Discovery Studio Visualizer简易教程
Discovery Studio官方教程--使用LibDock进行快速分子对接(虚拟筛选)

Discovery Studio LibDock教程Libdock –最快的分子对接技术介绍基于结构的药物设计技术在药物研发中起着非常重要的作用。
在药物分子产生药效反应的过程中,药物分子要与靶标相互结合,首先需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,才能得到一个稳定的复合物构象。
基于结构的药物设计主要采用的是分子对接技术。
分子对接就是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补以及化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。
分子对接是从整体上考虑配体与受体结合的效果,能够较好避免其他方法中容易出现的局部作用较好而整体结合欠佳的情况。
在药物设计中,分子对接方法主要用来从小分子数据库中搜寻与受体生物大分子有较好亲和力的小分子,进行药理测试,从中发现新的先导化合物;或者用于解释药靶之间的作用机制,并在得到作用模式的基础上指导化合物结构改造。
LibDock是Discovery Studio中的其中一种对接方法。
该对接方法首先会针对受体活性位点计算得到热区图,该热区图包含极性和非极性部分;接着不同构象的配体分子分别刚性地叠合至热区图以形成比较合适的相互作用;然后进行能量优化;最后保留打分较高的对接构象。
本教程将采用LibDock将一组配体分子对接到胸苷激酶(thymidine kinase)中,包括:•准备分子对接体系•执行分子对接计算•分析配体对接结果准备分子对接体系在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions| 1kim_prot.dsv文件。
该蛋白将在一个新的分子窗口中出现,该蛋白已经预处理过,且活性位点也已定义好。
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Define and Edit Binding Site,依次点击Show/Hide Residues Outside Sphere和Show/Hide Sphere。
esko studio visualizer使用方法

ESKO STUDIO VISUALIZER使用方法
ESKO STUDIO VISUALIZER是一款功能强大的可视化工具,可以帮助用户快速创建高质量的图形和可视化内容。
以下是使用ESKO STUDIO VISUALIZER的一些基本步骤:
1.打开ESKO STUDIO VISUALIZER软件,选择要创建的图形类型,例如条形
图、饼图、散点图等。
2.在弹出的窗口中,选择要使用的数据源。
您可以选择现有的Excel、CSV
或其他数据文件,或连接到数据库。
3.在数据源中选择要使用的数据列,并设置相应的图表属性,例如标题、坐
标轴标签、图例等。
4.根据需要调整图表的外观和样式,例如颜色、字体和标记等。
5.单击“预览”按钮以查看图表的最终效果。
如果需要更改图表,请返回上
一个窗口并修改相应的属性。
6.单击“保存”按钮以保存图表。
您可以选择保存为PNG、JPG、PDF或其他
格式的文件。
以上是使用ESKO STUDIO VISUALIZER的一些基本步骤。
通过掌握这些步骤,您可以轻松地创建高质量的图形和可视化内容,并将其用于报告、演示和其他用途。
dbvisualizer使用技巧

dbvisualizer使用技巧DBVisualizer使用技巧DBVisualizer是一款功能强大的数据库管理工具,提供了许多方便实用的功能和技巧,让数据库操作更加高效。
本文将介绍一些DBVisualizer的使用技巧,帮助您更好地利用这个工具。
连接数据库•在DBVisualizer的主界面,点击”创建连接”按钮,打开连接设置页面。
•在连接设置页面,填写数据库的连接信息,包括数据库类型、主机名、端口、数据库名、用户名和密码等。
•确认连接信息后,点击”测试连接”按钮,验证连接是否成功。
•点击”保存连接”按钮,保存连接信息。
•在主界面点击已保存的连接,即可连接到数据库。
数据库浏览•在DBVisualizer的主界面,选择一个已连接的数据库,可以查看数据库中的表、视图、过程等对象。
•双击一个表名,可以查看表的结构和数据。
•在表视图中,右键点击表名,可以进行各种操作,如查看表数据、编辑表结构、执行SQL脚本等。
搜索和过滤•在DBVisualizer的表视图中,可以使用搜索和过滤功能,快速定位到需要的数据。
•在搜索框中输入关键字,DBVisualizer会自动过滤显示匹配的数据。
•可以使用过滤条件,精确地查找需要的数据。
SQL编辑和执行•在DBVisualizer中,可以编写和执行SQL脚本,进行数据查询、更新和删除等操作。
•在主界面点击”新建SQL编辑器”按钮,打开一个新的SQL编辑器。
•在SQL编辑器中,可以编写SQL语句,并点击”执行”按钮执行SQL语句。
•执行结果将在结果面板中显示,可以查看查询结果或执行日志。
数据导入和导出•DBVisualizer提供了数据导入和导出的功能,可以方便地将数据从数据库导出为文件,或将文件中的数据导入到数据库中。
•在表视图中,右键点击表名,选择”导出数据”,可以将表数据导出为文件。
•在表视图中,右键点击表名,选择”导入数据”,可以将文件中的数据导入到表中。
数据库管理•DBVisualizer支持对数据库的管理操作,如创建表、修改表结构、执行数据库备份等。
Discovery Studio官方教程(Help-Tutorials) 分子对接前的准备受体和配体

Discovery Studio基本操作介绍在使用软件进行课题研究前,我们首先应该了解并掌握该软件使用的一些基本操作。
为后续的体系处理做好准备工作。
这个教程包括:●小分子配体准备●蛋白文件的处理小分子配体准备在Discovery Studio(DS)中,可以直接构建分子结构,也可以将在其它画图软件中画好的结构直接拷贝到DS中,本教程演示如何在DS中构建小分子结构。
1. 调用Sketching功能从View菜单下,打开Toolbars,选择Sketching。
Toolbars中将显示各种Sketching的工具,这些工具可以用来构建化合物的初始结构。
2. 利用Sketching构建化合物的3D空间构象打开一个分子显示窗口(Molecule Window),菜单栏File|New|Molecule Window。
注:DS中有四种窗口模式,包括Molecule window(显示分子结构),Protein Sequence Window (显示蛋白序列),Nucleotide Sequence Window(显示核酸序列),Script Window(显示脚本语言),因此我们需要根据载入的文件类型选择窗口。
DS中构建化合物的3D空间构象非常容易,也非常灵活。
本教程以以下化合物为示例,以图示的方法演示如何构建化合物的结构。
NHCl OOSOHNNH选择,在窗口中画出结构1。
点击(可以通过菜单栏View|Toolbars|Sketching调出)将其选中,然后选择菜单栏Chemistry|Bond|Aromatic得到结构2。
选择,鼠标指于芳环单键处并单击,构建稠环结构3。
选择,构建连接单键,再选择,鼠标指于C原子处并单击构建环状结构,最后得到结构4。
选择和构建单键和环状结构,选择再次点击相应的键就可以构建双键结构,最终可得到结构5。
更换元素类型,选中某个碳原子,选择菜单栏Chemistry|Element更换相应元素即可,最后得结构6。
Discovery Studio 讲义中文

附录:准备一个PDB文件作为同源性建模模板同源建模的基本原理是,你映射的一个未知的蛋白质序列一种已知蛋白质的结构。
因此,如果你没有已知的蛋白质,或模板,你将无法建立模型。
模板的共同来源是蛋白质在结构生物信息学研究的实验室数据银行(目标)。
该网站RCSB是HTTP:/ /。
org /。
蛋白质数据库(PDB)可能是世界上领先的公共源三维生物分子数据(1)。
截至七月2006,超过37000项可在PDB。
每个月都有更多的人加入。
X射线衍射仪和其它固态技术占大多数的结构。
然而,超过5500的核磁共振结构还可用。
这些沉积的结构包括蛋白质,肽,核酸,碳水化合物,这些分子的配合物。
在发现工作室环境中工作的这些分子是一个关键的过程给你的建模工作。
这个练习如何准备一个PDB文件作为一个模板同源建模项目。
你将在课程中学习如何:•加载PDB文件直接从蛋白质数据银行,•生产和检验蛋白质报告,•清除晶体单元电池,•分裂分子,•删除不需要的组件,和保存已完成的文件。
1、开始发现工作室2PDB3D HDAC8抑制剂,684个晶体的水,和几个离子。
抑制剂是曲古抑菌素A,一个很好的研究组蛋白去乙酰化酶抑制剂()。
的结构。
3、检查蛋白质报告在深入了解蛋白质,我们应该花一点时间来看看有什么。
一种快HTML1t64连锁。
但我们要清楚我们几个非原子以及。
4、删除单元电池晶体单元单元在分子力学计算中的意义不大。
有时它只是更容易5这个文件是由多个部分组成的。
我们可以通过层次结构查看组件其他练习窗口。
然而,我们实际上可以分开的组件的PDB文件更快的四的成分是蛋白质(1t64_a和1t64_b),配体(1t64_nonprotein),和结晶水(1t64_water)。
拆分命令的三个选项允许在如何处理对象的操作中有一定的灵活性。
所有打出的层次结构视图中所有链和非蛋白结构为独立的对象,并列出每个氨基酸序列为序列单独序列。
蛋白打出所有的蛋白链结构为独立的对象层次结构中的每个视图和列表的氨基酸序列为序列单独序列。
Discovery-Studio-讲义中文

附录:准备一个PDB文件作为同源性建模模板同源建模的基本原理是,你映射的一个未知的蛋白质序列一种已知蛋白质的结构。
因此,如果你没有已知的蛋白质,或模板,你将无法建立模型。
模板的共同来源是蛋白质在结构生物信息学研究的实验室数据银行(目标)。
该网站RCSB是HTTP:/ /。
org /。
蛋白质数据库(PDB)可能是世界上领先的公共源三维生物分子数据(1)。
截至七月2006,超过37000项可在PDB。
每个月都有更多的人加入。
X射线衍射仪和其它固态技术占大多数的结构。
然而,超过5500的核磁共振结构还可用。
这些沉积的结构包括蛋白质,肽,核酸,碳水化合物,这些分子的配合物。
在发现工作室环境中工作的这些分子是一个关键的过程给你的建模工作。
这个练习如何准备一个PDB文件作为一个模板同源建模项目。
你将在课程中学习如何:•加载PDB文件直接从蛋白质数据银行,•生产和检验蛋白质报告,•清除晶体单元电池,•分裂分子,•删除不需要的组件,和保存已完成的文件。
1、开始发现工作室2在3D窗口中显示的结构是两人HDAC8蛋白的晶体结构在复杂的抑制剂,684个晶体的水,和几个离子。
抑制剂是曲古抑菌素A,一个很的结构。
3、检查蛋白质报告在深入了解蛋白质,我们应该花一点时间来看看有什么。
一种快连锁。
但我们要清楚我们几个非原子以及。
4、删除单元电池晶体单元单元在分子力学计算中的意义不大。
有时它只是更容易这个文件是由多个部分组成的。
我们可以通过层次结构查看组件其他练习窗口。
然而,我们实际上可以分开的组件的PDB文件更快的四的成分是蛋白质(1t64_a和1t64_b),配体(1t64_nonprotein),和结晶水(1t64_water)。
拆分命令的三个选项允许在如何处理对象的操作中有一定的灵活性。
所有打出的层次结构视图中所有链和非蛋白结构为独立的对象,并列出每个氨基酸序列为序列单独序列。
蛋白打出所有的蛋白链结构为独立的对象层次结构中的每个视图和列表的氨基酸序列为序列单独序列。
discovery简明操作手册

一、软件安装:(Discovery installation)安装:安装软件考在D盘(或非软件盘,比如E盘),打开文件夹GNT→打开文件夹ggx2003.1→双击图标Install(安装)→出现界面,选择第一个→Next→选中我接受,单击Next→填入用户名,单击Next →选中典型安装,单击Next→选中米制(第二个),单击Next→单击Install(安装)→单击Finish→单击EXIT→单击内部退出安装系统。
(此时桌面上出现两个图标)安装之后打补丁(支持安装):双击GXR20031SU1图标(补丁)→单击Finish;双击GXR20031SU2图标(补丁)→单击Finish。
拷贝gxdb.db至C盘,(Documents and settings→All users→Application Data→Geographix→data→templates→project→粘贴。
)双击桌面上的油藏描述软件(地球图标)→正常情况下出现一个选择框,选择第三个(如果没有狗或临时许可证过期,则无法进行下一步),单击NEXT→在GNT文件夹中选中GGXLIC(许可证),单击NEXT →出现对话框,选择Activate an existing project …,选中一个,Next,则激活了一个存在的工区。
(如果不是初次使用,则选择Activate the last active project ,将自动进入近期使用过的工区。
隐含文件的建立:Tools→Folder options。
未显示地球图标时:打开ACDSEE.6→TOOLS→file Association(文件关联) →ZCO中的对钩去掉→单击OK。
重新打开即可。
二、建立工区目录在D盘上建立一个子目录做为工区目录。
建立过程:打开油藏描述软件(桌面中的小地球)→选中FILE中的NEW,再单击HOME,出现一个对话框,在该对话框中选中D盘,单击新建文件夹,取名,确定。
Discovery Studio 2.5操作教程

蛋白质结构预测技术简介简介蛋白质结构的解析对其功能的理解至关重要。
然而,由于技术手段的限制,利用实验方法(主要为X-ray,NMR)解析蛋白质结构投入大、周期长、风险大。
对于某些膜蛋白,只利用现有技术条件,其结构甚至无法解析。
另一方面,随着分子生物学技术的成熟及高通量测序技术的发展,越来越多的基因序列可以轻松被找到。
这造成了现代蛋白质科学中一个奇怪的现象:蛋白质序列数据的累积量及积累速度远远超过蛋白质结构。
这种序列与结构间不平衡的现象极大地限制了我们对蛋白质功能及其相关作用机理的理解。
所以我们需要一种能够简单、快速且相对准确的技术来确定蛋白质的空间结构。
蛋白质建模技术可以很好的解决上面的问题。
该方法利用信息技术的手段,可以直接从蛋白的一级结构(氨基酸序列)预测蛋白质的高级结构(主要为三级结构)。
根据最新一届国际建模大赛(CASP)的分类,目前主要的蛋白质建模方法包括两种:基于模板的建模(Template-based Modeling)和自由建模(Free Modeling)。
前者又包括两种方法:同源建模法(Homology Modeling)和“穿线法”(Threading)。
后者主要以从头计算法(ab initio)为主。
所有的建模方法中,以同源建模法(Homology Modeling)使用最为广泛,预测结果的准确性最大。
同源建模的理论基础为蛋白质三级结构的保守性远远超过一级序列的保守性。
因此,人们可以通过使用一个或多个已知结构的蛋白(模板蛋白,template)来构建未知结构蛋白(目标蛋白,target)的空间结构。
其主要的步骤包括:1.搜索用于建模的template(s)2.将target与templates进行比较3.将步骤(2)中的比较信息用于建模Discovery Studio为用户提供了一整套利用Homology Modeling方法自动预测蛋白质空间结构的工具。
用户只需要提供蛋白质的氨基酸序列就可以轻松完成模型构建及模型可信度评估的工作。
Discovery Studio 讲义中文

附录:准备一个PDB文件作为同源性建模模板同源建模的基本原理是,你映射的一个未知的蛋白质序列一种已知蛋白质的结构。
因此,如果你没有已知的蛋白质,或模板,你将无法建立模型。
模板的共同来源是蛋白质在结构生物信息学研究的实验室数据银行(目标)。
该网站RCSB是HTTP:/ /。
org /。
蛋白质数据库(PDB)可能是世界上领先的公共源三维生物分子数据(1)。
截至七月2006,超过37000项可在PDB。
每个月都有更多的人加入。
X射线衍射仪和其它固态技术占大多数的结构。
然而,超过5500的核磁共振结构还可用。
这些沉积的结构包括蛋白质,肽,核酸,碳水化合物,这些分子的配合物。
在发现工作室环境中工作的这些分子是一个关键的过程给你的建模工作。
这个练习如何准备一个PDB文件作为一个模板同源建模项目。
你将在课程中学习如何:•加载PDB文件直接从蛋白质数据银行,•生产和检验蛋白质报告,•清除晶体单元电池,•分裂分子,•删除不需要的组件,和保存已完成的文件。
1、开始发现工作室我们必须有发现工作室开始行使。
2PDB现在,我们将直接从目标通过Discovery Studio界面加载PDB文件。
3D HDAC8抑制剂,684个晶体的水,和几个离子。
抑制剂是曲古抑菌素A,一个很好的研究组蛋白去乙酰化酶抑制剂(2)。
的结构。
3、检查蛋白质报告在深入了解蛋白质,我们应该花一点时间来看看有什么。
一种快速的方法完成这项任务是使用蛋白质报告。
HTML1t64。
正如我们有多个分子在视图中,该报告从一个分子列表。
连锁。
但我们要清楚我们几个非原子以及。
4、删除单元电池晶体单元单元在分子力学计算中的意义不大。
有时它只是更容易删除单元电池。
5这个文件是由多个部分组成的。
我们可以通过层次结构查看组件其他练习窗口。
然而,我们实际上可以分开的组件的PDB文件更快的分割命令。
四的成分是蛋白质(1t64_a和1t64_b),配体(1t64_nonprotein),和结晶水(1t64_water)。
Discovery Studio官方教程--使用Flexible Docking进行柔性分子对接

Discovery Studio Flexible Docking教程Flexible Docking –全柔性受体-配体对接技术介绍柔性分子对接是指在对接过程中,蛋白的侧链和配体分子构象都可以自由变化,多用于精确考查分子间的结合模式,但由于计算量和计算时间的缘故,在实际应用中,一般只将活性位点处残基侧链定义为柔性,可以考虑其构象的改变。
在Flexible Docking中,配体将按如下步骤对接到考虑活性部位氨基酸残基侧链柔性的受体中:•选择受体柔性氨基酸残基,使用ChiFlex(CHARMm)通过改变侧链构象计算出一组蛋白构象•使用LibDock将柔性配体对接到每一个受体构象的活性部位•使用ChiRotor(CHARMm)在刚性配体存在的情况下优化选定的蛋白侧链•使用CDOCKER优化最后的配体对接构象在本教程中研究的两个蛋白分子为同一个HIV-RT受体,但由于所含的配体不同,两蛋白PDB编号不同,分别为1s1x和1fk9。
配体结构的差别会引起蛋白构象的改变,这也是柔性对接的理论基础。
本教程将1fk9中结合的配体对接到1s1x中。
具体步骤包括:•准备对接的分子体系•执行分子对接计算•分析对接结果•计算对接结果的RMSD值准备对接的分子体系1.蛋白和配体文件的准备在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions| 1s1x_prot.dsv文件。
该蛋白将在一个新的三维窗口中打开。
(图1)此蛋白文件已经过处理:已赋予CHARMm力场,所以在对接之前不需再进行力场的赋予;此外,结合位点也已识别,窗口中只显示该结合位点周围的8个残基及其相应的残基名,在对接过程中将会考虑这8个残基的侧链柔性,上述8个残基已被定义为一组并命名为1s1x_res8,系统视图中已显示。
图1 打开蛋白文件在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions|1fk9_lig.sd文件。
使用DS Visualizer3.5在PPT中动态展示分子三维结构
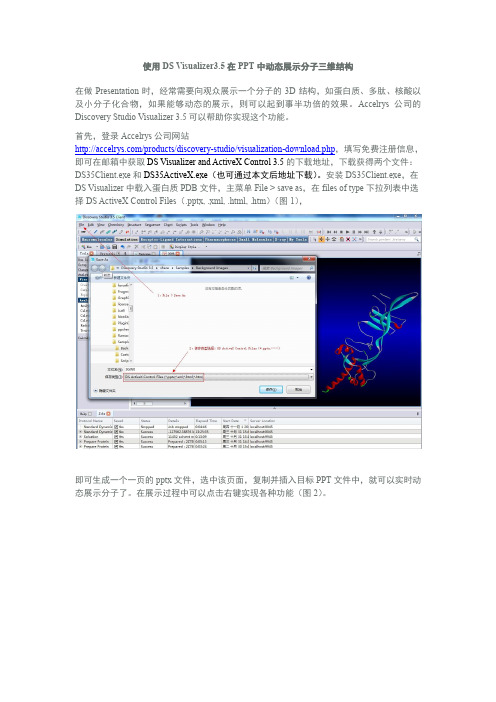
使用DS Visualizer3.5在PPT中动态展示分子三维结构在做Presentation时,经常需要向观众展示一个分子的3D结构,如蛋白质、多肽、核酸以及小分子化合物,如果能够动态的展示,则可以起到事半功倍的效果。
Accelrys公司的Discovery Studio Visualizer 3.5可以帮助你实现这个功能。
首先,登录Accelrys公司网站/products/discovery-studio/visualization-download.php,填写免费注册信息,即可在邮箱中获取DS Visualizer and ActiveX Control 3.5的下载地址,下载获得两个文件:DS35Client.exe和DS35ActiveX.exe(也可通过本文后地址下载)。
安装DS35Client.exe,在DS Visualizer中载入蛋白质PDB文件,主菜单File > save as,在files of type下拉列表中选择DS ActiveX Control Files(.pptx, .xml, .html, .htm)(图1),即可生成一个一页的pptx文件,选中该页面,复制并插入目标PPT文件中,就可以实时动态展示分子了。
在展示过程中可以点击右键实现各种功能(图2)。
如需在其他电脑中播放该PPT,只需将安装DS35ActiveX.exe(31MB)即可。
需要注意的是,Powerpoint的版本要在2007以上方可,否则会提示无法生成xml文件。
该功能十分实用,且所用到的DS Visualizer3.5和DS ActiveX Control 3.5均为免费软件。
DS Visualizer3.5:/share/link?shareid=102833&uk=235436026DS ActiveX Control 3.5:/share/link?shareid=102836&uk=235436026江大-大牙兔2012/11/5。
Discovery Studio官方教程--ADMET预测药物代谢动力学

使用Discovery Studio进行化合物ADMET性质预测教程介绍ADMET性质是指分子在有机体内的吸收、分布、代谢、排泄和毒性等性质。
如果在药物研发的早期阶段就能依据化合物的ADMET性质对先导化合物进行有针对性的选取和优化改造,这对提高药物研发的成功率以及减少药物研发后期过程中由于ADMET性质问题所造成的资金浪费问题,是非常必要的。
ADMET描述符可以有助于及早排除ADMET性质不好的化合物从而避免后期耗资巨大的结构改造,同时也可以评价结构优化的效果,是否确实改善了ADMET属性,从而避免合成所支出的过多资源。
本教程主要介绍了在Discovery Studio中如何进行化合物ADMET性质的预测,以及预测结果的分析。
本教程包括:•运行ADMET性质计算流程•分析ADMET性质预测结果Discovery Studio中可以计算的ADMET性质包括:•25摄氏度下水溶解度(aqueous solubility)•血脑屏障通透性(Blood brain barrier penetration,BBB)•细胞色素P450 2D6抑制性(Cytochrome P4502D6 inhibition)•肝毒性(hepatotoxicity)•人类肠道吸收性(human intestinal absorption,HIA)•血浆蛋白结合率(plasma protein binding)运行ADMET性质计算流程1. 导入小分子化合物文件在文件浏览器(Files Explorer)中,展开Samples | Tutorials | QSAR,双击打开pk-test.sd文件。
在表格浏览器中可以看到一共有20个丙酮酸盐激酶抑制剂。
2. 选择计算性质,运行计算流程在工具浏览器(Tools Explorer)中,展开Small Molecules | Calculate Molecular Properties,点击ADMET Descriptors,打开ADMET Descriptors对话框。
Discovery Studio官方教程(Help-Tutorials) 分子动力学模拟

Discovery Studio Molecular Dynamics教程Molecular Dynamics –分子动力学方法介绍分子动力学(Molecular Dynamics, MD)是分子模拟中最常用的方法之一。
该方法基于分子力场,能够动态的描述分子的运动状况,继而描述生命的动态过程。
分子动力学在生命科学领域中的应用非常广泛,如蛋白质折叠的机理研究、酶催化反应的机理研究、与功能相关蛋白质的运动研究、生物大分子大范围构象变化的研究等等。
近几十年来,分子动力学方法已经成功的运用于大分子体系低能量构象的模建、X射线晶体衍射以及NMR实验结果的处理。
[1,2]现在分子动力学方法已经成了理论生物学研究中必不可少的方法之一。
[3,4]分子动力学模拟主要包括如下几个步骤:1.模拟体系升温过程(Heating Stage)2.模拟体系平衡过程(Equilibrium Stage)3.模拟体系采样过程(Production Stage)4.分析体系的目标性质(Analysis)然而,由于一般的待模拟体系的初始结构或多或少的存在缺陷(比如,初始结构不完整或存在不合理的结构区域),所以我们往往需要对初始结构进行预处理才能进行分子动力学模拟。
一个完整的分子动力学模拟过程应包括如下几个步骤:1.初始结构检查及预处理2.给模拟体系赋力场参数3.考虑溶剂效应4.初始结构能量最小化5.动力学模拟(包括升温、平衡、采样)6.结果分析目前,常用的分子动力学模拟软件都基于Unix(Linux)操作系统。
实施每一个模拟的步骤都需要特定的命令来调用相关的程序对模拟体系进行处理。
同时,模拟者还需要熟知每个参数的意义并定义相关的参数值。
最终分析过程也需要配合其他软件才能完成。
这对初级分子动力学模拟者而言是非常困难的(模拟者不仅需要掌握分子动力学软件的使用命令,还需要掌握操作系统相关的命令)。
为了解决这样的问题,Discovery Studio为用户提供了基于窗口的分子动力学模拟工具。
DiscoveryStudio2.1中文教程

DiscoveryStudio2.1中⽂教程DS2.1教程介绍●⽤户界⾯和⿏标UI and mouse●打开并游览数据opening and viewing a data组合库设计●枚举组合库enumerate library●Pareto optimization of a combinatorial subset library药效团Pharmacophore●创建药效团(基于结构)creating pharmacophores (structure-based)●配体分析器ligand profiler●通⽤特征药效团⽣成common feature pharmacophore generation●创建和使⽤基于⽚段的药效团creating and using fragment-based pharmacophores●创建特定特征creating custom features蛋⽩质同源模建protein modeling●同源模建细胞外淀粉酶●Loop 构建Looper with antibodies●ZDOCK定量构效关系QSAR●建⽴⼀个QSAR⽅程building a QSAR equation受体配体相互作⽤Receptor-ligand interaction●使⽤libdock完成⼩分⼦对接Docking small molecules with libdock模拟Simulation●带有限制体⼩肽的模拟simulation of a small peptide with restraints (protein simulation)●在最⼩化之前之后,使⽤能量计算协议(QM-MM)计算能量去测定活⼒,rank order pose和⽐较energies using the calculate energy (QM-MM)protocol to determine energies,rank order poses and compare energies before and after minimization静电势计算Electrostatics●泊松玻尔兹曼——有限差分●计算蛋⽩质离⼦化和残基的pK●计算电势●计算溶剂化能开发客户端Developer Client⽤户界⾯和⿏标教程需要数据⽂件:1TPO.pdb 和1BVN.pdb背景可视化和基本的结构分析⼯具在⽣物和⽣化系统中是很重要的。
DiscoveryStudioVisualizer简易教程

DiscoveryStudioVisualizer简易教程Discovery Studio Visualizer精简教程
哈尔滨医科⼤学
解鸿波
PDB: 3K9F
⼯具栏⼯具导航栏
主窗⼝
Ctrl+H
氨基酸残基
氨基酸相应性质
Ctrl+D
线型显⽰必须先棍型显⽰选择原⼦
球棍显⽰
条带
改变背景颜⾊
View > Toolbars > View View > Toolbars > Sketching 区旋平放画分⼦⼯具域转移⼤(不推荐使⽤)选缩
*不同部分蛋⽩(或DNA
等⼤分⼦)
点加号+
查看侧链
所含有氨
基酸残基
点加号+
查看氨基酸所
含有原⼦
氨基酸及编号
字母重新循环配体分⼦去除对勾
表⽰隐藏其他分⼦
例如⽔等
显⽰配体和周围形成的氢键作⽤
2. Structure > Monitor > HBonds
1. 仅显⽰配体
分⼦,并双击
选中,分⼦显⽰为黄⾊。
3. 显⽰和配体可以形成氢键的氨基酸序号及相应原⼦将氨基酸以棍状形态显⽰,Stick
默认氢键为绿⾊size调整为0.10和配体以⽰区别,并
不显⽰其带状⼆级结构
选中分⼦,查看其他相互作⽤Tools > Analyze Complexes > Show 2D Diagram
Structure > Surface > Add
Structure > Monitor > Distance 选中两个原⼦,测距离单位为(?)
选中两根化学键,测⾓度查看蛋⽩序列。
Visualizer模块快速入门教程

Visualizer模块快速入门教程Visualizer 模块快速入门教程下面的教程将示范如何使用 Visualizer。
z 管理 Project: 介绍 MS Modeling 中的 Project,并示范如何使用它们管理工作流文件。
z 绘制简单分子: 介绍用于绘制链和环的绘制工具,编辑键级和元素类型并且测量不同的几何性质。
z 绘制卟啉分子: 显示如何操纵片断并使用Display Style 对话框。
z 绘制有机金属结构: 介绍片断浏览并示范如何使用 Find Symeetry 工具。
z 将分子对接到表面: 介绍了用于连接晶体结构的表面模建。
z 使用聚合物模建工具: 介绍了构建多种类型聚合物结构的聚合物模建工具。
z 使用层模建工具: 先是如何使用层模建工具来构造一个界面和金属-聚合物金属分层结构。
z 使用晶体模建工具: 介绍用于构造并可视化 3D 周期结构的晶体模建工具。
1.管理 Project背景当你正在运行一些高级的操作,例如Discover 或者Amporphous Cell 工作的时候,会生成具有不同文件名的不同文档。
要使管理这些文档变得更简单,MS Modeling 有一个叫做Project Explorer 的文件管理器。
它与一些高级程序程序语言包如 VC++的文档管理系统相似。
与程序可以生成文档和文件夹相同,你也可以生成自己的文件夹和文档来订制文档组织并帮助你纪录所作的工作。
介绍本教程分为两个部分。
第一部分描述了一个简单的Project,在这里你会看到如何执行计算,例如能量的最小化。
第二和以后的部分将通过生成自己的 Project 来进行指导。
因为处理 Project 是 MS Modeling 中的一个基本部分,也是进行其它教程的参考部分。
(1).例子Project 的示范性细目分类此部分由一个例子Project 的细目分类组成。
只用于示范目的,不牵涉到任何要完成的工作。
Discovery Studio官方教程--药物毒理学性质预测及结果分析

使用Discovery Studio进行化合物毒理性质预测(TOPKAT)教程介绍毒理学性质预测流程TOPKAT基于分子的2D结构对化合物的毒性及环境效应进行快速且准确地计算预测并加以验证。
该流程采用了一系列强大且经交叉验证过的定量结构毒性关系(QSTR)模型来评估各种毒性预测结果。
基于该流程,我们可以完成以下工作:•快速评估有机化合物一系列毒理学性质•检查子结构以及结构改造的结构毒性关系以及相关的作用机制•分子排名以用于实验测试或者进一步的开发•设计新的化合物因此采用TOPKAT流程进行毒性预测在节省药物上市时间,减少动物实验,评估人体健康风险以及药物研发流程的战略性部署等方面都起了非常重要的作用。
本教程主要介绍了在Discovery Studio中如何进行小分子化合物毒理学性质的预测,以及预测结果的分析。
本教程包括:•运行TOPKAT流程•分析预测结果Discovery Studio中计算的毒理学性质包括:•潜在发育毒性(Developmental Toxicity Potential ,DTP)•致突变性(Mutagenicity (Ames test))•啮齿动物致癌性(Rodent Carcinogenicity)——包括NTP及FDA数据集•大鼠长期口服最低毒副反应水平(Rat Chronic Oral Lowest Observed Adverse EffectLevel,LOAEL)•皮肤致敏性(Skin Sensitization (GPMT) )•皮肤刺激性(Skin Irritancy)•大鼠口服LD50(Rat Oral LD50)•最大耐受剂量(Maximum Tolerated Dosage)•黑头呆鱼LC50(Fathead Minnow LC50)•大型蚤EC50(Daphnia magna EC50)•VlogP•眼刺激性(Ocular Irritation)•吸入LC50(Inhalational LC50)•好氧生物降解性能(Aerobic Biodegradability)运行TOPKAT毒性预测流程1. 导入小分子化合物文件在文件浏览器(File Explors)中,展开Samples | Receptor-Ligand Interaction,双击打开1fvv_ligand.dsv文件,可以看到1个化合物显示在Molecule Window中。
DS_Libdock教程

Discovery Studio LibDock教程Libdock–最快的分子对接技术目的:采用Libdock,以一组配体及一个明确活性位点的蛋白质为实例,示范分子对接及结果分析的操作过程。
所需功能和模块:Discovery Studio Visualizer client, DS LibDock, DS Catalyst Conformation。
所需数据文件:1kim.pdb和kinase_ligands.sd。
所需时间:0.5小时介绍基于结构的药物设计技术在药物研发中起着非常重要的作用。
在药物分子产生药效反应的过程中,药物分子要与靶标相互结合,首先需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,才能得到一个稳定的复合物构象。
基于结构的药物设计主要采用的是分子对接技术。
分子对接就是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补以及化学环境互补的原则来实时评价配体与受体相互作用好坏,并找到两个分子之间最佳的结合模式。
分子对接是从整体上考虑配体与受体结合的效果,能够较好避免其他方法中容易出现的局部作用较好而整体结合欠佳的情况。
在药物设计中,分子对接方法主要用来从小分子数据库中搜寻与受体生物大分子有较好亲和力的小分子,进行药理测试,从中发现新的先导化合物。
本教程中,一组配体分子将被对接到胸苷激酶(thymidine kinase)中,本教程包括:∙准备分子对接体系,执行分子对接计算∙分析配体对接构象准备分子对接体系,执行分子对接计算1.定义蛋白为受体分子图1 蛋白质三维结构示意图在Files浏览器中找到数据文件1kim.pdb,鼠标双击,该蛋白将在三维窗口中出现。
(图1)点击选择下拉菜单Chemistry | Hydrogens | Add给蛋白添加氢。
展开Receptor-Ligand Interactions工具栏,在Tools浏览器中打开Define and Edit Binding Site 工具面板,点击“Define Receptor”,将前面选择的蛋白分子1kim定义为受体分子,供下一步使用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
默认氢键为绿色
选中分子,查看其他相互作用 Tools > Analyze Complexes > Show 2D Diagram
选中分子,给分子增加表面 Structure > urface > Add
Structure > Monitor > Distance 选中两个原子,测距离 单位为(Å)
去除对勾 表示隐藏
氨基酸及编号 点加号+ 查看氨基酸所 含有原子
显示配体和周围形成的氢键作用
1. 仅显示配体 分子,并双击 选中,分子显 示为黄色。
2. Structure > Monitor > HBonds
3. 显示和配体可以形成氢键 的氨基酸序号及相应原子
将氨基酸以棍状形态显示,Stick size调整为0.10和配体以示区别,并 不显示其带状二级结构
No.2
索拉菲尼(Sorafenib)
HMG-CoA还原酶 PDB code: 1HWI
No.3
氟伐他汀(Fluvastatin)
神经氨酸酶 PDB code: 4KS1
No.4
达菲(Tamiflu)
青霉素结合蛋白 PDB Code: 3ITA
No.5
氨苄西林(Ampicillin)
Structure > Monitor > Torsion 选中两根化学键,测角度
查看蛋白序列
查看蛋白拉氏图 Chart > Ramachandran Plot
选择原子 > 右键 > Attributes of … 查看原子坐标
Build and Edit Nucleic Acid工具
Build and Edit Protein工具 (不是蛋白三维结构,需要做从头预测)
Create Pharmacophore Manully工具 (根据已知分子构建药效团,但不实用)
写字板打开*.pdb文 件查看信息,内附 原子坐标,原子类 型及氨基酸序号等 信息。
HIV-1蛋白酶 PDB code: 3OXC
No.1
沙奎那韦(Saquinavir)
p38 MAP激酶 PDB code: 3GCS
条带
改变背景颜色
View > Toolbars > View View > Toolbars > Sketching
区旋平放
域转 移 大
选
缩
择
小
画分子工具 (不推荐使用)
代表晶格
按字母顺 序排列 *不同部分 字母重新 循环
蛋白(或DNA 等大分子)
配体分子 其他分子 例如水等
PDB编号
侧链编号 点加号+ 查看侧链 所含有氨 基酸残基
Discovery Studio Visualizer精简教程
PDB: 3K9F
哈尔滨医科大学 解鸿波
工具栏
工具导航栏 主窗口
Ctrl+H
氨基酸残基
Ctrl+T
氨基酸相应性质
Ctrl+D
编辑原子
必须先 选择原 子
线型显示 棍型显示 球棍显示
编辑蛋白
必须先 选择原 子
线型显示 棍型显示 线型条带