引物设计的标准操作规程
PCR引物设计详细步骤

PCR引物设计详细步骤引言PCR(聚合酶链式反应)是一种在分子生物学中常用的技术,用于放大DNA片段。
在PCR过程中,引物的选择非常重要,因为引物的设计质量直接影响到PCR反应的效率和准确性。
本文将详细介绍PCR引物设计的步骤。
步骤1. 确定目标序列首先,需要确定所要放大的目标序列。
这可以是任何你感兴趣的DNA片段,如某个基因的编码区域,特定的DNA序列等。
2. 提取目标序列从已有的DNA样本中提取目标序列。
可以通过DNA提取试剂盒等方法进行提取,确保获得纯净的DNA。
3. 序列比对使用BLAST等工具将目标序列与已知的序列数据库进行比对,以确认目标序列的唯一性和可能存在的变异。
4. 引物设计原则根据目标序列,设计符合以下原则的引物:•引物长度通常在18-25个碱基对之间。
•碱基组成均匀,避免引物中存在大量的G或C碱基,以及连续多个重复的碱基。
•引物之间的互补性尽量避免,以防止二聚体的形成。
•避免引物末端存在碱基的互补序列,以防止非特异性扩增。
5. 引物设计工具使用引物设计工具,如Primer3、NCBI Primer-BLAST等,在目标序列中选择合适的引物。
这些工具可以根据给定的参数,自动设计合适的引物。
6. 引物评估对设计的引物进行评估,包括检查引物的反向互补性、引物的Tm值(熔解温度)、引物的二聚体和自身结构等。
确保引物的质量达到实验要求。
7. 引物合成将设计好的引物发送给合成公司进行合成。
确保引物的纯度和浓度符合要求。
8. PCR反应使用合成的引物进行PCR反应,按照标准的PCR反应体系和条件进行。
根据实验需求调整PCR反应的温度、时间等参数。
9. PCR产物验证通过凝胶电泳等方法验证PCR反应产物的大小和纯度。
确保PCR反应成功,并且没有非特异扩增的产物。
结论PCR引物设计是PCR反应成功的关键。
通过遵循引物设计的原则,结合引物设计工具的辅助,可以设计出合适的引物,弥补PCR技术在DNA放大中的巨大优势,为实验研究提供有效的工具。
idt设计引物步骤

idt设计引物步骤1. 确定目标序列:首先,我们需要明确实验的目标,即我们想要扩增、克隆或测序的DNA序列。
可以是一个基因、一个特定的DNA 片段或者是整个基因组。
2. 引物长度:接下来,我们需要确定引物的长度。
引物是一小段DNA序列,通常由20-30个核苷酸组成。
引物的长度应该足够短,以便在PCR扩增或测序过程中能够特异性地结合到目标序列上。
3. 引物设计原则:在设计引物时,有一些原则需要遵循。
首先,引物的GC含量应在40-60%之间,这可以增加引物与目标序列的特异性结合。
其次,引物的3'末端应以C或G为主,这有助于避免引物的5'末端的退火。
此外,引物的Tm(退火温度)应在50-65℃之间,以确保引物能够稳定地结合到目标序列上。
4. 引物序列分析:在设计引物之前,我们需要对目标序列进行一些分析。
可以使用一些生物信息学工具,如BLAST,来搜索类似的序列并进行比对。
这可以帮助我们确定引物与目标序列的特异性。
5. 引物设计工具:现在有很多在线工具可以帮助我们设计引物。
这些工具可以根据我们提供的目标序列自动生成合适的引物。
其中一些工具还可以根据特定的实验要求,如PCR扩增、测序或突变分析,进行引物设计。
6. 引物合成:设计好引物后,我们需要将其发送给DNA合成公司进行合成。
在合成引物时,我们需要提供引物的序列、长度和纯度要求。
DNA合成公司将根据我们的要求合成引物,并将其以干燥的形式提供给我们。
7. 引物验证:在使用引物进行实验之前,我们需要对其进行验证。
可以使用一些实验方法,如聚丙烯酰胺凝胶电泳、比色法或测序等,来验证引物的纯度和特异性。
总结起来,设计引物是进行各种生物学实验的重要步骤之一。
通过确定目标序列、确定引物长度、遵循引物设计原则、进行引物序列分析、使用引物设计工具、合成引物和验证引物等步骤,我们可以设计出高质量的引物,从而保证实验的成功。
希望本文对你对IDT 设计引物有所了解。
引物设计的原理和程序

1 引物的设计以及初步筛选引物的设计与初步筛选基本上通过一些分子生物学软件和相关网站来完成的,目前运用软件Primer Premier 5 或美国 whitehead 生物医学研究所基因组研究中心在因特网上提供的一款免费在线PCR引物设计程序 Primer 3来设计引物,再用软件Oligo 6进行引物评估,就可以初步获得一组比较满意的引物。
但是对于初学者来说,运用软件和程序来设计引物好象无从着手,其实只要我们掌握了引物设计的基本原则和注意事项,所有问题便迎刃而解。
因为无论是软件还是程序,都是以这些基本原则和注意事项为默认标准来进行引物设计的。
所以,我们在进行引物设计的时候大可不必在软件和程序的参数上花费过多的时间来思考,如果没有特殊要求我们完全可以把一些参数设为默认值。
下面我们主要讨论一下引物设计的原则和注意事项。
①引物的长度一般为15-30 bp,最好在18~24 bp,因为太短易形成错配(F alse priming) 降低特异性,而太长也会降低特异性,并且降低产量[21。
②引物在模板内最好具有单一性,也就是说在模板内部没有错配。
特别是3’端,一定要避免连续4个以上的碱基互补错配。
③引物序列的GC 含量最好在40%一60%,且上下游引物序列GC含量的差异不要太大,3’端最后5个碱基最好不要富含GC,特别是连续3个的G或C。
④DNA双链形成所需的自由能AG,应该以5’端向3’端递减,3’端AG最好不要高于9.0 keaf mol[31。
⑤避免形成稳定的引物二聚体(Dimer and Cross DimeO 和发夹结构(Hairp in),AG高于4.5 keal/mol时易引发上述两种结构的产生。
⑥引物所在的模板区域应该位于外显子区,最好跨越一个内含子区,这样便于对扩增出来的片段进行功能鉴定和表型分析。
⑦如果以DNA为模板设计引物,产物长度在100—600 bp比较理想。
而以m RNA为模板设计引物时,产物长度在150—300 bp比较理想。
qPCR引物设计原则及具体操作步骤

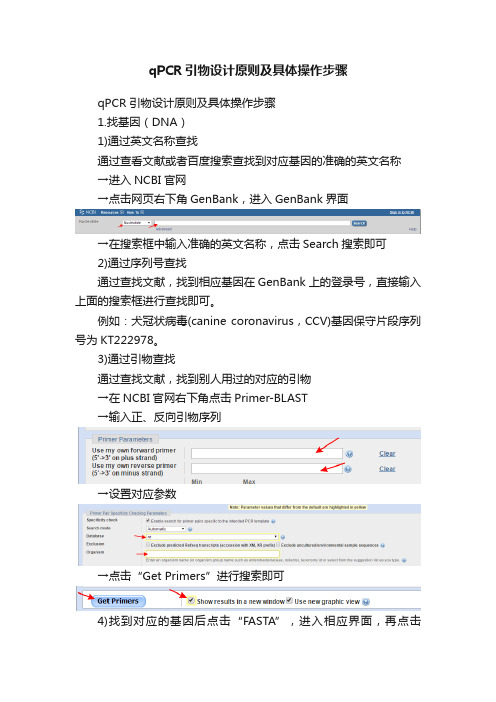
qPCR引物设计原则及具体操作步骤1.找基因(DNA)1)通过英文名称查找通过查看文献或者百度搜索查找到对应基因的准确的英文名称→进入NCBI官网→点击网页右下角GenBank,进入GenBank界面→在搜索框中输入准确的英文名称,点击Search搜索即可2)通过序列号查找通过查找文献,找到相应基因在GenBank上的登录号,直接输入上面的搜索框进行查找即可。
例如:犬冠状病毒(canine coronavirus,CCV)基因保守片段序列号为KT222978。
3)通过引物查找通过查找文献,找到别人用过的对应的引物→在NCBI官网右下角点击Primer-BLAST→输入正、反向引物序列→设置对应参数→点击“Get Primers”进行搜索即可4)找到对应的基因后点击“FASTA”,进入相应界面,再点击“Send to”选择相应格式,保存序列。
2.qPCR引物和TaqMan探针的设计1)引物设计注意事项a)引物长度17bp-25bp为佳。
太短的引物容易导致扩增效率降低;太长的引物会导致出现引物高级结构的几率增加。
两者都会干扰定量结果的准确性b)扩增片段长度为:90-150 bp(最低不能超过70,最高不能超过180)c)引物的Tm值为:最小57℃,最大63℃,最适为60℃,两条引物之间退火温度得差距不超过1℃,推荐使用Primer Premier 5进行Tm值计算;d)引物A、G、C、T整体分布尽量要均匀,避免使用GC或者TA含量高的区域,尤其是3’端,必须避开GC含量不均匀的区域。
e)引物设计时请尽量避开TC或者AG的连续结构。
f)3’端不能超过3个以上碱基互补,自互补碱基数不超过3;3’端最后一个碱基绝对不能搭上g)特异性要有保证,与非特异模板3’端互搭碱基数不超过3,不连续出现4个及以上的GC互搭h)引物3’端最后五个碱基不能包含超过2个以上的G或者Ci)引物的GC含量控制在40%-60%之间为好,最佳为45%-55%之间j)正向或者反向引物应尽量接近探针序列但是不能和探针序列有重合区域k)在Primer-BLAST设计时,在Organism 处选择相应物种l)需跨外显子设计,避免基因组污染2)TaqMan探针设计指南a)探针序列应尽量接近正向或者反向引物,但是不能与之有重合区域;一般相隔1~5个碱基(一般10个以内,最好是1个碱基)。
引物设计的详细步骤

一、引物设计step by step1、在NCBI上搜索到目的基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy 该编码序列作为软件查询序列的候选对象.2、用Primer Premier5搜索引物①打开Primer Premier5,点击File—New—DNA sequence,出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
②此窗口可以链接到“引物搜索"、“引物编辑"以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度.在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择 300~500bp。
③点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
④对于引物的序列,可以简单查看一下,避免出现下列情况: 3'不要出现连续的3个碱基相连的情况,比如GGG或CCC,否则容易引起错配.此窗口中需要着重查看的包括:T m应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的T m值最好不要相差太多,大概在2度以下较好.该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置.若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好.但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。
引物设计的详细步骤

引物设计是PCR(聚合酶链式反应)技术中的关键步骤,以下是引物设计的详细步骤:选择合适的引物长度:通常选择18-30个核苷酸长度的引物。
引物太短可能降低特异性,
而太长则可能导致非特异性结合。
选择合适的引物GC含量:通常选择40%-60%的GC含量。
GC含量过高或过低都可能
影响PCR的效率。
避免引物二聚体和发夹结构:这些结构可能导致引物自身结合,从而影响PCR的效率。
可以使用软件工具检查引物的这种可能性。
避免引物间的互补:引物之间互补的序列可能导致引物结合,从而影响PCR的效率。
选择合适的引物位置:引物应位于目标基因的特异区域,通常选择基因的编码区。
此外,应避免选择有高突变率的区域,这可能影响引物的特异性。
使用软件进行引物设计:有许多在线和离线软件可以帮助设计PCR引物,如Primer3、Oligo 等。
这些软件可以根据输入的基因序列自动设计和选择最佳的引物。
实验验证:即使通过软件设计的引物看起来很好,也需要在实验中进行验证,以确保其特异性、有效性和可靠性。
引物浓度和退火温度的优化:引物的浓度和退火温度也是PCR的重要参数,需要针对特定的反应条件进行优化。
请注意,对于具体的实验和目的,可能需要更具体和详细的设计考虑,建议咨询相关领域的专家或具有丰富经验的实验员。
标准操作规程(SOP)——流感禽流感病毒 RT-PCR 引物制备

RT-PCR 引物制备,用于流感/禽流感病毒RT-PCR 检测,确保RT-PCR 检测结果的可靠性。
二、范围
适用于中国国家流感中心所有技术人员对疑似流感/禽流感病毒感染标本进行RT-PCR 检测。
三、程序
(一)引物合成
六合通(大连宝生物)或同类公司合成,PAGE 纯化。
(二)引物配制
1.10×储存液配制:
(1)将新合成的引物在开盖前短暂离心(12000rpm ,15s )。
(2)用RNase Free Water 溶解,加水量为10×总摩尔数,充分混匀,此时引物浓度为100pmol/μL ,即100μM 。
(例如:合成引物2OD ,9.5nmol ,则加水量为10×9.5=95μL 。
)
2.引物使用浓度配制:将100μM 的引物浓度再10倍稀释,配成浓度为10μM ,即可直接用于PCR 反应。
(三)引物保存
1.引物未溶解前4℃保存。
2.引物溶解之后-20℃或-20℃以下保存。
注意:引物配制前,先将引物管短暂离心,以免引物丢失。
一、目的标准操作规程(SOP 毒RT-PCR。
引物设计的详细步骤

引物设计的详细步骤详细步骤如下:步骤一:了解引物设计的基本原理引物设计是指为特定的DNA序列设计一对合适的引物,以便在PCR反应中扩增目标DNA序列。
引物是PCR反应的关键组成部分,引物的选择和设计对于PCR扩增的成功率和特异性非常重要。
因此,了解引物设计的基本原理对于有效设计合适的引物至关重要。
步骤二:确定PCR反应的目标序列在设计引物之前,我们需要确定PCR反应的目标序列,即我们需要扩增的DNA区域。
这个目标序列可以是已知的基因序列,也可以是未知的区域。
确定目标序列后,我们可以继续设计引物。
步骤三:确定引物的一些基本参数在设计引物之前,我们需要确定一些基本的参数,以便帮助我们选择合适的引物。
这些参数包括引物的长度、GC含量、Tm值以及避免二聚体形成等。
引物长度:通常来说,引物的长度应在18-25个核苷酸之间。
过长的引物可能导致不特异的扩增产物的形成,而过短的引物则可能导致低扩增效率。
GC含量:引物的GC含量对于引物的稳定性和特异性有影响。
在正常情况下,引物的GC含量应在40%-60%之间。
Tm值:引物的Tm值是指引物在PCR反应中的解离温度。
Tm值过低可能导致非特异的扩增产物的形成,而Tm值过高则可能导致低扩增效率。
避免二聚体形成:在设计引物时,我们还需要考虑引物之间的互补性以及避免引物形成二聚体。
引物之间的互补性可能导致引物形成二聚体,从而降低PCR反应的效率和特异性。
步骤四:选择合适的引物设计工具目前有很多在线引物设计工具可供选择,例如NCBI Primer-BLAST、OligoAnalyzer等。
这些工具可以根据输入的目标序列帮助我们快速选择合适的引物。
此外,还可以使用一些商业引物设计软件,如Primer Premier等。
步骤五:进行引物特异性分析设计好引物后,我们需要进行引物特异性分析,确保引物只扩增目标序列而不扩增其他非特异性产物。
这可以通过BLAST或其他相似性工具来完成。
特异性分析的目的是排除可能存在的非特异性扩增产物,以确保PCR反应的准确性和特异性。
设计引物操作

1.PubMed Nucleotide下载PCR基因所需序列,下载格式为FASTA/nuccore2.可以批量打开下载的引物序列,批量设计引物点击Analyze - Primer searchSearch Parameters:一般情况search range为全长Primer parameters:Tm:溶解温度,63-67(ideally 64),Tm=2(A+T)+4(G+C)Ta:退火温度(比Tm一般低1-2℃),一般设置60±2℃(PS:如果设计小鼠鉴定的DNA PCR引物,可以将温度范围设宽,跑PCR时可以自己设置退火温度,但是Takara RT-PCR的退火温度为60℃,所以设计引物尽量让退火温度在60℃附近)Ta:退火温度(比Tm低1-2℃)3.Length:18-30bp4.Amplication length:80-150bp5.如果search不到所需要的引物,可以适当放宽条件6.Cross dimer(上下游引物形成二聚体),△G(负的越大,越稳定),比如△G=-3比-1稳定,当为-3时就比较容易形成二级结构,此时也需看形成二聚体的结构能否被PCR出来,如果仅仅是在5’端有些许碱基配对,不会形成产物也没关系。
引物设计好,Blast进行比对/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC =blasthomecopy sense, copy antisense,分开进行序列比对Database: Mouse genomic + transcriptBlast: show results in a new window,E value越小,特异性越高。
(除了对比出mRNA,有可能对比出DNA,如果上下游引物对比得到DNA结构也仅仅在5’或3’端有配对,不形成长的产物,也没关系,要保证自己的样品没有DNA污染。
引物设计具体步骤

引物设计一、引物设计简介引物设计是以一小段单链DNA或RNA,作为DNA复制的起始点,在核酸合成反应时,作为每个多核苷酸链进行延伸的起点而起作用的多核苷酸链。
我们根据根据蛋白的基因序列及所选取的目的片段,设计引物。
二、引物设计的一般原则(一)抗原性:引物设计在完整的Domain区域或者抗原表位集中区域;(二)PCR产物长度:PCR产物长度不应过长,最佳长度为500bp左右。
;(三)长度:15-30bp,其有效长度[Ln=2(G+C)+(A+T)]一般不大于38,否则PCR 的最适延伸温度会超过Taq酶的最佳作用温度(74℃),从而降低产物的特异性;(四)GC含量:应在40%-60%之间,PCR扩增中的复性温度一般是较低Tm 值引物的Tm值减去5℃;(五)碱基分布的随机性:应避免连续出现4个以上的单一碱基。
尤其是不应在其3’端出现超过3个的连续G或C,否则会使引物在G+C富集序列区错误引发;(六)互补、错配、二级结构:引物自身不能含有自身互补序列,否则会形成发夹样二级结构。
两个引物之间不应有多于4个互补或同源碱基,不然会形成引物二聚体,尤应避免3’端的互补重叠;(七)引物的3’端很大程度上影响Taq酶的延伸效应,应尽量避免3’端发生错配。
而且尽可能地避免选用T,尤应避免连续出现2个以上的T;(八)特异性:与非特异扩增序列的同源性应小于70%,或少于连续8个的互补碱基。
三、常用软件DNAman5,Primer Preimer5,DNAStar/EditSeq、Snapgene四、信息检索常用数据库(一)uniprot(二)ncbi五、引物设计流程以蛋白Actin Beta的Met1-Phe375为例,说明引物设计具体流程:(一)根据蛋白名称或uniprot等信息,点开uniprot数据库;(二)在uniprot数据库中最左侧的菜单栏中,点击“sequence”,找到氨基酸序列;注:若此蛋白有多个isoform,一般以isoform1的序列为准设计引物(三)在“sequence databases”中找到这个蛋白对应的NM号;注:每一个isoform对应唯一一个NM号(四)点击NM号,进入NCBI数据库,找到对应的CD S序列,图中棕色部分即为此蛋白的碱基序列,根据研究需要,截取对应片段长度,如氨基酸片段为1-375,则碱基序列为1-1125;(五)打开Primer Preimer5引物设计软件,输入选取的碱基序列,点击“Primer”示anti-sense;再点击“Edit Primers”进行编辑;(七)当信息栏显示的发夹结构、二聚体、错配等信息为“None”时,初步认为此引物为最佳选择;(八)进入NCBI数据库,点击“BLAST”,选择“Primer-BLAST”,再次分析确认此引物是否为最佳引物。
简述pcr引物设计的基本步骤

简述pcr引物设计的基本步骤
PCR引物设计是PCR技术中至关重要的一步,它直接影响到PCR 反应的特异性和效率。
以下是PCR引物设计的基本步骤:
1. 确定目标序列,首先需要确定要扩增的目标DNA序列,这可以是基因、片段或者其他特定的DNA区域。
2. 引物长度,一般来说,PCR引物的长度应在18-25个碱基对之间,太短会影响特异性,太长则会影响引物的合成效率。
3. 引物的GC含量,引物的GC含量应在40-60%之间,这有助于提高引物与模板DNA的亲和力。
4. 引物特异性,引物应该与目标DNA序列高度特异性地结合,避免与其他非特异性DNA结合。
5. 引物序列的避让,避免引物序列中出现相互补的碱基对,以免引物之间发生非特异性结合。
6. 引物的末端,引物的末端应该避免出现多余的碱基对,以免
影响PCR扩增的效率。
7. 引物的Tm值,引物的熔解温度(Tm值)应该相似,一般来说,它们之间的差异不应超过5摄氏度。
在进行PCR引物设计时,以上这些基本步骤可以帮助确保PCR 反应的特异性和效率。
同时,也可以利用一些生物信息学工具来辅助引物设计,如NCBI的Primer-BLAST、IDT的PrimerQuest等。
PCR引物设计的好坏直接关系到PCR扩增的成功与否,因此在实验前务必进行充分的设计和验证。
引物设计步骤与要点

引物设计步骤与要点引物(primer)是在 DNA 或 RNA 聚合酶链式反应(PCR)或逆转录聚合酶链式反应(RT-PCR)中使用的短的 DNA 或 RNA 片段。
引物通过与目标序列的互补配对,为 PCR 或 RT-PCR 提供起始点,使得复制过程能够在目标序列上进行。
引物的设计是 PCR 或 RT-PCR 的关键步骤,影响其特异性和效率。
下面将介绍引物设计的步骤与要点。
引物设计的步骤如下:1.确定目标序列:首先要明确所需扩增的目标DNA或RNA序列。
例如,目标序列可以是特定基因的编码区域,或者是需要检测的病原体的DNA片段。
2. 引物长度:引物的长度通常在 18-30 bp 之间。
长度较长的引物可能会导致非特异性扩增,而较短的引物可能会导致不够稳定,产生非特异性扩增产物。
在设计引物时,应注意避免引物间或引物与模板间的互相互补性。
3.GC含量:引物的GC含量应在40-60%之间。
GC含量过高可能导致引物之间的二聚体形成,而GC含量过低可能导致引物的稳定性不足。
4.特异性:引物应与目标序列的特定部分互补配对,以确保特异性扩增。
在设计引物时,通常选择序列中的保守区域作为互补匹配的区域,以确保其在各物种或基因型中的适用性。
此外,可以通过使用在线工具,如NCBIBLAST,对引物进行特异性检测,以避免与非目标序列互补匹配。
5. 引物之间的互补配对:在 PCR 扩增中,引物通常成对使用,所以引物之间不应存在互补配对,以避免二聚体形成。
另外,引物对之间的距离应合适,通常在 100-300 bp 之间。
6.引物的末端设计:引物的末端设计直接影响PCR的效率和特异性。
在设计引物时,应注意避免末端的一些特定的串扰序列,如GGGG、CCCC、AAAA、TTTT等。
此外,引物的末端可以添加一些特定的序列,如引物标记和引物序列的识别序列,以便进一步的实验操作。
引物设计的要点如下:1.使用专业软件或在线工具进行辅助设计:可以使用一些专业的引物设计软件或在线工具来辅助引物的设计。
引物的配置标准操作规程

引物的配置标准操作规程引物的配置是分子生物学实验中常用的一项操作,它用于扩增目标DNA片段。
引物的配置标准操作规程主要包括以下几步:第一步:设计引物在进行引物的配置之前,首先需要根据目标DNA序列进行引物的设计。
引物通常由18-25个碱基组成,需要具备以下特点:1. 引物的长度应在18-25个碱基之间,以确保扩增效果的最优化。
2. 引物的GC含量应在40%-60%之间,以确保引物的稳定性和互补性。
3. 引物的Tm值(两股DNA解离中点温度)应在50-65℃之间,以确保合适的扩增温度。
第二步:引物的合成完成引物设计后,需要将引物送至合成顺序。
引物的合成可以委托给专业的合成公司进行,也可以通过自己的实验室进行合成。
合成的引物需要进行纯度检测,确保引物没有杂质和污染物。
第三步:引物的稀释将合成好的引物溶解在无菌纯水中,制备成一定浓度的存储液。
根据具体实验需求,通常可以将引物配置成100μM的浓度。
第四步:引物的保存配置好的引物需要进行适当的保存,以确保引物的稳定性和活性。
通常可以将引物储存在-20℃的冰箱或冻存管中,避免光照和长时间的暴露。
第五步:引物的稀释和配对在进行PCR反应时,需要将引物稀释至适当的浓度,通常可以将100μM的引物稀释成10μM的工作浓度。
同时,需要进行正反向引物的配对,确保引物能够正确结合和扩增目标DNA。
第六步:引物的PCR反应条件设置根据不同的实验目的和目标DNA,需要设置适当的PCR反应条件。
包括扩增温度、时间和循环次数等参数。
一般情况下,引物的扩增温度设置在50-65℃之间,时间设置在30-60s,循环次数根据目标DNA的需求,通常为25-40个循环。
第七步:引物的PCR反应验证配置好引物的PCR反应系统后,需要进行验证实验,确保引物的可靠性和扩增效果。
通过运行PCR反应并进行凝胶电泳分析,观察扩增产物的带型和大小,判断引物配置的准确性和可行性。
最后,根据实验的具体需求和引物的配置情况,可以通过调整PCR反应条件和引物设计来进一步优化引物的效果和稳定性。
详尽的引物设计心得及具体流程操作

详尽的引物设计⼼得及具体流程操作本⽂作者:张慧慧(长沙湘雅附⼆)本⽂系参加解螺旋段⼦⼿招募作品查看更多段⼦⼿招募详情,请回复“悬赏”1 克隆载体: 表达载体⼀般需要的是编码序列(起始密码⼦ATG到终⽌密码⼦TAA/TGA/TGA的序列),也就是NCBI的 coding sequence(CDS)。
⼀般情况是全长序列,直接取ATG开始的20bp为上游引物,TAA/ TGA/ TAG端的20bp的反向互补序列反向引物。
根据载体的酶切位点、读码框的要求在引物的5’端碱基即可。
要注意的是: ①选⽤的酶切位点在编码序列上是否存在,存在就选择其他酶切位点或者选⽤同尾酶。
②载体的标签实在插⼊序列的上游还是下游,在上游编码序列的终⽌密码就要留,在下游编码序列的终⽌密码就要去掉。
③读码框是否正确,有些酶切位点会破坏插⼊序列的读码顺序,这个时候在引物上添加碱基使其正常读码即可。
例如构建EGFP-C1-P53质粒。
① P53序列 绿⾊表⽰选取的引物位置(EGFP-C1的GFP标签在插⼊序列的上游,所以终⽌密码保留。
) 引物序列是:上游引物(与绿⾊标⽰的序列⼀致)5'-ATGGAGGAGCCGCAGTCAGA-3';下游引物(与绿⾊标⽰的序列反向互补)5'-TCAGTCTGAGTCAGGCCCTT-3' ②载体多克隆位点(MCS)分析 我选择酶的原则是价格便宜,常⽤的酶,例如XhoI/BamHI/HindIII等,这样的酶在很多载体中都会有,有时候可以通⽤。
分析p53的序列中是否有XhoI/BamHI两个酶切位点,结果是没有,那就可以放⼼的⽤这两个酶了。
于是引物可以变成 上游引物5’-CTCGAGATGGAGGAGCCGCAGTCAGA-3’(XhoI) 下游引物5’-GGATCCTCAGTCTGAGTCAGGCCCTT-3’(BamHI) 酶在发挥作⽤的时候要有个保护碱基,我认为就是给它⼀个空间让它发挥作⽤,保护碱基在百度百科可以查到,于是引物就会变成 上游引物5’-ccgCTCGAGATGGAGGAGCCGCAGTCAGA-3’ 下游引物5’-cgGGATCCTCAGTCTGAGTCAGGCCCTT-3’③读码分析 XhoI的序列CTC GAG与载体的读码框不⼀致,载体的读码是 TCT CGA GCT,当⽤XhoI/BamHI双酶切载体时,最后的CT需要插⼊序列的AT来代替,就会使插⼊的序列移码,这时候,只要在插⼊序列ATG的前⾯加CT即可,所以引物序列就变成上游引物5’-ccgCTCGAG CTATGGAGGAGCCGCAGTCAGA-3’下游引物5’-cgGGATCCTCAGTCTGAGTCAGGCCCTT-3’ 所以当你给公司发引物序列时就是上游引物5’-ccgCTCGAG CTATGGAGGAGCCGCAGTCAGA-3’下游引物5’-cgGGATCCTCAGTCTGAGTCAGGCCCTT-3’ 因为是全长序列,就表⽰引物已经定了,没有选择。
qPCR引物设计原则及具体操作步骤
qPCR引物设计原则及具体操作步骤qPCR引物设计原则及具体操作步骤1.找基因(DNA)1)通过英文名称查找通过查看文献或者百度搜索查找到对应基因的准确的英文名称→进入NCBI官网→点击网页右下角GenBank,进入GenBank界面→在搜索框中输入准确的英文名称,点击Search搜索即可2)通过序列号查找通过查找文献,找到相应基因在GenBank上的登录号,直接输入上面的搜索框进行查找即可。
例如:犬冠状病毒(canine coronavirus,CCV)基因保守片段序列号为KT222978。
3)通过引物查找通过查找文献,找到别人用过的对应的引物→在NCBI官网右下角点击Primer-BLAST→输入正、反向引物序列→设置对应参数→点击“Get Primers”进行搜索即可4)找到对应的基因后点击“FASTA”,进入相应界面,再点击“Send to”选择相应格式,保存序列。
2.qPCR引物和TaqMan探针的设计1)引物设计注意事项a)引物长度17bp-25bp为佳。
太短的引物容易导致扩增效率降低;太长的引物会导致出现引物高级结构的几率增加。
两者都会干扰定量结果的准确性b)扩增片段长度为:90-150 bp(最低不能超过70,最高不能超过180)c)引物的Tm值为:最小57℃,最大63℃,最适为60℃,两条引物之间退火温度得差距不超过1℃,推荐使用Primer Premier 5进行Tm值计算;d)引物A、G、C、T整体分布尽量要均匀,避免使用GC或者TA 含量高的区域,尤其是3’端,必须避开GC含量不均匀的区域。
e)引物设计时请尽量避开TC或者AG的连续结构。
f)3’端不能超过3个以上碱基互补,自互补碱基数不超过3;3’端最后一个碱基绝对不能搭上g)特异性要有保证,与非特异模板3’端互搭碱基数不超过3,不连续出现4个及以上的GC互搭h)引物3’端最后五个碱基不能包含超过2个以上的G或者Ci)引物的GC含量控制在40%-60%之间为好,最佳为45%-55%之间j)正向或者反向引物应尽量接近探针序列但是不能和探针序列有重合区域k)在Primer-BLAST设计时,在Organism 处选择相应物种l)需跨外显子设计,避免基因组污染2)TaqMan探针设计指南a)探针序列应尽量接近正向或者反向引物,但是不能与之有重合区域;一般相隔1~5个碱基(一般10个以内,最好是1个碱基)。
引物设计的详细步骤

引物设计的详细步骤引物设计是一项关键的实验技术,用于在分子生物学实验中扩增目标DNA片段。
该技术的成功与否直接影响到实验结果的准确性和可靠性。
以下是引物设计的详细步骤:1.确定目标DNA序列:首先,确定需要扩增的目标DNA序列。
这可以通过已知的参考序列、文献调研或基因数据库进行。
2.定位扩增区域:根据目标DNA序列,确定需要扩增的特定区域。
通常选择在该区域中的保守性较高的片段,以确保引物的特异性。
3. 确定引物长度:引物长度通常为18-25个核苷酸(nt),最好不超过30nt。
引物长度的选择是为了确保引物在反应温度下的特异性和稳定性,同时不会引起非特异扩增。
4.碱基组成与G/C含量:引物的碱基组成应平衡,避免过多的同质二聚物和结构异常。
G/C含量一般在40-60%之间,过高或过低的G/C含量可能会导致引物与模板DNA结合的效力降低。
5.特异性:使用基因序列数据库或引物设计软件进行引物BLAST比对,确保引物与目标DNA序列的独特性。
6. 避免互补引物间的二聚体形成:引物间不能有太多相互衔接的序列,以免引物自身形成二聚体。
通常应避免引物间的结合自由能低于-9 kcal/mol。
7.避免引物内部二聚体的形成:通过引物设计软件计算引物的内部二聚体结合自由能,避免过多的二聚体形成。
8.引物末端设计:通常引物的末端应设计在较保守的区域,以确保扩增的特异性。
9.引物的副产物与杂交:避免引物自身产生副产物以及与其他非特异目标DNA序列杂交。
10.引物设计验证:使用引物设计软件对设计的引物进行验证,包括引物特异性、二聚体和杂交等。
11.引物合成:通过合成引物的商业公司进行引物合成,选择信誉好的厂家。
12.引物纯化:使用聚丙烯酰胺凝胶电泳等方法对引物进行纯化。
13.引物浓度测定:使用紫外分光光度计等方法测定引物的浓度。
总之,引物设计是一项细致而复杂的步骤,需要考虑多个因素,如目标DNA序列,引物长度,碱基组成,特异性和二聚体等。
pcr引物设计操作流程

pcr引物设计操作流程
PCR引物设计是PCR技术中非常重要的一步,引物的设计质量直接影响到PCR反应的效果和结果。
下面是PCR引物设计的操作流程:
1. 确定目标序列:首先需要确定要扩增的目标序列,这个序列可以是基因、DNA片段或RNA序列等。
2. 选择引物设计工具:选择合适的引物设计工具,比如NCBI Primer-BLAST、Primer3等在线工具或者专业的引物设计软件。
3. 设定引物设计参数:根据实验需求设定引物设计的参数,比如引物长度、GC含量、Tm值等。
4. 引物设计:根据目标序列和设定的参数,使用引物设计工具进行引物设计。
通常需要设计一对引物,一个用于扩增目标序列的前端,一个用于扩增目标序列的后端。
5. 引物评估:对设计出的引物进行评估,包括检查引物的特异性、二聚性、自身互补性等。
6. 引物合成:将设计好的引物提交给引物合成公司进行合成。
通常引物的长度在18-25个碱基对之间。
7. PCR反应:将合成好的引物与DNA模板、PCR反应缓冲液、
DNA聚合酶等混合,进行PCR反应。
根据引物的设计,可以选择不同的PCR条件进行扩增。
8. PCR产物分析:对PCR反应产物进行分析,比如琼脂糖凝胶电泳、测序等,确认扩增的目标序列是否正确。
总的来说,PCR引物设计是PCR技术中至关重要的一步,需要仔细设计和评估引物,确保PCR反应的准确性和可靠性。
通过以上的操作流程,可以有效地设计出高质量的引物,为PCR实验的成功提供保障。
引物设计的详细步骤

引物设计的详细步骤引物设计步骤如下:1.目标序列选择:根据研究目的选择需要扩增或检测的目标DNA序列。
这个目标序列可能是基因的特定区域、启动子区域、外显子、cDNA序列等。
选择一个合适的目标序列对于引物设计至关重要,因为它将决定引物的特异性和扩增产物的大小。
一般而言,目标序列具有良好的保守性和特异性。
2.引物长度和Tm计算:引物通常是15-30个核苷酸长度。
引物长度的选择取决于目标序列的特点以及所使用的实验条件。
合适的引物长度应该综合考虑引物的特异性和扩增效率。
引物的熔解温度(Tm)是指DNA链的两个链断裂开所需要的温度,它是引物设计中一个重要的参数。
Tm可以通过计算引物的碱基组成、盐度和引物浓度等因素来估计,通常约为55-65℃。
3. 引物特异性检查:使用生物信息学工具,如BLAST(Basic Local Alignment Search Tool)或NCBI(National Center forBiotechnology Information)数据库来检查所设计引物的特异性。
确保引物不会扩增与目标序列不匹配的区域,避免非特异性扩增和假阳性结果。
4.引物序列设计:根据目标序列和引物长度选择合适的引物序列。
引物设计的要求包括:GC含量约为40-60%、避免重复序列和目标序列内部的局部重复、避免碱基偏差和突变等。
此外,引物的碱基组成应该是均匀的,避免多个连续G或C碱基的存在。
6.引物的合成:将设计好的引物交给合成公司进行合成,通常采用化学合成方法。
引物的质量控制非常重要,合成的引物应进行质控检测,如毛细管电泳或质谱分析。
推荐文献:2. Untergasser A, et al. (2024) Primer3--new capabilities and interfaces. Nucleic Acids Res. 40(15): e115.。
引物设计是分子生物学研究中必不可少的技术之一、通过遵循上述步骤和参考推荐文献,可以设计出特异性高、效率好的引物,为后续实验的顺利进行提供支持。
PCR引物流程设计详解

PCR引物流程设计详解PCR(Polymerase Chain Reaction)引物流程设计是在进行PCR反应过程中引物的设计。
PCR反应是一种体外的DNA复制技术,可在短时间内扩增特定DNA序列。
引物在PCR反应中起到了至关重要的作用,因此设计合适的引物是成功进行PCR反应的关键。
1.目标序列选择:首先需要明确PCR反应的目标序列,即要扩增的特定DNA序列。
选定目标序列后,需要使用相应的软件分析该序列的特性,如GC含量、碱基组成、互补性等。
这些特性将有助于引物的设计和优化。
2. 引物长度:引物的长度通常在18-30bp之间。
较短的引物能提高PCR反应的特异性,但较长的引物能提高PCR反应的特异性和效率。
引物长度不宜超过30bp,以免在PCR反应过程中产生副产物。
3. 引物序列设计:PCR反应通常需要设计两个引物,一个称为前向引物(forward primer),另一个称为反向引物(reverse primer)。
两个引物应该在目标序列两侧的互补区域上设计,以确保引物能够结合在目标序列的两端。
为了提高特异性,引物的3'端应尽可能与目标序列互补,而5'端则可根据需要进行一定的修改,如添加限制性酶切位点、引入Tm值调整等。
4.引物Tm值计算:Tm值可用于估计引物与目标序列结合的稳定性。
Tm值是引物在PCR反应中的解链温度,通常在50-60°C之间。
使用软件计算引物的Tm值时需要考虑引物的长度、碱基组成和浓度等因素,确保引物的Tm值相近。
5.引物特异性检验:根据引物设计的序列,使用引物设计软件进行特异性检验,确保引物只结合在目标序列上而不结合在其他非特定序列上。
特异性检验可通过引物序列的BLAST分析和二聚体结构预测等方法进行。
6.引物修饰:在一些情况下,可以根据需要对引物进行特定的修饰,以增强PCR反应的效果。
常见的修饰方法包括添加引物标记(如荧光标记)、引物末端修饰(如磷酸化)等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
引物设计的标准操作规程(编号:004)
1、软件使用
1.1 推荐软件:Primer Premier 5.0
1.2 优点:操作简单、显示各种参数改变和二聚体、异二聚体、发夹结构等。
1.3 本地同类软件:DNAClub;Oligo 6.22;Vector NTI Suit;Dnasis;Omiga;Dnastar;DNAMAN (Lynnon Biosoft, Quebec, Canada)。
1.4 网上同类软件:Primer3、JaMBW(European Molecular Biology Laboratory of Heidelberg 开发)。
http://210.7
2.11.60网站已引进并调试好这两种软件。
2、推荐操作
引物搜索:Primer Premier 5.0、引物评价:Oligo 6.22
3、引物设计的原则
首先引物要跟模板紧密结合,其次引物与引物之间不能有稳定的二聚体或发夹结构存在,再次引物不能在别的非目的位点引起DNA聚合反应(即错配)。
围绕这几条基本原则,设计引物需要考虑诸多因素,如引物长度(primer length)、产物长度(product length)、序列Tm值(melting temperature)、ΔG值(internal stability)、引物二聚体及发夹结构(duplex formation and hairpin)、错误引发位点(false priming site)、引物及产物GC 含量(composition),有时还要对引物进行修饰,如增加限制酶切点,引进突变等。
以使用Oligo 软件分析设计引物为例,笔者总结出以下的要点:3.1 引物的长度一般为18-25bp,引物长度过长会导致延伸温度过高,从而影响DNA聚合酶的效率,上下游引物长度差别最好不要大于3bp。
3.2 引物最好在模板DNA的保守区内设计。
DNA序列的保守区是通过物种间相似序列的比较确定的。
可在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。
3.3 引物3′端不能选择A,最好选择T。
引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3′端最好选择T。
3.4 引物的GC含量一般为40-60%。
上下游引物的GC含量不能相差太大。
Tm值以接近72℃为宜,上下游引物Tm值同样不应相差过大。
3.5 引物3′端要避开密码子的第3位。
如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。
14。