flask. The boron trifluoride dissolves in the water, and the other gases are led into a good hood (Note 4). The p-carbethoxybenzenediazonium fluoborate is placed in the decomposition flask and heated at its upper edge with a Bunsen flame. When the white fumes of boron trifluoride commence to appear, the flame is removed and the decomposition is permitted to proceed spontaneously. The heat is applied as necessary, and finally the flask is strongly heated to complete the decomposition and melt the solid. Some of the ethyl ester, b.p. 105–106°/25 mm., of p-fluorobenzoic acid is collected in the receiver, where it is carried by the gases, but the larger part is left in the decomposition flask. The ester is washed from the decomposition flask and the receiver with ether, and the ether is distilled from a steam bath. The residue is refluxed for one hour on a steam bath with a solution of 56 g. (1 mole) of potassium hydroxide in 80 cc. of 95 per cent ethyl alcohol and 120 cc. of water. The solution is then filtered while still hot. The p-fluorobenzoic acid is precipitated by adding concentrated hydrochloric acid to the hot filtrate until the mixture is acid to Congo paper. After the mixture has cooled, the solid is filtered and allowed to dry. For purification, the p-fluorobenzoic acid is dissolved in a hot solution of 40 g. of potassium carbonate in 400 cc. of water; the solution is treated with Norite and filtered hot. Hydrochloric acid is added with stirring to precipitate the fluorobenzoic acid, which is then cooled, filtered, and dried.When 85 g. (0.32 mole) of p-carbethoxybenzenediazonium fluoborate is thus decomposed, there is obtained 38–40 g. of p-fluorobenzoic acid (84–89 per cent of the theoretical amount based on the fluoborate; 63–69 per cent based on the ester of p-aminobenzoic acid). The melting point of the purified acid is 186°. The crude acid melts at 183–184°.2. Notes1. Ethyl p-aminobenzoate can be prepared by reduction of the corresponding nitro compound as described in Org. Syn. Coll. Vol. I, 1941, 240, or by esterification of p-aminobenzoic acid: 274 g. (2 moles) of p-aminobenzoic acid is added to 1.4 kg. of ethyl alcohol which has been saturated with dry hydrogen chloride. The reaction mixture is refluxed for twenty-four hours, poured into water, neutralized with sodium carbonate, and the insoluble ester is separated by filtration. The yield is 80 per cent of the theoretical amount.2. Hydrofluoric acid causes extremely painful burns. Exposed parts of the body must be protected when working with this pare Note 3, p. 297.For preparing the fluoboric acid a lead jar may be conveniently used instead of the beaker lined with paraffin-wax. By using a lead stirrer of the usual shape, mechanical stirring may be substituted. The stirrer should be thrust through a hole in a lead cover of sufficient size to prevent spattering of the hydrofluoric acid.Forty per cent fluoboric acid is now available commercially; 200 g. of the commercial product may be used in the procedure above.3. It is very important that the fluoborate be dry. If this solid is wet, the decomposition is very violent, tar is formed, and the yield is lowered.4. A simpler apparatus for the decomposition is a 2-l. round-bottomed short-necked flask connected by means of a wide (2-cm.) bent tube to a 1-l. flask containing 500 cc. of water. The gases from the second flask are led to a good hood.3. Discussionp-Fluorobenzoic acid has been prepared by the oxidation of p-fluorotoluene with chromic acid in dilute sulfuric acid at 160°;1 by the oxidation of p-fluorotoluene with potassium permanganate;2 by the electrolytic oxidation of p-fluorotoluene;3 by heating p-carboxybenzenediazonium chloride with fuming hydrofluoric acid;4 by the oxidation of p-fluorobenzaldehyde;5 by the oxidation of p,p'-difluorostilbene with potassium permanganate;6 by the oxidation of 4,4'-difluorobiphenyl with nitric acid;7 and by the oxidation of 4,4'-difluorobiphenyl with chromic acid.8 The above method is adapted from that of Balz and Schiemann.9References and Notes1.Wallach, Ann. 235, 263 (1886).2.Slothouwer, Rec. trav. chim. 33, 324 (1914); Holleman, ibid. 25, 332 (1906); Holleman andSlothouwer, Proc. K. Akad. Wetensch. Amsterdam 19, 497, 500 (1910) (Chem. Zentr. 1911, I,74); Koopal, Rec. trav. chim. 34, 152 (1915); Meyer and Hub, Monatsh. 31, 933 (1910).3.Fichter and Rosenzweig, Helv. Chim. Acta 16, 1154 (1933).4.Schmitt and Gehren, J. prakt. Chem. (2) 1, 394 (1870); Paterno and Oliveri, Gazz. chim. ital. 12,87 (1882).5.Rinkes, Chem. Weekblad 16, 206 (1919).6.Meyer and Hoffmann, Monatsh. 38, 154 (1917).7.v. Hove, Bull. acad. roy. Belg. [5] 12, 801 (1926) (Chem. Zentr. 1927, I, 884).8.Schiemann and Roselius, Ber. 62, 1813 (1929).9.Balz and Schiemann, ibid. 60, 1186 (1927).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)fluoboric acidethyl alcohol (64-17-5)potassium carbonate (584-08-7)sulfuric acid (7664-93-9)hydrogen chloride,hydrochloric acid (7647-01-0)methyl alcohol (67-56-1)ether (60-29-7)nitric acid (7697-37-2)potassium permanganate (7722-64-7)sodium carbonate (497-19-8)sodium nitrite (7632-00-0)nitrous acid (7782-77-6)hydrofluoric acid (7664-39-3)Norite (7782-42-5)potassium hydroxide(1310-58-3)chromic acid (7738-94-5)boric acid (10043-35-3)boron trifluoride (7637-07-2)fluorobenzoic acid (445-29-4)Ethyl p-aminobenzoate (94-09-7)4,4'-DIFLUOROBIPHENYL (398-23-2)p-aminobenzoic acid,ester of p-aminobenzoic acid (150-13-0)p-Fluorobenzoic acid,Benzoic acid, p-fluoro- (456-22-4)p-carbethoxyaniline hydrochloridep-Carbethoxybenzenediazonium fluoboratep-fluorotoluene (352-32-9)p-carboxybenzenediazonium chloridep-fluorobenzaldehyde (459-57-4)p,p'-difluorostilbeneCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应 氨基变卤素 o-CHLOROBROMOBENZENE

Organic Syntheses, Coll. Vol. 3, p.185 (1955); Vol. 24, p.22 (1944).o-CHLOROBROMOBENZENE[Benzene, 1-bromo-2-chloro-]Submitted by Jonathan L. HartwellChecked by H. R. Snyder and Zeno Wicks, Jr..1. ProcedureA mixture of 127.5 g. (1 mole) of a good commercial grade of o-chloroaniline and 300 ml. (2.5 moles) of 48% hydrobromic acid(Note 1) in a 2-l. flask set in an ice bath is cooled to 0° by the addition of ice. A solution of 70 g. (1 mole) of sodium nitrite in 125 ml. of water is added rapidly, with stirring, the temperature being kept below 10° by the addition of small pieces of ice. When only about 5 ml. of the sodium nitrite solution remains, further additions are made cautiously until an excess of nitrous acid remains after the last addition (Note 2).In the meantime, a mixture of 79 g. (0.55 mole) of cuprous bromide(Note 3) and 80 ml. (0.6 mole) of 48% hydrobromic acid(Note 1) is heated to boiling in a 5-l. round-bottomed three-necked flask, equipped with a condenser set for distillation and provided with a 2-l. receiving flask, a steam inlet tube closed by a screw clamp, and a separatory funnel. About one-fourth of the diazonium solution is transferred to the separatory funnel, without filtration, and immediately run into the cuprous bromide-hydrobromic acid solution, which is kept boiling over a free flame, at such a rate that boiling is continuous. When the separatory funnel is nearly empty a further portion of the cold diazonium solution is transferred to it without interrupting the addition. All the diazonium solution is added in this way over a period of about 30 minutes, during which time much of the product steam-distils. When the addition is complete, the stopcock in the separatory funnel is closed, the screw clamp in the steam line is opened, and a vigorous current of steam is passed through the mixture until no more organic material distils. About 1–1.5 l. of distillate is collected.The heavy organic layer is separated from the distillate and washed with 10-ml. portions of concentrated sulfuric acid until the acid becomes only slightly colored during the washings; four washings usually suffice. The oil is then washed with one 100-ml. portion of water, two 50-ml. portions of 5% aqueous sodium hydroxide, and finally with one 100-ml. portion of water. The product is dried over about 3 g. of calcium chloride and distilled from a 250-ml. distilling flask. The yield of pure, colorless o-chlorobromobenzene, boiling at 199–201°/742 mm., is 170–183 g. (89–95%) (Note 4) and (Note 5).2. Notes1. When 40% hydrobromic acid is used in both the diazotization and Sandmeyer reaction the yield is only about 75%.2. Free nitrous acid causes an immediate blue color at the point of contact with starch-iodide test paper.A delayed color or a color around the periphery of the wetted area is of no significance. At all timesthere must be an excess of mineral acid (blue color on Congo paper).3. The submitter used commercial cuprous bromide. The checkers prepared cuprous bromide by dissolving 600 g. (2.4 moles) of commercial copper sulfate crystals and 350 g. (3.4 moles) of sodium bromide in 2 l. of warm water; the solution was stirred while 151 g. (1.2 moles) of solid sodium sulfite was added over a period of 10 minutes. Occasionally a little more sodium sulfite was required to discharge the blue color. The mixture was cooled, and the solid collected on an 8-in. Büchner funnel, washed once with water, pressed nearly dry, and then dried in the air overnight. The yield of cuprous bromide was 320 g. (93%).4. Runs 3 times this size give proportional yields.5. The checkers have prepared the following bromides by the same procedure: m-chlorobromobenzene(b.p. 191–194°) from m-chloroaniline in 91–94% yields; m-dibromobenzene (b.p. 215–217°) from m-bromoaniline in 80–87% yields; and o-bromoanisole (b.p. 114–116°/29 mm.) from o-anisidine in 88–93% yields. In the preparation of o-bromoanisole the washing with sulfuric acid was omitted.3. Discussiono-Chlorobromobenzene has been prepared by the diazotization of o-bromoaniline followed by replacement of the diazonium group by chlorine;1 by the elimination of the amino group from 3-chloro-4-bromoaniline;2 by the chlorination of bromobenzene in the presence of thallous chloride,3aluminum chloride,4 or ferric chloride;4 by the bromination of chlorobenzene without a catalyst5 or in the presence of aluminum,4iron,4ferric bromide,4 or aluminum-mercury couple;6 by the diazotization of o-chloroaniline followed by replacement of the diazonium group with bromine;4,7 from o-chlorophenylmercuric chloride by the action of bromine;8 and by treatment of silver o-chlorobenzoate with bromine.9This preparation is referenced from:z Org. Syn. Coll. Vol. 3, 200z Org. Syn. Coll. Vol. 6, 36References and Notes1.Dobbie and Marsden, J. Chem. Soc., 73, 254 (1898).2.Wheeler and Valentine, Am. Chem. J., 22, 266 (1899).3.Thomas, Compt. rend., 144, 33 (1907).4.Vander Linden, Rec. trav. chim., 30, 305 (1911).5.Van Loon and Wibaut, Rec. trav. chim., 56, 815 (1937).6.Sen and Bhargava, J. Indian Chem. Soc., 25, 277 (1948).7.Narbutt, Ber, 52, 1028 (1919).8.Hanke, J. Am. Chem. Soc., 45, 1321 (1923).9.Dauben and Tilles, J. Am. Chem. Soc., 72, 3185 (1950).AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)cuprous bromide-hydrobromic acidcalcium chloride (10043-52-4)sulfuric acid(7664-93-9)sodium sulfite (7757-83-7)sodium hydroxide (1310-73-2)iron (7439-89-6) HYDROBROMIC ACID (10035-10-6) bromine (7726-95-6)sodium bromide (7647-15-6)copper sulfate (7758-98-7)sodium nitrite (7632-00-0)nitrous acid (7782-77-6)aluminum (7429-90-5)chlorobenzene (108-90-7)aluminum chloride (3495-54-3)chlorine (7782-50-5)bromobenzene (108-86-1)cuprous bromide (7787-70-4)ferric chloride (7705-08-0)Benzene, 1-bromo-2-chloro-,o-Chlorobromobenzene (694-80-4) 3-chloro-4-bromoaniline (21402-26-6)thallous chlorideferric bromide (10031-26-2)aluminum-mercurym-chloroaniline (108-42-9)o-chloroaniline (95-51-2)o-bromoaniline (615-36-1)m-bromoaniline(591-19-5)o-bromoanisole (578-57-4)m-chlorobromobenzene (108-37-2)m-dibromobenzene (108-36-1)o-anisidine (90-04-0)o-chlorophenylmercuric chloridesilver o-chlorobenzoate Copyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
重氮化反应氨基变硝基1,4-DINITRONAPHTHALENE

重氮化反应氨基变硝基1,4-DINITRONAPHTHALENEOrganic Syntheses, Coll. Vol. 3, p.341 (1955); Vol. 28, p.52 (1948).1,4-DINITRONAPHTHALENE[Naphthalene, 1,4-dinitro-]Submitted by H. H. Hodgson, A. P. Mahadevan, and E. R. Ward.Checked by C. C. Price and Sing-Tuh Voong.1. ProcedureTen grams (0.14 mole) of powdered sodium nitrite is dissolved in 50 ml. of concentrated sulfuric acid (sp. gr. 1.84) contained in a 1-l. beaker placed in an ice bath. A solution of 10 g. of 4-nitro-1-naphthylamine (0.053 mole) (p. 664) in 100 ml. of glacial acetic acid is prepared by heating, and the well-stirred solution is cooled below 20°. Some crystals separate. The resulting thin slurry is dropped slowly into the cold solution of nitrosylsulfuric acid with mechanical stirring. Throughout the addition, and for 30 minutes thereafter, the temperature is kept below 20°. Seven hundred milliliters of dry ether is added slowly with stirring, and the temperature of the mixtur e is kept at 0° for 1 hour. At the end of this period, the precipitation (aided byscratching) of the crystalline 4-nitronaphthalene-1-diazonium sulfate is complete (Note 1). This precipitate is collected, washed with ether and then with 95% ethanol until all the acid is removed, and finally dissolved in 100 ml. of iced water.A saturated aqueous solution containing 50 g. of crystalline copper sulfate is treated with a similar solution of 50 g. of crystallized sodium sulfite. The greenish brown precipitate is collected, washed with water (Note 2), and then stirred into a solution of 100 g. (1.45 moles) of sodium nitrite in 400 ml. of water contained in a 2-l. beaker provided with an efficient mechanical stirrer.The cold aqueous solution of the diazonium salt is then added slowly to the decomposition mixture. Considerable frothing occurs, and 4–5 ml. of ether is added from time to time to break the foam. After stirring for 1 hour (Note 3), the mixture is filtered and the crude dark brown precipitate of the 1,4-dinitronaphthalene is washed several times with water, then with 2% aqueous sodium hydroxide, and again with water. The precipitate is dried and extracted three times with boiling 95% ethanol (450 ml. in all). The extract is concentrated to 75 ml.; most of the 1,4-dinitronaphthalene separates and is collected. Additional amounts can be obtained by further concentration. The resulting product melts at 130–132° and weighs 6.0–7.0 g. (52–60%). The product can be purified either by steam distillation (Note 4) or by recrystallization from aqueous ethanol. Pale yellow needles melting at 134° are obtained (Note 5) and (Note 6).2. Notes1. This precipitate is sometimes sticky. It can be made granular by treating it with a small amount of 95% ethanol (after the removal of the supernatant liquid). Alternatively, it maysuffice to keep the ethereal diazotized solution cold and scratch the sides of the beaker with a glass rod.2. The cupro-cupri sulfite of this variety is more efficient as a decomposition reagent than the red-violet precipitate obtained by treating a hot solution of copper sulfate with a solution of ammonium sulfite saturated with sulfur dioxide and subsequently heating the mixture for 10 minutes at 90°.3. The decomposition appears to be immediate, and at the end of 1 hour most of the inorganic material has passed into solution.4. Steam distillation of 1,4-dinitronaphthalene is very slow. However, the cupro-cupri sulfite method1 isa general one for the replacement of the diazonium group by the nitro group, and steam distillation is preferable whenever the product is readily volatile.5. The solution is decolorized with charcoal in the course of the recrystallization. The checkers obtained5 g. (43%) of golden needles after three recrystallizations.6. 1,2-Dinitronaphthalene may be obtained similarly from 2-nitro-1-naphthylamine, or less satisfactorily from 1-nitro-2-naphthylamine; 1,6- and 2,6-dinitronaphthalenes can be prepared from the 5-nitro- and 6-nitro-2-naphthylamines, respectively, by a modification of the process. Since the solubility of these amines in glacial acetic acid is slight, it is preferable to prepare the diazonium sulfate as follows: Ten grams of the amine is dissolved in 50 ml. of sulfuric acid (sp. gr. 1.84), and the solution is mixed with one of 10 g. of sodium nitrite in 50 ml. of sulfuric acid (sp. gr. 1.84). This mixture is stirred into 200 ml. of glacial acetic acid. The temperature is maintained below 20° throughout these operations. After 30 minutes, the diazonium sulfate isprecipitated at 0° by the addition of 200–500 ml. of ether as previously described.3. Discussion1,4-Dinitronaphthalene has been prepared previously from diazotized 4-nitro-1-naphthylamine by a modified Sandmeyer procedure,2,3 from 5,8-dinitrotetralin by dehydrogenation,4 by the deamination of 1,4-dinitro-2-naphthylamine,5 and by the decomposition of 4-nitro-1-naphthalenediazonium cobaltinitrite.6 The method described above has been published.1References and Notes1.Hodgson, Mahadevan, and Ward, J. Chem. Soc., 1947, 1392.2.Vesely and Dvorak, Bull. soc. chim. France, 33, 319 (1923).3.Contardi and Mor, Rend. ist. lombardo sci., 57, 646 (1924)[C. A., 19, 827 (1925)].4.Chudozilov, Collection Czechoslov. Chem. Commun., 1, 302 (1929) [C. A., 23, 4212 (1929)].5.Hodgson and Hathway, J. Chem. Soc., 1945, 453.6.Hodgson and Ward, J. Chem. Soc., 1947, 127.AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)diazonium sulfatecupro-cupri sulfite1,6- and 2,6-dinitronaphthalenes5-nitro-and 6-nitro-2-naphthylamines4-nitro-1-naphthalenediazonium cobaltinitriteethanol (64-17-5)sulfuric acid (7664-93-9)acetic acid (64-19-7)ether (60-29-7)sodium sulfite (7757-83-7)sodium hydroxide (1310-73-2)sulfur dioxide (7446-09-5)copper sulfate (7758-98-7)sodium nitrite (7632-00-0)charcoal (7782-42-5)nitrosylsulfuric acid (7782-78-7)1,4-Dinitronaphthalene,Naphthalene, 1,4-dinitro- (6921-26-2)4-nitro-1-naphthylamine (776-34-1)4-nitronaphthalene-1-diazonium sulfateammonium sulfite (10196-04-0)1,2-Dinitronaphthalene2-nitro-1-naphthylamine (607-23-8)1-nitro-2-naphthylamine5,8-dinitrotetralin1,4-dinitro-2-naphthylamine Copyright ? 1921-2005, Organic Syntheses, Inc. All Rights Reserved。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Organic Syntheses, Coll. Vol. 7, p.508 (1990); Vol. 60, p.121 (1981).m-TRIFLUOROMETHYLBENZENESULFONYL CHLORIDE[Benzenesulfonyl chloride, m-(trifluoromethyl)-]Submitted by R. V. Hoffman1Checked by G. Saucy, G. P. Roth, and J. W. Scott.1. ProcedureCaution! All operations should be carried out in a hood! m-Trifluoromethylbenzenesulfonyl chloride is a lachrymator. Spills should be treated with saturated sodium carbonate.α,α,α-Trifluoro-m-toluidine(m-aminobenzotrifluoride) (96.7 g, 0.6 mol) (Note 1) is added in one portion to a mixture of concentrated hydrochloric acid (200 mL) and glacial acetic acid (60 mL) in a 1000-mL beaker arranged for efficient mechanical stirring (Note 2). The white hydrochloride salt precipitates (Note 3). The beaker is placed in a dry ice-ethanol bath and, when the temperature of the stirred mixture has reached −10°C, a solution of sodium nitrite (44.8 g, 0.65 mol) in water (65 mL) is added dropwise at such a rate that the temperature does not exceed −5°C (Note 4). After all the sodium nitrite solution has been added, the mixture is stirred for 45 min while the temperature is maintained between −10°C and −5°C (Note 5).While the diazotization is being completed, glacial acetic acid (600 mL) is placed in a 4000-mL beaker and stirred magnetically. Sulfur dioxide is introduced by a bubbler tube with a fritted end immersed below the surface of the acetic acid until saturation is evident (Note 6). Cuprous chloride (15 g) (Note 7) is added to the solution. The introduction of sulfur dioxide is continued until the yellow–green suspension becomes blue-green. Most of the solids dissolve during this time (20–30 min). The mixture is then placed in an ice bath and cooled with stirring. When the temperature approaches 10°C, the diazotization reaction mixture (Note 8) is added in portions over a 30-min period to the sulfur dioxide solution. Considerable foaming occurs after each addition, and this can be disrupted with a few drops of ether. The temperature rises during the addition, but it should not exceed 30°C. After all the diazonium salt mixture has been added, the mixture is poured into ice water (1 : 1, 2000 mL), stirred magnetically until the ice has melted, and added to a 4000-mL separatory funnel. The product separates as a yellow oil that is drawn off. The reaction mixture is extracted with 200-mL portions of ether until the ether washings are colorless (Note 9), and these washings are added to the initial product. The combined organic fraction is washed with saturated aqueous sodium bicarbonate until neutral (Note 10), then with water, and is then dried with magnesium sulfate. The solvent is removed with a rotary evaporator, and the residue is distilled (bp 54–55°C, 0.1 mm) through a 10-cm vacuum-jacketed Vigreux column to give m-trifluoromethylbenzenesulfonyl chloride (100–115 g, 68–79%) as a colorless or slightly yellow, clear liquid (Note 11), (Note 12).2. Notes1. α,α,α-Trifluoro-m -toluidine was obtained from Aldrich Chemical Company, Inc. The checkers distilled this material prior to use (bp 187–189°C).2. A chain beaker clamp is very satisfactory for supporting the beaker, as it can later be used as a handle to pour the diazonium solution. For efficient stirring the blade of the stirrer was made by trimming the ends of a large Teflon stirring paddle to the diameter of the beaker. The paddle was inverted (straight edge on bottom) and should rotate 1–1.5 cm from the bottom of the beaker.3. If solid amines are used, they should be thoroughly crushed in a mortar and pestle before adding to the acid mixture.4. Temperature control during the sodium nitrite addition is essential to the success of the preparation. The temperature can go as low as −15°C but must not exceed −5°C. The addition takes ca. 1 hr. At temperatures greater than −5°C, dark-red by-products form which lower the yield.5. Temperature control is conveniently accomplished by raising and lowering the dry-ice bath. It does not seem to matter if longer reaction times are employed, but the temperature should be lowered to −10°C or below after 45 min.6. Saturation, which requires 15–30 min, is conveniently noted by observing that most sulfur dioxide bubbles reach the surface of the acetic acid .7. The original literature 2 suggests that copper(II) chloride dihydrate can be used as a catalyst, since it is reduced by the sulfur dioxide to copper(I). It has been noted on several occasions that catalytically inactive mixtures result. If copper(II) chloride dihydrate is used, it is expedient to add copper(I) chloride (1 g) to ensure efficient catalysis in the early stages of reaction.8. This mixture should be a pale tan suspension, and it should be cooled between additions.9. The first portion of ether may be larger (400 mL) since much dissolves in the aqueous mixture. A total of 1000 mL of ether is usually sufficient. 10. A considerable amount of acid is present in the ether extracts, so vigorous gas evolution occurs during the sodium bicarbonate extraction. Caution must be exercised at this point. 11. The product is sufficiently pure for most purposes. A second distillation affords a colorless product (lit.3 bp 88–90°C, 6 mm). 12. With many anilines used as precursors in this reaction, the sulfonyl chloride product is a solid and an alternate workup procedure is used. After the reaction is quenched with ice water, the solid product is filtered with suction and washed copiously with cold water. The crude product tends to occlude water and copper salts, which may be detrimental in later reactions. A good washing protocol involves rinsing the solid on the filter with water (200 mL), then suspending the solid in cold water (1000 mL), stirring briskly, and filtering with suction. The latter process should be repeated three times. The final water wash should be only very slightly yellow. After air drying the product can be recrystallized from an appropriate solvent.3. Discussionm -Trifluoromethylbenzenesulfonyl chloride has been prepared by treatment of m -trifluoromethylbenzenediazonium chloride with sulfur dioxide and hydrochloric acid 4 and by conversion of benzotrifluoride to m -trifluoromethylbenzenesulfonic acid with oleum, followed by chlorination with phosphorus pentachloride .3 Derivatives of this compound, such as esters and amides, are quite useful in that they display reactivities similar to p - and m -nitrobenzenesulfonyl compounds but have greatly improved solubilities.The described procedure essentially follows that described by Meerwein et al.2 as modified slightly by Yale and Sowinski.4 This same method can be used for a great variety of substituted anilines with good results. As evident in Table I, good yields are obtained in most cases, and the reaction works better for anilines with electron-withdrawing substituents. The identical procedure has been used to prepare many other examples, such as m -F, o -F, 3,5-di-CF 3.5 This method readily provides many unavailable arylsulfonyl chlorides; it is experimentally straightforward, and the products are isolated without complications.There are two general routes to arylsulfonyl chlorides. The first involves the conversion of analready sulfur-substituted aromatic compound to the sulfonyl chloride. Thus arylsulfonic acids or their alkali metal salts yield sulfonyl chlorides by treatment with a variety of chlorinating agents such as phosphorus pentachloride , thionyl chloride , phosgene , and chlorosulfonic acid . Alternatively, substituted thiophenols or aryl disulfides can be oxidized by chlorine-water to the sulfonyl chloride.6The second route utilizes the introduction of the chlorosulfonyl substituent directly onto the aromatic nucleus. The reaction of substituted benzenes with chlorosulfonic acid gives good yields of arylsulfonyl chlorides; however, the aryl substituent dictates the position of attachment of the chlorosulfonyl function in this electrophilic aromatic substitution.7 The method described herein allows replacement of a diazotized amine function by the chlorosulfonyl group. The ready availability of substituted anilines makes this the method of choice for the preparation of arylsulfonyl chlorides.Arylsulfonyl chlorides are pivotal precursors for the preparation of many diverse functional types including sulfonate esters,8 amides,4 sulfones,9 sulfinic acids,10 and others.11 Furthermore, sulfonyl fluorides are best prepared from sulfonyl chlorides.12 The sulfonyl fluorides have many uses, among which is their utilization as active site probes of chymotrypsin and other esterases.13 The trifluoromethyl group also plays valuable roles in medicinal chemistry.14References and Notes1.Department of Chemistry, Box 3C, New Mexico State University, Las Cruces, NM 88003.2.Meerwein, H.; Dittmar, G.; Gollner, R.; Hafner, K.; Mensch, F.; Steinfort, O. Chem. Ber . 1957,90, 841–852.3.Yagupolskii, L. M.; Troitskaya, V. I. Zh. Obsch. Khim . 1959, 29, 552–556; Chem. Abstr . 1960,54, 356f.4.Yale, H. L.; Sowinski, F. J. Org. Chem . 1960, 25, 1824–1826.5.Gerig. J. T.; Roe, D. C. J. Am. Chem. Soc . 1974, 96, 233–238.6.Muth. F., In Houben-Weyl, "Methoden der Organischen Chemie," Müller, E., Ed.; Georg ThiemeVerlag: Stuttgart, Germany, 1955; Vol. 9 pp. 563–585.7.Gilbert, E. E. "Sulfonation and Related Reactions"; Interscience: New York, 1965; pp. 84–87.8.See: Coates, R. M.; Chen, J. P. Tetrahedron Lett . 1969, 2705–2708 and references cited therein. 9.Truce, W. E. In "Organic Chemistry of Sulfur," Oae, S., Ed.; Plenum Press: New York, 1977; pp.532–536.10.Truce, W. E.; Murphy, A. M. Chem. Rev . 1951, 48, 68–124.11.Trost, B. M. Acc. Chem. Res . 1978, 11, 453–461.12.Davis W.; Dick, J. H. J. Chem. Soc . 1932, 483–484; DeCat, A., Van Poucke, R.; Verbrugghe, M.J. Org. Chem . 1965, 30, 1498–1502.TABLE IC ONVERSION OF ARYLAMINES TOARYLSULFONYL CHLORIDESAmine, XC 6H 4NH 2, X =Yield (%) of XC 6H 4SO 2Clm -CF 372 p -NO 268 m -NO 286 p -Cl90p -CO 2CH 390 3,5-di-NO 281 m -CH 371 H53p -OCH 3 2713.See 5 for a good introduction.14.Yale, H. L. J. Med. Pharm. Chem. 1959, 1, 121.AppendixChemical Abstracts Nomenclature (Collective Index Number);(Registry Number)sulfonyl chlorideaminep- and m-nitrobenzenesulfonylhydrochloric acid (7647-01-0)acetic acid (64-19-7)ether (60-29-7)chlorosulfonic acid (7790-94-5)phosphorus pentachloride (10026-13-8)thionyl chloride (7719-09-7)sodium bicarbonate (144-55-8)sodium carbonate (497-19-8)sulfur dioxide (7446-09-5)sodium nitrite (7632-00-0)phosgene (75-44-5)cuprous chloride,copper(I) chloride (7758-89-6)magnesium sulfate (7487-88-9)copper(I)benzotrifluoride (98-08-8)copper(II) chloride dihydrate (10125-13-0)m-aminobenzotrifluoride,α,α,α-Trifluoro-m-toluidine(98-16-8)m-Trifluoromethylbenzenesulfonyl chloride,Benzenesulfonyl chloride, m-(trifluoromethyl)- (777-44-6)m-trifluoromethylbenzenediazonium chloridem-trifluoromethylbenzenesulfonic acidCopyright © 1921-2005, Organic Syntheses, Inc. All Rights Reserved。