全基因组关联分析-基于全基因组重测序
全基因组重测序数据分析详细说明

全基因组重测序数据分析详细说明全基因组重测序(whole genome sequencing, WGS)是一种高通量测序技术,用于获取个体的整个基因组信息。
全基因组重测序数据分析是指对这些数据进行处理、分析和解读,以获得有关个体的遗传变异、基因型、表达和功能等信息。
下面详细说明全基因组重测序数据分析的过程和方法。
首先,全基因组重测序数据的质量控制是必不可少的。
这一步骤包括对测序数据进行质量评估、剔除低质量序列,并进行去除接头序列和过滤序列等预处理操作,以确保后续分析的准确性和可靠性。
接下来,需要对全基因组重测序数据进行序列比对,将读取序列与参考基因组进行比对,以确定每个读取序列在参考基因组上的位置。
常用的比对工具包括Bowtie、BWA、BLAST等。
比对的结果将提供每个读取序列的基因组位置信息。
在序列比对完成后,就可以进行个体的变异检测。
变异检测的目的是识别个体的单核苷酸多态性(single nucleotide polymorphisms, SNPs)、插入缺失变异(insertions/deletions, indels)和结构变异(structural variations, SVs)等基因组变异。
通常,变异检测分为两个步骤:变异发现和变异筛选。
变异发现即根据比对结果,通过一定的算法和统计学原理,找到潜在的变异位点。
然后,利用临床数据库、已知变异数据库和基因功能注释数据库等,进行变异筛选,剔除假阳性和无功能变异,筛选出最有可能的致病变异。
接着,对筛选出的变异位点进行基因型確定。
基因型的确定可以通过直接从比对结果中读取碱基信息,或者通过再次测序来获取高度精确的基因型,以获得更可靠的变异信息。
随后,对变异位点进行注释和功能预测。
注释是指对变异位点进行功能和可能影响的基因、基因组区域和调控元件等进行注释。
常用的注释工具包括ANNOVAR、SnpEff、VEP等。
功能预测则是根据变异位点的位置和可能影响的功能进行预测,如是否影响蛋白质功能、是否在编码序列、是否在启动子或增强子区域等。
全基因组重测序数据分析

全基因组重测序数据分析1. 数据质量控制:对测序数据进行质量控制,包括去除低质量的碱基、过滤含有接头序列和接头污染的序列等。
这一步骤可以使用各种质控工具,例如FastQC、Trim Galore等。
2. 比对到参考基因组:将经过质控的测序数据与参考基因组进行比对。
参考基因组一般是已知的物种的基因组序列,在人类研究中通常使用人类参考基因组。
比对工具主要有BWA、Bowtie等。
3. 变异检测:从比对结果中检测出样本与参考基因组之间的差异,称为变异检测。
这包括单核苷酸变异(SNV)、插入/缺失(Indel)、结构变异(SV)等。
常用的变异检测工具有GATK、SAMtools、CNVnator等。
4. 注释和解读:对检测到的变异进行注释和解读,以确定其对基因功能和疾病相关性的影响。
注释可以包括基因、转录本、蛋白质功能、通路、疾病关联等信息。
常用的注释工具包括ANNOVAR、Variant Effect Predictor等。
5.结果可视化:将分析结果以图表或图形的形式展示出来,以便研究人员更好地理解和解释结果。
常用的可视化工具包括IGV、R软件等。
除了上述步骤,全基因组重测序数据分析还可以应用于其他研究领域,例如种群遗传学、复杂疾病研究、药物研发等。
在进行这些研究时,可能还需要其他分析方法和工具来完成特定的研究目标。
总之,全基因组重测序数据分析是一个复杂而关键的过程,它可以帮助研究人员了解个体的基因组特征,并揭示与疾病发生和发展相关的重要信息。
在不断发展的测序技术和分析方法的推动下,全基因组重测序数据分析将在基因组学领域中发挥越来越重要的作用。
全基因组关联分析剖析

对家系数据进行检查,排 除样本混淆、亲子关系 错误等问题,控制家系关 系的正确性。
全基因组关联分析的结果验证
验证检查
对于全基因组关联分析的结果,需要进行严格的验证检查,以确保结果的可靠性和重复性。
重复实验
在不同的人群或样本中重复实验,比较结果是否一致进一步的功能实验,探讨基因变异与表型之间的机制。
全基因组关联分析的统计方法
统计分析
全基因组关联分析通常采用统计模型对遗传标记与表型之间的关联进行测试,如线性回归、logistic 回归等。
多重检验校正
由于基因组级别的大量比较检验,需要采用Bonferroni、FDR等方法进行多重检验校正,以控制I型错 误风险。
机器学习方法
近年来,全基因组关联分析也开始采用机器学习技术,如Ridge回归、Lasso回归等方法,以提高检测 能力。
全基因组关联分析的研究 热点
1 复杂疾病研究
全基因组关联分析被广 泛应用于探索复杂疾病 如糖尿病、心血管疾病 、肿瘤等的遗传学基础 。
3 交互作用研究
多基因、基因-环境等交 互作用的研究是全基因 组关联分析的重要方向 。
2 药物反应预测
全基因组分析有助于识 别影响药物反应的基因 变异,助力个体化精准医 疗。
生物学解释
从统计上显著关联的遗 传位点到生物学功能解 释存在鸿沟,需要更深入 的研究。
跨人群适用性
现有大多数研究集中于 欧美人群,如何推广到其 他人群是一大挑战。
全基因组关联分析的研究进 展
多组学整合
研究者正在探索将全基因组 关联分析与转录组学、表观 遗传学等多种组学数据相结 合的方法,以更全面地了解 复杂疾病的遗传学机制。
新型统计方法
学者们不断开发基于机器学 习、贝叶斯统计等的创新分 析方法,以提高检测复杂遗 传变异和基因-环境相互作 用的能力。
全基因组关联分析

“全基因组关联分析”资料合集目录一、全基因组关联分析在作物农艺性状研究中的应用二、玉米12个农艺性状的全基因组关联分析及玉米氮响应相关基因的鉴定三、全基因组关联分析在水稻遗传育种中的应用和研究进展四、支气管哮喘的全基因组关联分析研究进展五、水稻苗期稻瘟病抗性的全基因组关联分析六、全基因组关联分析的进展与反思七、甘蓝型油菜分枝角度和株高全基因组关联分析八、基于SNP芯片和全测序数据的奶牛全基因组关联分析和基因组选择研究九、桃基因组及全基因组关联分析研究进展全基因组关联分析在作物农艺性状研究中的应用一、引言在过去的十年中,随着基因测序技术的飞速发展,全基因组关联分析(Genome-wide Association Study,GWAS)已成为研究作物农艺性状的重要工具。
作物农艺性状是指作物在生长发育过程中表现出的形态、生理和产量等特征,这些性状通常受到多个基因的控制,并且会受到环境因素的影响。
通过GWAS,我们可以识别与特定农艺性状相关的基因变异,进一步理解作物生长发育的规律,并为作物育种提供重要的指导。
二、全基因组关联分析的原理和方法GWAS的基本原理是利用单核苷酸多态性(SNP)作为分子标记,通过比较不同品种或群体中SNP位点的差异,来寻找与特定农艺性状相关的基因变异。
在作物研究中,常用的方法包括基因组重测序和基因组扫描。
基因组重测序是对作物种质资源进行全基因组测序,以获取高精度的基因型信息。
基因组扫描则是利用已发表的SNP数据和农艺性状数据,进行大规模的关联分析。
三、全基因组关联分析在作物农艺性状研究中的应用1、作物产量:通过GWAS,研究者已经识别了许多与作物产量相关的基因变异。
例如,在玉米中,与产量相关的基因变异被发现与植物生长和发育的多个阶段有关,包括叶片大小、节间长度和花粉传播等。
这些发现为提高作物产量提供了重要的理论依据。
2、作物抗病性:GWAS也被广泛应用于研究作物的抗病性。
例如,在小麦中,研究者发现了一种与对白粉病抗性相关的基因变异。
人类基因组学中的全基因组关联分析

人类基因组学中的全基因组关联分析人类基因组学是近年来生物学领域最为热门的研究方向之一。
全基因组关联分析(Genome-wide association study, GWAS)是遗传学中的一种重要方法,用来探究人类基因组与疾病等特定性状之间的关联。
随着高通量测序技术的进步,全基因组关联分析越来越受到关注。
一、全基因组关联分析的意义全基因组关联分析是一种通过大规模筛选人群基因组变异,并将其与临床症状、生物活动和药物反应等特定生理表现联系起来的方法。
全基因组关联分析可揭示基因多态性和疾病之间的关联,并有可能为疾病治疗和预防提供新的目标和方法。
二、全基因组关联分析的流程1.选定样本:全基因组关联分析的第一步是确定所要研究的样本。
对于常见疾病,通常需要至少数千例患者和对照组,以便确定基因与疾病之间的关联。
2.基因组测序:接下来需要对样本进行基因组测序,通常是通过芯片或高通量测序仪等设备来完成。
这样可以得到基因组上数百万个单核苷酸多态性(Single Nucleotide Polymorphisms, SNP)的信息。
3.数据分析:数据分析是全基因组关联分析的核心步骤。
所有SNP都必须进行质量控制以去除低质量的SNP。
然后,需要将SNP与基因组坐标对应以快速找到SNP在哪个基因里。
接着,通过计算每个SNP与临床表现之间的关联程度,确定SNP是否与疾病或特定性状有关联。
4.验证和功能鉴定:通过验证关联SNP的结果,确定SNP是否真正能影响疾病发生和发展,同时研究其功能机制。
三、全基因组关联分析的优点和不足优点:1.大规模化:全基因组关联分析可同时分析数百万个SNP的数据,为基因异质性和疾病之间的关联提供了新的视角。
2.高效性:全基因组关联分析的流程更高效,大大缩短了研究时间。
3.现实性:全基因组关联分析涵盖各种各样的基因,容易从大规模人群中识别与疾病有关的基因变异。
不足:1.解释性:全基因组关联分析结果不是绝对的,需要进一步解释其生理和药物学意义。
全基因组关联分析

全基因组关联分析全基因组关联分析(GWAS)是一种用于探究基因和人类疾病之间关联的方法。
它是一种统计分析方法,通过比较大样本的疾病患者与健康个体的基因组数据,寻找与疾病相关的基因变异。
GWAS的目标是通过研究人类基因组的变异与各种疾病之间的关系,找出与疾病风险相关的遗传变异。
GWAS的实施过程是:首先收集大样本的疾病患者和对照组个体的基因组数据,其中疾病患者组是有特定疾病(如癌症、心血管疾病、精神疾病等)的个体,而对照组则是与疾病患者组相近的健康个体。
然后通过基因芯片或次代测序等技术,测量并比较两组个体的基因组中单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)。
最后,利用特定的统计方法,分析基因组上的这些变异与疾病风险之间的关系。
GWAS的结果能够帮助科学家确定与疾病风险相关的遗传变异。
通过在整个基因组中寻找与疾病风险相关的SNPs,GWAS研究可以揭示有助于疾病发生和发展的遗传因素。
基于GWAS的研究结果,可以进行功能注释和生物信息学分析,从而深入了解这些SNPs对基因功能和表达的影响。
GWAS的研究已经取得了一些重要的突破。
例如,GWAS已经发现了与多种疾病相关的SNPs。
其中最著名的研究之一是发现了与乳腺癌风险相关的BRCA1和BRCA2基因的突变。
此外,GWAS还发现了与糖尿病、高血压、哮喘等疾病相关的SNPs。
这些研究结果不仅有助于我们更好地理解疾病的遗传基础,也对疾病的预防、治疗和个体健康管理提供了新的思路。
GWAS的未来发展可能会面临一些挑战。
首先,由于基因组上的SNPs数量巨大,需要收集大量的样本来获得统计意义上有力的结果。
这需要联合多个研究团队进行合作,共享样本和数据。
其次,GWAS的结果仅仅是发现与疾病风险相关的SNPs,但无法确定这些SNPs对基因功能和表达的影响机制。
因此,需进一步进行功能注释和机制研究,来解析这些遗传变异的具体影响。
动植物重测序
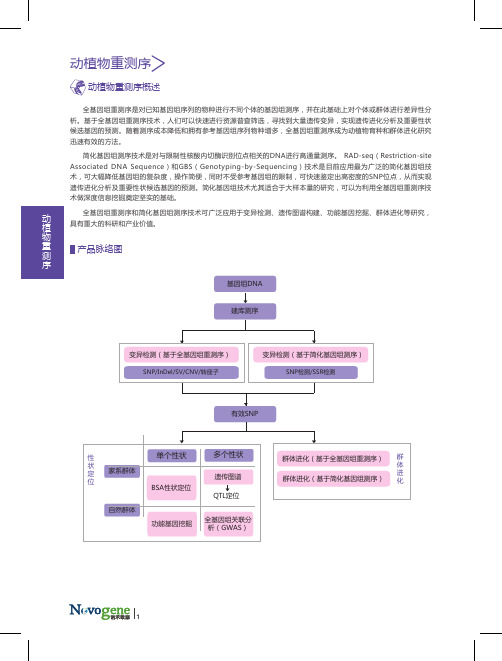
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS(Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图动植物重测序建库测序单个性状家系群体自然群体SNP/InDel/SV/CNV/转座子基因组DNA有效SNP性状定位群体进化群体进化(基于简化基因组测序) 群体进化(基于全基因组重测序) 变异检测(基于简化基因组测序)SNP检测/SSR检测遗传图谱全基因组关联分析(GWAS)功能基因挖掘变异检测(基于全基因组重测序) QTL定位BSA性状定位多个性状动植物重测序动植物重测序概述SNP检测、注释及统计基因组DNA350 bp小片段文库HiSeq PE150测序数据质控与参考基因组比对利用全基因组重测序技术对某一物种个体或群体的基因组进行测序及差异分析,可获得SNP、InDel、SV、CNV、PAV、转座子等大量的遗传多态性信息,建立遗传多态性数据库,为后续揭示进化关系、功能基因挖掘等奠定基础。
基于高通量测序的全基因组关联分析

基于高通量测序的全基因组关联分析随着基因测序技术的不断进步,全基因组关联分析(GWAS)已成为大规模研究人类疾病遗传因素的重要手段之一。
与传统的家系研究相比,GWAS可以更全面地探索单个基因和多个基因间的相互作用,对于发现人类遗传变异和疾病的新机制具有重要的意义。
而高通量测序技术的出现使得GWAS的研究范围更加广泛,应用于更多的生物样本和研究对象。
一、高通量测序技术的发展与应用高通量测序技术(High-throughput sequencing,HTS),也称为下一代测序技术,是指一种高效且自动化的测序方式。
目前,常见的高通量测序技术包括Illumina HiSeq、PacBio、Oxford Nanopore等。
这些技术的出现大大提高了测序效率,降低了测序成本,缩短了测序周期,使得全基因组测序成为可能。
举个例子,Illumina HiSeq 2500平台可以同时测序多个样本,并对每个样本产生上亿条的短序列,比起以前的Sanger测序方法,它的测序深度更高,更加准确,能够更好地保证数据的可靠性。
基于这种高效、准确、经济的测序技术,全基因组关联分析的研究得以快速地推进和深入。
二、全基因组关联分析的原理和方法全基因组关联分析通过对单个核苷酸多态性(Single Nucleotide Polymorphisms, SNPs)的基因型数据进行分析,寻找与相关表型(如疾病、性状等)存在关联的遗传变异。
GWAS通常包括三个主要步骤:样本分组、基因型分析和关联分析。
其中,样本分组包括病例组和对照组的设计,基因型分析包括测序、数据预处理和质量控制,而关联分析则是通过计算基因型频率和表型之间的相关性来进行的。
在这个过程中,全基因组关联分析可以使用许多不同的方法来确定SNP与表型之间的关联。
最经典的方法是使用线性回归模型,通过计算每个SNP在不同表型下的频率和表型之间的相关性来寻找关联SNP。
此外,GWAS还可以使用逻辑回归、Cox回归、贝叶斯分析等方法。
全基因组重测序数据分析

全基1. 简通过变(d 的功况,dise 比较实验(1)(2)基因组重测序简介(Introduc 过高通量测序识deletioin, du 功能性进行综合杂合性缺失ease (cance 较基因组学,群验设计与样本Case-Contr)家庭成员组序数据分析ction)识别发现de plication 以及合分析;我们(LOH )以及r )genome 中群体遗传学综ol 对照组设计组设计:父母novo 的som 及copy numb 们将分析基因及进化选择与中的mutation 综合层面上深计 ;-子女组(4人matic 和germ ber variation 因功能(包括与mutation 之n 产生对应的深入探索疾病基人、3人组或m line 突变,)以及SNP miRNA ),重之间的关系;以的易感机制和基因组和癌症多人);结构变异-SN 的座位;针对重组率(Rec 以及这些关系功能。
我们将症基因组。
NV ,包括重排对重排突变和combination )系将怎样使得将在基因组学排突SNP)情在学以及初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
全基因组关联分析

全基因组关联分析全基因组关联分析(Genome-wideAssociationStudies,GWAS)是一种比较新的研究方法,它可以帮助研究人员更好地理解基因与疾病、特定外貌特征等之间的关系。
该类研究技术利用大量遗传数据,结合大规模测序技术,在数据量比较大的情况下获得特定基因变异位点与相应疾病之间的显著相关性。
全基因组关联分析技术的开发始于八十年代,但在2005年到2008年之间,该技术发展迅速,成为最为重要的基因组学研究方法之一。
GWAS的开展使得研究者有机会探索数以百万计的位点与基因组变异之间的关系,而这种探索又能更准确地揭示复杂疾病和特定表型的遗传基础,从而为疾病的预防、诊断、治疗和基因编辑等提供一些科学依据。
GWAS的基础是以单碱基多态性(Single Nucleotide Polymorphism, SNPs)为基础的基因关联分析,通过基因组中的SNP (单核苷酸多态性)来检测与特定表型之间的关联。
SNPs是位于DNA 中不同定量位点的变异,它们的存在可以在基因组中的各个位点上被发现,并有助于研究疾病的发生发展机制以及它们与基因及基因组变异之间的关系。
GWAS帮助研究者更全面地探索复杂疾病的遗传遗传基础,发现复杂疾病的重要基因组区域,并发现其中潜在的遗传因素。
这种研究方法可以迅速查明某些疾病的遗传学与致病机制,这些疾病包括糖尿病、心脏病、癌症等,从而为临床治疗和预防提供依据。
此外,GWAS也可以应用于研究特定外貌特征,开展人类群体中的遗传多样性研究,并发现重要的外貌相关基因。
GWAS的最终目的是结合其他研究方法,有助于临床和公共卫生领域的实际应用,为治疗和预防疾病提供更准确的信息。
GWAS技术被广泛用于分子遗传学研究,其优势在于可以对超过十万个基因位点进行检测,从而更全面地探索基因与特定相关性的关联,例如疾病的发生机制和外貌特征的形成机理等。
GWAS的进展使得科学家可以更全面准确地研究基因与表型之间的关系,为治疗疾病和预防疾病提供有价值的科学信息。
动植物重测序的多种最新分析方法
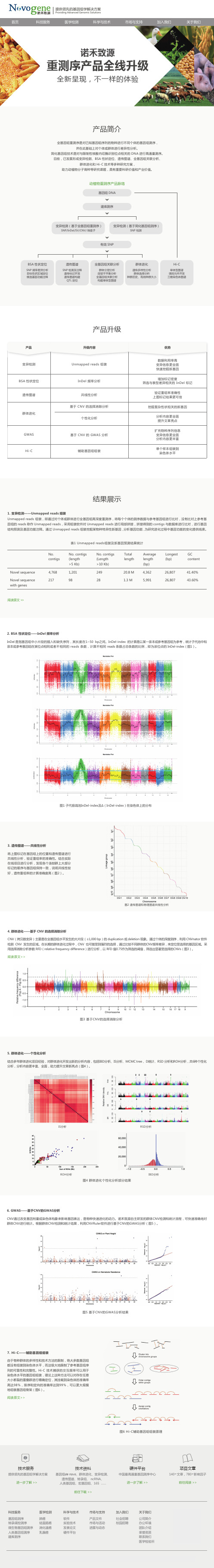
BSA 性状定位
InDel 频率分析Leabharlann 遗传图谱共线性分析
基于 CNV 的选择消除分析 群体进化 个性化分析
GWAS
基于 CNV 的 GWAS 分析
Hi-C
辅助基因组组装
结果展示
1. 变异检测—— Unmapped reads 组装 Unmapped reads 组装,即通过对个体或群体进行全基因组高深度重测序,将每个个体的测序数据与参考基因组进行比对,没有比对上参考基 因组的 reads 称作 Unmapped reads,采用组装软件对 Unmapped reads 进行局部拼接,拼接得到的 contigs 与数据库进行比对,进行基因 结构预测及基因功能注释。通过 Unmapped reads 组装挖掘某物种特异性新基因, 分析基因功能, 为研究进化过程中基因功能的变化提供线索。
Novel sequence Novel sequence with genes
4,768 217
1,201 98
20.8 M 1.3 M
4,362 5,991
26,807 26,807
41.40% 43.60%
阅读原文 > >
2. BSA 性状定位——InDel 频率分析 InDel 是指基因组中小片段的插入和缺失序列,其长度在1~50 bp之间。InDel-index 的计算是以某一亲本或参考基因组为参考,统计子代池中和 亲本或参考基因组在某位点相同或者不相同的 reads 条数,计算不相同 reads 条数占总条数的比例,即为该位点的 InDel-index(图1)。
图3 基于CNV的选择消除分析
5. 群体进化——个性化分析 结合多年群体进化项目经验,对群体进化开发出新的分析内容,包括IBD分析、IS分析、MCMC tree 、D统计、RSD 分析和ROH分析,共6种个性化 分析,分析内容更丰富、全面,助力提升文章新亮点(图4)。
全基因组关联分析

全基因组关联分析
全基因组关联分析(GWAS)是一种统计学方法,可用来检测特定群体的基因与特定的疾病或行为的关联。
此方法可用于识别可能存在的一个或多个不同位点间的关联,并且可以确定特定基因和特定疾病或行为之间的“直接”关联。
这种方法主要通过比较不同位点之间的基因型,从而推断该位点及其邻近位点是否与特定疾病或行为有关。
全基因组关联分析(GWAS)主要由三个步骤组成:一是建立组,二是收集数据,三是使用统计分析方法检测特定基因如何与特定疾病或行为有关。
首先,研究者需要创建一个研究组,该组必须由病人和正常人组成,以便比较两者之间的基因组差异。
然后,这些数据必须收集,以确定研究的基础。
其次,为了检测具体基因和指定疾病或行为之间的关联,必须使用统计分析方法。
GWAS带来的主要优势在于它可以帮助科学家们更好地了解特定病症,以及特定疾病或行为之间的关联。
这些知识可以用于进一步开展临床研究。
例如,通过GWAS可以更加准确地识别特定基因与特定疾病或行为之间的关联,从而有助于开发新的药物,新的治疗方案,和新的预防策略,以减轻疾病的负担。
GWAS也有一些弊端,例如需要大量的计算机处理时间和精度。
此外,这项技术还受到数据质量、外部变量和拟合函数的限制。
因此,研究人员需要更多地关注这些因素,以确保数据的可靠性和结果的准确性。
尽管GWAS存在一些局限性,但它仍然是一种重要的统计技术,
可用于研究基因与特定疾病或行为之间的关联,从而帮助科学家们更好地了解这些关联。
另外,GWAS还可以帮助开发新的药物和治疗方案,使得相关的研究和应用能够得到更好的发展。
全基因组关联分析(GWAS)技术在家犬中的应用研究进展

繁育•犬病reeding-Disease全基因组关联分析(GWAS)是一种在关联分析的基础上利用群体的连锁不平衡,对全基因组范围内的遗传标记进行检测,以定位影响表型性状的遗传因素的分析方法。
随着二代基因组测序技术不断发展更新,测序费用逐步降低,为畜禽高密度芯片的开发以及全基因组重测序的应用奠定了基础。
组关联fW(EWA5)技Ttt 在家犬中的应用HF宕滋展万九生李静陈超邓卫东岳锐徐虎黎立光一、全基因组关联分析(GWAS)是目前滦度发掘自然群体物种复奈性状相关功能基因的高效手段全基因组关联分析(GWAS)是最早研究复杂性状和疾病遗传变异的有效方法,其核心是研究分子变异和目标表型性状之间的关联。
尤其是近几年来随着高通量测序和高分辨的代谢检测技术的不断发展,以及多种生物信息学技术和统计学方法发展,这些为复杂性状基因变异的精细定位提供基础。
2005年,Science 项目编号:公安部科研专项2019GABJC29,云南省基础研究项目(青年项目)2019FD025项目名称:昆明犬繁殖性状的全基因组关联分析研究中国工作犬业2021.02|39繁育•犬病reeding-Disease杂志首次报道了年龄相关性视网膜黄斑变性GWAS 结果,在医学界和遗传学界弓I起了极大的轰动,此后一系列GWAS陆续展开。
2006年,波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的GWAS结果,已经陆续报道了关于人类身高、体重、血压等主要性状,以及视网膜黄斑、乳腺癌、前列腺癌、白血病、冠心病、肥胖症、糖尿病、精神分裂症、风湿性关节炎等几十种威胁人类健康的常见疾病的GWAS结果,累计发表了近万篇论文,确定了一系列疾病发病的致病基因、相关基因、易感区域和SNP变异。
此外,复杂疾病GWAS方法学(如研究设计、统计分析、结果的解释)也取得了极大的进步,因此称为"GWAS 第一次浪潮”。
同时,动、植物中重要的经济性状、农艺性状关联分析也已经大量开展。
全基因组关联分析

全基因组关联分析(Genome-wide association study or GWAS)人类基因包含着百万种序列变异,它们对于疾病的形成或者对患者药物的反应程度有直接或间接的影响.全基因组关联分析是指在人类全基因组范围内找出存在的序列变异,即单核苷酸多态性(SNP),从中筛选出与疾病相关的部分。
此项技术能够一次性对疾病进行轮廓性概览,在全基因组层面上,开展多中心、大样本、反复验证基因与疾病的关联研究,全面揭示疾病发生、发展,以及与治疗相关的遗传基因。
随着人类基因组学的大幅度进步和基因测序的飞速进展,这种最新的研究方式开始大规模应用于筛选与人群复杂疾病和药物特异性相关的序列变异。
进行全基因组关联分析研究时,通过采集某类疾病患者与非患者两类人群的DNA,在基因芯片上读出DNA中的序列变异,然后用生物工程技术进行分析比较。
若某些基因变异在患者人群中非常普遍,则该序列变异是与此种疾病‘相关’的。
有了全基因组关联分析,今后从事疾病诊断,患者对药物的反应程度的研究,可以集中于这些与疾病‘相关’的序列变异,从而显著缩短研究时间,提高研究效率。
全基因组关联分析是研究人类复杂疾病的一项重大突破,其优势在于:1 高通量 --- 一个反应监测成百上千个序列变异;2 不只局限于“候选基因”,基因可以是“未知”的;3无需在研究之前构建任何假设。
2005年,Science杂志报道了第一项具有年龄相关性的黄斑变性全基因组关联分析研究,之后陆续出现有关冠心病、肥胖病、II型糖尿病、甘油三酯、精神分裂症以及相关表型的报道。
由此可见,全基因组关联分析研究作为一种全新的疾病研究方式,自人类基因测序大规模展开以来,就被医学界广泛接受和应用。
截止到2010年12月,世界范围内进行了超过1200项针对200多种疾病的全基因组关联分析研究,找到4000多个‘相关’的序列变异。
在全基因组关联分析研究中,SNP基因芯片(SNP array)扮演了非常重要的角色。
全基因组关联分析

全基因组关联分析以《全基因组关联分析》为标题,写一篇3000字的中文文章近几年,由于全基因组测序(whole-genome sequencing)和其他技术的出现,基因关联分析已成为科学家们在研究某类疾病的遗传基础方面最为重要的分析技术。
全基因组关联分析(GWAS)是一种研究特定病症,疾病和行为特性与关联基因的统计分析方法。
通过它,科学家们可以跨越整个基因组,在不同的关联位点上发现相关性,以及进行细粒度的分析,以更加准确地了解发病机制、预测病例风险以及发展治疗方案。
简言之,全基因组关联分析是一种利用大量基因组数据从而探索疾病发病机制的技术,它使研究者能够发现和映射病理机制的相关位点,并帮助实现精准预测和治疗。
首先,全基因组关联分析是一种能够将基因之间的复杂关系揭示出来的分析研究方法。
它以统计学和流行病学为基础,使研究者能够精确地检测基因与疾病之间的相关性,这种相关性能够进一步促进基因和疾病之间的显著关系以及病理机制的理解。
其次,全基因组关联分析是一种即能够拓展研究,又能够更深入地理解基因组的分析技术。
当研究人员对アナリタイプ(观察或测量的值)与遗传位点的相关性进行分析时,他们能够确定性状的遗传基础,从而突出关键的基因,使进一步的研究变得更容易。
此外,通过全基因组关联分析,研究者们可以更加深入地探究疾病发病机制,这对于指导药物开发和医学治疗有重大意义。
因此,开发新药物和治疗方案需要以全基因组关联分析作为基础,以明确靶向分子和在靶向分子活动中发挥作用的基因组元件。
再者,通过全基因组关联分析,研究人员可以推断出特定病症的风险,提高病人的诊断准确性和预测力。
通过研究基因的变异性,可以有效地指导治疗,而不仅仅是凭借病史和临床检查结果来指导治疗。
在获取精确的诊断之后,病人可以享受更有效、更快捷的治疗,从而达到更充分、更及时的治疗效果。
最后,通过全基因组关联分析,研究者们可以对人类的遗传和病理机制进行全面探索,从而获得更加丰富的知识和信息,因此能够高效地获取重要的知识,从而利用基因组研究的结果来更有效地管理公共卫生,预防和治疗重大疾病,使更多的病人受益。
mGWAS是什么,mGWAS专业解读

mGWAS是什么,mGWAS专业解读近期经常听到mGWAS这个词,好像很⾼⼤上,是个新兴技术,作为⼀个科研狗不知道mGWAS是不是out啦。
那mGWAS究竟是什么呢,今天我们就来⼀探mGWAS的真⾯⽬。
⼀、什么是mGWAS?mGWAS的“m”指的是“代谢组(metabolome)”,“GWAS”指的是“全基因组关联研究(Genome-Wide Association Study)”,字⾯意思来看mGWAS指的就是“代谢组+全基因组关联研究”。
1958年中⼼法则被提出,告诉我们遗传信息经DNA——RNA——蛋⽩质传递,最终影响表型,1977年桑格发明了双脱氧核苷酸终⽌法,基于这个实验,⼀代测序开始兴起,使⼈们了解⼈类基因组成为可能。
经过多年的发展,DNA 测序⼿段越来越成熟,成本越来越便宜,从DNA测序⼊⼿研究疾病发⽣原因及机制也成为⼤家⾮常认可的研究思路。
全基因组关联分析就是对多个个体在全基因组范围的遗传变异多态性进⾏检测,获得基因型,进⽽将基因型与可观测的性状即表型,进⾏群体⽔平的统计学分析,根据统计量或P值筛选出最有可能影响该性状的遗传变异,进⽽找到与关注表型相关的SNP位点,定位与性状相关的基因。
但传统GWAS分析存在⼀些不⾜,如难以确定具体功能突变,难以解析突变如何影响性状的具体机制,并且有些⾼阶表型难以定性定量。
代谢组学系统⽣物学的⼀种,区别于基因组学、转录组学、蛋⽩组学,它是最接近表型的组学。
代谢物数量远远⼤于表型种类,所以将全基因组关联分析的表型信息⽤代谢物信息替代,可以弥补传统GWAS 的不⾜,这就诞⽣了代谢物的全基因组关联分析——mGWAS。
代谢物的全基因组关联分析(metabolome Genome-Wide Association Study, mGWAS),是基于重测序的信息,如SNP 等,结合代谢组数据开展代谢物的全基因组关联分析,批量、精准定位疾病相关候选基因,挖掘代谢⽣理、表型相关的功能基因,阐释相关代谢通路分⼦⽣化机理。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
图2 重要性状GWAS结果
参考文献
[1] Chen W, Gao Y, Xie W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism [J]. Nature genetics, 2014, 46(7): 714-721.
对已有参考基因组的物种群体进行全基因组重测序,检测分布于全基因组范围内的SNP标记,基于它们与分析性状的连锁不平衡关系,通过各种统计分析方 法,获得与这些性状关联的候选基因或基因组区域。与简化基因组及芯片技术相比,全基因组重测序可以更全面的挖掘基因组的变异信息,开发更多的分子标 记,因此可更精确的找到与性状关联的候选基因或基因区域。
ቤተ መጻሕፍቲ ባይዱ
与参考基因组比对 群体SNP检测、统计与注释
构建系统进化树 群体主成分分析
连锁不平衡分析 性状关联分析
目标性状相关区域基因功能注释 构建单体型图谱
标准分析时间为120天,个性化分析需根据项目实际情况进行评估
案例解析
[案例一] 水稻代谢性状关联分析[1]
通过对有840种代谢产物的529份水稻进行全基因组重测序,结合 已知的950份水稻数据,获得6,428,770个SNP。通过群体分层分 析,分为Indica和Japonica两个亚群,对两个亚群水稻代谢性状 进行全基因组关联分析,鉴定出2947个与634个基因相关的主导 SNP位点。随后,在210个Indica的RILs群体中进行验证,定位 出36个候选基因与代谢相关。对36个候选基因进行实验验证,最 终确定了5个候选基因。
[案例二] 大豆驯化性状关联分析[2]
通过对302株大豆(62个野生大豆,130个地方品种和110个驯化 品种)进行高通量重测序,共发现979万个SNP,87.68万个 Indel,还有1614个CNV和6388个大片段缺失。通过构建系统进 化树以及主成分分析,发现本研究所选大豆群体明显可以聚成三 类——野生、驯化及改良。全基因组关联分析表明10个受选择区 域和9个驯化性状相关联,发现13个被注释为与油脂、株高等农 艺性状相关的位点。与之前QTL定位结果比较分析发现,230个 受选择区域中96个与调控油脂的QTL相关,21个区间内包含脂肪 酸合成关键基因。
XP–CLR
–log10 P
–log10 P
40
30
20
10
0
Chr. 1 0
2
3
4
5
10
20
30
40 Flavonoids Phenolamines Terpenoids
6
7
8
9 10
AA and NA ders
Others
11 12 , Unknown
图1 关联分析曼哈顿图
300
200
Oil23–2
技术参数适用范围 样品要求 类型 测序策略与深度 分析内容 项目周期
群体进化(基于全基因组重测序)
1. 已有参考基因组序列的动植物自然群体,建议样本数≥200个 2. 样本间无明显的亚群分化(如生殖隔离等) 3. 所研究表型性状遗传力较强
DNA样深度≥5X/个体
100 0
Oil20–2
1
2
24–34 28–2 28–3 Oil36–9
3
4
Oil23–1
Oil36–2 E1
36–7 Sd oil-prot1–1 Oil1–2 16–5 Oil34–7 Oil34–5
Sg1 Oil24–1
5
6
7
8
9
W1
Oil24–4
E2
Oil32–2
10 11 12 13
−log10P
GWAS on oil percent 10
8 6 4
2 0
5 10 15 20 25 30 35 40 45 Chromosome 3 (Mb)
−log10P
GWAS on oil percent 10
8 6 4 2 0
5 10 15 20 25 30 35 40 Chromosome 13 (Mb)
[2] Zhou Z, Jiang Y, Wang Z, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean [J]. Nature Biotechnology. 2015, 33(4):408-414.