67 Paclitaxel poliglumex药理药效研究 动物模型
药理学研究中的动物模型

药理学研究中的动物模型药理学是研究药物对生物系统的作用、药物相互作用及其在人体内的代谢和排泄等方面的科学。
而动物模型则是药理学研究的重要手段之一,是在药理学研究中广泛应用的实验方法。
本文将探讨动物模型在药理学研究中的运用以及存在的问题。
一、动物模型在药理学研究中的运用1.药效学研究药物剂量-反应关系是药一个药物治疗效果的关键参数。
通过对动物模型进行药效学研究,可以帮助研究者确定药物的剂量和给药方法,及其对特定疾病产生的治疗作用。
2.药代动力学研究药代动力学研究涉及药物在生物体内的吸收、分布、代谢及排泄等过程。
通过使用动物模型,研究者可以更好地理解人体内药物相互作用的机制,以及药物在体内的代谢及排泄吸收规律。
3.毒理学研究另外,动物模型也广泛应用于毒理学研究方面。
通过使用动物模型,研究者可以评估药物的毒性,并为药物的临床使用提供指导。
二、动物模型存在的问题1.模型转化性能的问题研究者对动物模型的应用需要深刻认识到动物模型本身具有的局限性。
动物模型无法完全反映人体内部各种细微变化的复杂性,因此,能否将动物的结果转化为人体的治疗方案存疑。
2.道德问题另外,动物模型研究也存在一定的道德问题。
因为动物在实验中往往会受到一定程度的折磨,所以必须确保实验的道德可接受,避免动物受到过度转化。
3.统计学意义上的问题最后,动物模型的应用还可能存在统计学意义上的问题,研究者必须严格控制实验中的各种环境因素,以确保研究结果的可靠性。
三、结语总体来说,动物模型在药理学研究中扮演着重要的角色。
尽管存在一些问题,但研究者仍需要认真对待这种研究方法,尽力避免它的局限性,改善其缺陷,并为临床应用提供可靠的依据。
同时,必须通过科学的伦理道德评估来确保研究过程的公正公平。
只有慎重对待,才能更好地补充人类已知的药理学知识,为发现从动物实验中发现的新药物奠定基础。
博来霉素诱导的肺纤维化动物模型

博来霉素诱导的肺纤维化动物模型
(一)实验动物
8周龄,体重为23g左右的C57BL/6小鼠
(二)实验材料
博来霉素、麻醉剂、手术剪、眼科镊、缝合线、1 ml注射器或微量注射器
(三)实验步骤
博来霉素(盐酸博来霉素或硫酸博来霉素均可,给药时需换算为博来霉素)使用生理盐水溶解,剂量为2-5 U/kg(PS:需根据实验动物的质量摸索造模剂量,不同来源的鼠有一定的差异)。
博来霉素给药采用器官内给药,成模率、成模速度较其他给药方式快。
具体操作如下:
小鼠按照麻醉剂剂量麻醉后,沿颈部中线开口(开口大小,小鼠0.5-1 cm左右)。
使用眼科镊分离出气管,此时需要轻柔操作,减少因手术操作造成的气管损伤,使用注射器(1ml或者微量注射器,胰岛素针不失为好的选择)将少量的博来霉素溶液(不超过0.2 ml/只,肺内大量积液会导致动物的死亡,溶液量极可能少)经气管注射进入器官内,缝合伤口后将动物直立1-3 min可稍微摇晃一下,保证博来霉素在肺内均匀分布。
(3天左右即会出现损伤,体重降低等症状,纤维化则与博来霉素的剂量和溶液体积有很大关系,一般在7-21天出现,时间越久模型组死亡率可能会比较高。
)
PS:博来霉素剂量当天配置当天使用,另外博来霉素诱导的肺纤维化模型在不同品系的动物上有很大的差异,C57Bl/6小鼠的敏感程度显著高于BALB/c小鼠。
动物死亡率可作为此模型药效指标。
博来霉素除经气管给药造成肺纤维化模型之外,还可以通过口服、腹腔等方式给药来造模[1]。
紫杉醇(PTX)在食管癌同期放化疗中的研究进展

紫杉醇(PTX)在食管癌同期放化疗中的研究进展邓家营【摘要】紫杉醇(paclitaxel,PTX)是食管癌化疗中最有活性的药物之一,因其独特的作用机制和良好的耐受性,被广泛与顺铂(cisplatin,DDP)、5氟尿嘧啶(fluorouracil,5 Fu)等联合应用.PTX还有一定的放射增敏作用,作为放射增敏剂之一常被用于同期放化疗.在临床上,PTX类药物有不同的制剂类型和用药方案,本文就其在食管癌放化疗中的研究进展,在不同方案中的作用以及应用中存在的问题作一综述.【期刊名称】《复旦学报(医学版)》【年(卷),期】2014(041)005【总页数】5页(P697-700,705)【关键词】食管肿瘤;同期放化疗;紫杉醇(PTX)【作者】邓家营【作者单位】复旦大学附属肿瘤医院放疗科上海200032;复旦大学上海医学院肿瘤学系上海200032【正文语种】中文【中图分类】R735.1食管癌是我国常见的恶性肿瘤之一,也是世界范围内常见的癌症死亡原因。
因其早期症状不明显,30%~40%的食管癌确诊时已属无法手术切除的局部晚期。
食管癌单纯放疗效果不佳,5年生存率约为20%,局部未控和复发可达60%~80%。
单纯的化疗又很少获得病理完全缓解,且缓解期较短。
所以临床上更多地应用同期放化疗来治疗局部区域性食管癌非手术的患者。
顺铂(cisplatin,DDP)联合5-氟尿嘧啶(5-fluorouracil,5-Fu)是经典化疗方案,但是该方案患者耐受性差且疗效不理想。
探索更加高效、低毒并有放射增敏作用的新化疗药物是进一步提高食管癌同期放化疗疗效的重要途径之一。
紫杉醇(paclitaxel,PTX)是一种二萜类生物碱,主要从红豆杉树皮中分离提取。
作为抗肿瘤药,PTX具有独特的作用机制,一方面它可与β-微管蛋白结合并促进其聚合,诱导有丝分裂阻滞于G2/M期,从而使细胞出现分裂性死亡;另一方面,它还可以影响信号通路诱发细胞凋亡。
动物模型在药物研究中的应用及其限制

动物模型在药物研究中的应用及其限制药物研究是一个复杂的过程,其中动物模型在药物研究的发展中起到了极为重要的作用。
动物模型可以模拟人体内的生理和病理过程,尤其是可以模拟人体内药物的代谢和副作用。
然而,动物模型也有其局限性。
本文将讨论动物模型在药物研究中的应用及其限制。
一、动物模型在药物研究中的应用1. 建立疾病模型疾病模型可以为药物研究提供一个可靠的模拟环境,疾病模型可以通过动物模型来建立。
例如,研究肿瘤药物可以使用小鼠肿瘤模型,研究糖尿病药物可以使用小鼠和大鼠糖尿病模型。
这些模型可以用于检测药物的疗效和副作用。
2. 药代动力学药代动力学是研究药物在人体内的吸收、分布、代谢和排泄的过程。
动物模型可以用于检测药物的药代动力学。
这些动物模型可以为研究人类体内的药物动力学提供一定的参考。
3. 安全性评估药物在人体内可以引起各种不良反应,例如中毒、过敏反应和疾病进展。
在药物研究的早期阶段,动物模型可以检测药物的安全性。
这些动物模型可以检测药物的副作用、毒性和耐受性等数据。
二、动物模型在药物研究中的限制尽管动物模型在药物研究中起着至关重要的作用,但是它们也有其局限性。
以下是动物模型在药物研究中的限制。
1. 种属差异不同的动物有不同的生理和代谢过程,因此药物在不同动物体内的代谢方式也有所不同。
例如,大鼠和小鼠在药物代谢方面存在明显差异。
因此,使用不同种类的动物进行药物研究可能会导致误判和误导。
2. 疾病模型不准确虽然动物模型可以模拟人类疾病的一些特征,但是它们仍然不能完全模拟人类内部环境。
例如,化学药物在动物体内的代谢方式和在人体内的代谢方式是不同的。
因此,在进行药物研究时需要谨慎评估动物模型的准确性和适用性。
3. 数据解释困难药物在动物体内和人体内的代谢方式和生理反应不尽相同。
因此,在动物模型中获得的数据需要进行解释和转化,以便用于预测人体内的药物代谢和生理反应。
这一转化过程可能会导致推理和缺乏可重复性的困难。
小细胞肺癌可视化PDOX肿瘤动物模型免疫表型Ki-67表达的研究进展

小细胞肺癌可视化PDOX肿瘤动物模型免疫表型Ki-67表达的研究进展作者:吴柳盛李小强来源:《中国医学创新》2021年第25期【摘要】小细胞肺癌(SCLC)是肺癌所有病理类型中预后最差的一种,患者生存周期极短,死亡率极高。
虽然SCLC对化疗药物敏感,临床上抗肿瘤药物治疗成为主流,但有循证医学研究发现在临床中不同的患癌个体之间对抗肿瘤药物的敏感性和耐受性存在着差异,这给临床治疗带来了困难和挑战。
因此,人们急需要一种能高度模拟体内肿瘤细胞生长微环境,能与患者进行同步化动态观察抗肿瘤药物应答的理想动物模型。
可视化PDOX肿瘤动物模型满足了这些需求,这也是目前国际上最前沿的肿瘤动物模型构建技术,避免了个体差异,为临床治疗提供了个性化思路,同时Ki-67免疫表型表达的监测为早期SCLC的诊断提供了有效帮助。
现对最近国内外SCLC可视化PDOX肿瘤动物模型构建技术和免疫表型Ki-67表达监测的研究进展以及该模型对临床上不同个体之间抗肿瘤药物应答差异做全面综述。
【关键词】小细胞肺癌可视化PDOX肿瘤动物模型 Ki-67[Abstract] Small cell lung cancer (SCLC) has the worst prognosis among all pathological types of lung cancer. The survival cycle of patients is very short and the mortality rate is very high. Although SCLC is sensitive to chemotherapy drugs and anti-tumor drug therapy has become the mainstream in clinical practice, evidence-based medicine studies have found that there are differences in the sensitivity and tolerance of anti-tumor drugs among different cancer patients in clinical practice, which brings difficulties and challenges to clinical treatment. Therefore, there is an urgent need for an ideal animal model that can highly simulate the microenvironment of tumor cell growth in vivo and can dynamically observe the response to antitumor drugs in synchronization with patients. Visualized PDOX tumor animal model meets these needs, which is also the most advanced tumor animal model construction technology in the world at present. It avoids individual differences and provides personalized ideas for clinical treatment. Meanwhile, the monitoring of Ki-67 immunophenotype expression provides effective help for the diagnosis of early SCLC. In this paper,the research progress of visualized PDOX tumor animal model construction technology and immunophenotype Ki-67 expression monitoring of SCLC at home and abroad, as well as the clinical differences of anti-tumor drug response among different individuals in this model are comprehensively reviewed.[Key words] SCLC Visualized PDOX tumor animal model Ki-67First-author’s address: Peking University Shenzhen Hospital, Shenzhen 518036, Chinadoi:10.3969/j.issn.1674-4985.2021.25.042小細胞肺癌(small cell lung cancer,SCLC)占全部肺癌的15%~17%[1],病情恶化快,可迅速出现远处内脏肿瘤转移,生存寿命短,死亡率极高,同时在临床上不同肿瘤患者个体之间存在着抗肿瘤药物的耐受差异,给临床治疗与疗效评估带来了困难。
动物模型在药物研究中的应用

动物模型在药物研究中的应用药物研究是一个综合性强、需要借助各种手段和方法才能完成的科学研究。
其中,动物模型是一个重要的研究手段,在药物研究中应用广泛。
动物模型可以帮助研究人员更好地了解药物的药理作用、剂量、毒性等方面的信息,提高药物研究的效率和成功率。
本文将探讨动物模型在药物研究中的应用,并分析其优缺点。
一、1. 药物的药理作用研究动物模型可以帮助研究人员更好地了解药物的药理作用。
例如,针对肿瘤的化疗药物,研究人员可以通过动物模型来评估药物的抗肿瘤作用、毒性和耐受性。
动物模型可以模拟人体中的肿瘤环境,通过观察动物体内的肿瘤变化,了解药物的药理作用和剂量范围,为药物的临床应用提供依据。
2. 药效学研究动物模型可以帮助研究人员进行药效学研究。
例如,对于心血管疾病的药物,研究人员可以通过动物模型来评估药物的心血管效应、有效剂量、安全剂量等信息。
通过动物实验,研究人员可以获取药物在动物体内的体内药效学参数,了解药物的药效学特性,为药物的临床应用提供依据。
3. 毒理学研究动物模型可以帮助研究人员进行毒理学研究。
例如,在新药研究过程中,需要对药物的毒性进行评估。
通过动物模型,研究人员可以获取药物的毒性数据,包括剂量效应关系、生化毒性、组织学和病理学损伤等信息。
这些数据可以为药物的临床应用提供依据,帮助研究人员了解药物的毒性水平,以及如何使用药物时避免毒性损害。
4. 药物代谢动力学研究动物模型可以帮助研究人员进行药物代谢动力学研究。
例如,在新药研究过程中,需要了解药物的代谢途径、代谢产物和半衰期等信息。
通过动物实验,研究人员可以获得药物代谢动力学参数,如药物清除率、药物代谢酶的活性等,为药物的临床应用提供依据。
二、动物模型在药物研究中的优缺点1. 优点(1)相对真实:动物模型的研究结果相对比较真实,因为它可以模拟人体生理环境,给人类疾病的研究提供一定的可靠性。
(2)有利于筛选药效:动物模型有助于筛选药物的药效,检索药物的安全性、有效性等,从而为药物的研发和临床应用提供依据。
预测肿瘤药物临床试验效果的动物模型新进展

预测肿瘤药物临床试验效果的动物模型新进展余飞;丁慧【期刊名称】《中国比较医学杂志》【年(卷),期】2015(000)006【摘要】基于人体试验的实际应用及伦理方面的考虑,合适的动物模型对于肿瘤药物研发至关重要。
制药公司和研究机构在肿瘤治疗新药的开发过程中消耗大量资源,最佳动物体内模型的选择可以改进或缩短研发进程。
在技术复杂性方面,肿瘤遗传工程小鼠模型( GEMM)已逐步完善,并且GEMM能够准确重建人类肿瘤的同源发生,为加快肿瘤药物的开发提供机遇。
本文主要综合比较预测肿瘤药物临床试验效果的不同类型动物模型,探讨其优劣,并对体内模型的评估方法及与临床转化等进行简述,为肿瘤药物临床前试验提供参考。
%Due to practical and ethical concerns associated with human experiments, animal models have been essential in cancer research.Vast resources are expended during the development of new cancer therapeutics, and selection of optimal in vivo models should improve this process.Genetically engineered mouse models ( GEMM) of cancer have progressively improved in technical sophistication and, accurately recapitulating the human cognate condition, have provided opportunities to accelerate the development of cancer drugs.In this article we consider the different types of animal models used for predicting the results of clinical trials of cancer drugs, and discuss the strengths and weaknesses of each in this regard.In addition, the methods of predicting in vivo models and clinical translation are discussed.【总页数】6页(P65-69,70)【作者】余飞;丁慧【作者单位】南京大学医学院附属鼓楼医院,南京 210008;南京大学医学院附属鼓楼医院,南京 210008【正文语种】中文【中图分类】R-33【相关文献】1.新冠肺炎疫情下抗肿瘤药物临床试验现状与紧急应对策略 [J], 刘小保; 高素彬; 衡建福; 刘洋; 杨农; 李坤艳; 王静; 肖亚洲2.新冠肺炎疫情下抗肿瘤药物临床试验中受试者访视管理的紧急应对 [J], 蒋云; 刘小保; 汤清涛; 杨农; 黄钢; 王静; 李坤艳3.某院抗肿瘤药物临床试验方案偏离的回顾性研究 [J], 叶丽君;蔡淑帆;林能明;王飞4.通过基于风险评估统计模型的中心化监查优化抗肿瘤药物临床试验质量管理 [J], 胡劲;徐炎;周高超;殷悦;金迪蒂5.影像组学在抗肿瘤药物临床试验疗效评估中的应用和挑战 [J], 李佳铮;唐磊因版权原因,仅展示原文概要,查看原文内容请购买。
淋巴瘤药理药效研究动物模型

弥漫大B细胞淋巴瘤(DLBCL)
DLBCL是一种大B淋巴细胞弥漫性增生 所组成的肿瘤,瘤细胞核至少2倍于正 常小淋巴细胞核或大于巨噬细胞核
DLBCL临床特点
☞ 最常见的恶性淋巴瘤 ☞ 任何年龄均可发生,高峰年龄60~69岁,
男性稍多 ☞ 淋巴结无痛性增大,40%可原发于结外 ☞ 可原发或继发于其它小B细胞淋巴瘤 ☞ 50%患者临床分期Ⅲ或Ⅳ期 ☞ 总的5年生存率46%
► 11、滤泡性淋巴瘤 -胃肠道滤泡性淋巴瘤 -儿童滤泡性淋巴瘤 -“原位”滤泡性淋巴瘤 12、结内边缘带B细胞淋巴瘤 13、套细胞淋巴瘤 14、弥漫大B细胞淋巴瘤 -弥漫大B细胞淋巴瘤,非特殊类型 T细胞/组织细胞丰富的大B细胞淋巴瘤 老年人EBV阳性的弥漫大B细胞淋巴瘤 慢性炎症相关的弥漫大B细胞淋巴瘤 -脓胸相关淋巴瘤 -慢性骨髓炎相关淋巴瘤 -植入物相关淋巴瘤
CLL/SLL临床特点
☞ 老年人,中位年龄65岁,男∶女=2∶1 ☞ 无症状或表现无力,自体免疫性贫血,易
感染,骨髓和周围血大多累及,可伴脾脏 肿大
☞ 5%患者进展为DLBCL(Richter综合征) ☞ 5年生存率51%
CLL/SLL形态学
肿瘤细胞
小圆形淋巴细胞,幼淋巴细胞和副免疫 母细胞
组织结构
(占GI的85%)。以下依次为肺、头颈 部、眼眶、皮肤、甲状腺和乳腺等
☞ 临床分期大多为Ⅰ或Ⅱ期 ☞ 幽门螺杆菌胃炎史或自体免疫性疾病史 ☞ 5年生存率74%
MALTL形态学
肿瘤细胞
边缘区(中心细胞样)细胞,单核 细胞样B细胞,小淋巴细胞,IB样和 CB样细胞
组织结构
滤泡的边缘区和/或滤泡间区分布 滤泡植入 淋巴上皮病变
ki-67阳性细胞>30% 者预后 较差
帕金森病动物模型的制作的详细步骤及方法

帕金森病动物模型的制作的详细步骤及方法一、通过耗竭单胺类神经递质制作PD模型(一)利血平模型利血平是一种生物碱。
通过不可逆地封闭囊内单胺运输消耗中枢和外周单胺,从而影响细胞内囊泡的单胺再循环,于20世纪50年代被用于制作大鼠PD模型。
当啮齿类动物注射利血平后,囊内摄取的DA、5-羟色胺(5-HT)和去甲肾上腺素(NA)被封闭,在胞质内被降解,快速降低单胺水平,导致肌肉僵硬等PD的症状。
制备方法:应用雄性Wistar大鼠,腹腔内注射一定剂量的利血平后即可使其出现骨骼肌僵硬、震颤、姿势异常。
该模型制作方法简单。
其对人类大的贡献在于揭示出左旋多巴(L-DOPA)可作为治疗PD的潜在药物。
缺点是只能短暂模拟PD的症状,不能完全复制自发性PD 的病理变化。
(二)甲丨基丨苯丨丙丨胺模型苯丨丙丨胺丨类药物是一类具有潜在成瘾性的神经兴丨奋丨剂,具有促DA释放作用,大剂量应用对啮齿类动物和非人类的灵长类动物具有神经毒性,像利血平一样,它能够导致DA能神经元末梢DA的耗竭,对黑质神经元损伤作用较小。
Boirean等研究表明,该药可通过DA转运体被摄入细胞内,兴奋性氨基酸受体拮抗剂MK.801可阻断其毒性作用,体外实验提示能量代谢障碍、氧化应激和兴奋性氨基酸均与其毒性作用有关。
制作方法:用成年大鼠,按体重10~25mg/kg 皮下注射甲丨基丨苯丨丙丨胺,每天一次,连续4天。
用药后可出现短暂活动过多,随之是活动减少。
4~5d后可有“幻觉样”行为,表现为颤抖、舔毛、咬皮毛。
1周后检测即可发现黑质和纹状体中酪氨酸羟化酶(tyrosine hydroxylase, TH)活性降低,DA含量减少,但黑质 DA能神经元丢失不明显。
该模型制作方法简单,是一个急性损伤模型,症状的持续时间及稳定性方面尚不理想,也无PD的特征性组织病理学改变。
主要用于研究PD多巴胺耗竭后的纹状体生理、生化学改变,也可用于神经保护方面的研究。
二、神经毒素制作PD模型对PD患者大脑的研究显示,在氧化应激过程中存在线粒体复合物Ⅰ功能的抑制,因而这可能是黑质DA神经退行性变的重要组成部分。
药理学研究中的动物模型选择

药理学研究中的动物模型选择概述:动物模型在药理学研究中发挥着重要的作用。
通过合适的动物模型可以更好地理解药物的机制、效果以及安全性。
然而,在选择适当的动物模型时需要考虑多个因素,包括相似性、成本和可行性等。
本文将探讨药理学研究中动物模型选择的原则和常见的应用。
一、相似性1.1 物种相似性:选择与人类生理相似度较高的动物作为研究对象,如大鼠和小鼠。
因为这些动物在生命活动、器官结构和代谢途径等方面与人类有较好的相似性,能够提供更可靠的数据。
1.2 疾病模型:根据所研究的药物治疗目标来选择与之相关的疾病模型。
例如,在心血管领域,可以使用高胆固醇饮食诱导小鼠产生高血压或者缺血再灌注损伤等模型来评估药物对这些情况下心脏功能改善效果。
二、成本和可行性2.1 成本:动物模型的建立和维护需要耗费大量资金,因此在选择时需要考虑经济实用性。
多个相关研究中常用的小鼠是较经济实惠的选择。
2.2 可行性:动物模型的选取还要根据实验室条件、技术设备和人员配备等方面进行考虑。
对于一些高度特化的模型,如果缺乏相关资源,则不宜选择。
三、常见应用3.1 药效学评价:通过动物模型可以评估药物在生物体内的药效学参数,包括药代动力学和药效学等。
这些数据对于了解药物吸收、分布、代谢和排泄过程非常重要。
3.2 治疗策略验证:在新药开发阶段,动物模型常被用来验证治疗策略的有效性。
例如,在癌症治疗研究中,使用小鼠移植瘤模型可以评估抗肿瘤药物的疗效。
3.3 安全评价:动物模型能够帮助检测潜在毒副作用,并评估药物在各种毒理学指标上的影响。
这对于药物安全性评价以及制定适当的用药指南至关重要。
四、局限性和新技术4.1 物种差异:尽管动物模型在探索药理学的研究中有很大帮助,但人类与动物之间仍然存在一定的生理、代谢差异。
因此,不能直接将动物实验结果推广到人体。
4.2 替代方法:随着科技的发展,出现了许多替代动物模型的技术,如体外细胞培养和计算机模拟等。
这些新技术可以减少对动物实验的需求,并提供更快速、精确、经济高效的研究手段。
药物在不同动物模型中的药效研究

药物在不同动物模型中的药效研究药物的研发是一项复杂而关键的任务,而了解药物在动物模型中的药效则是评价药物疗效和安全性的重要一环。
动物模型的使用可以帮助科学家评估药物的有效性、毒性和副作用。
本文将探讨药物在不同动物模型中的药效研究。
一、小鼠模型中的药物研究小鼠是广泛使用的动物模型之一,由于其生命周期短、繁殖能力强等特点,使得小鼠成为药物研究的理想选择之一。
小鼠模型主要用于研究药物的毒性、疗效和药物代谢等方面。
在小鼠模型中,科学家通常将药物以不同的途径给予小鼠,如口服、腹腔注射、静脉注射等,观察不同途径给药后药物的疗效。
此外,科学家还会检测小鼠血液中的药物浓度以及药物在体内的代谢情况,以便更好地评估药物的药代动力学。
二、大鼠模型中的药物研究大鼠模型也是常用的药物研究动物模型之一。
大鼠与小鼠类似,在药物研究中也具备许多优势,如体型较大、解剖结构与人类相似等。
大鼠模型主要用于研究药物的剂量反应关系、药物的毒性以及慢性疾病模型的建立。
在大鼠模型中,研究者通常会根据研究目的选择适当的大鼠品系,如Sprague-Dawley大鼠、Wistar大鼠等,并通过不同的给药途径给予药物,观察不同剂量药物的疗效。
此外,大鼠模型的可移植性较强,使得科学家能够进一步验证药物的疗效和安全性。
三、猪模型中的药物研究猪是较大型的动物模型,在某些药物研究领域也被广泛使用。
猪的器官结构与人类较为相似,使得猪模型成为研究某些重大疾病的理想选择。
猪模型主要用于研究心血管药物、外科手术和组织工程等方面。
在猪模型中,科学家常常会进行心脏导管插入、组织活检等操作,以评估药物对心血管系统的影响。
此外,猪模型还可用于进行手术模拟和组织移植等研究,以提高药物的生物利用度和治疗效果。
四、其他动物模型中的药物研究除了上述提到的小鼠、大鼠和猪模型外,还有其他动物模型也被广泛应用于药物研究中。
例如,狗模型主要用于研究药物对心血管系统的影响;兔模型则常用于研究药物在动脉粥样硬化治疗中的效果。
药理药效研究 动物模型subsequent erlotinib

Lung Cancer 73 (2011) 211–216Contents lists available at ScienceDirectLungCancerj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /l u n g c anDuration of prior gefitinib treatment predicts survival potential in patients with lung adenocarcinoma receiving subsequent erlotinibKazuhiro Asami a ,∗,Masaaki Kawahara b ,Shinji Atagi a ,Tomoya Kawaguchi a ,Kyoichi Okishio aa Kinki-chuo Chest Medical Center,1180Nagasone-cho,Kita-ku,Sakai City,Osaka 591-8555,Japan bOtemae Hospital,1-5-34Otemae,Chuo-ku,Osaka,Japana r t i c l ei n f oArticle history:Received 9August 2010Received in revised form 19October 2010Accepted 18December 2010Keywords:Gefitinib ErlotinibEGFR mutationResistance to EGFR-TKIsTime to progression of gefitiniba b s t r a c tPurpose:We investigated survival potential in patients receiving erlotinib after failure of gefitinib,focus-ing on response and time to progression (TTP)with gefitinib.Methods:We retrospectively reviewed lung adenocarcinoma patients who received erlotinib after expe-riencing progression with gefitinib.Our primary objective was to evaluate the prognostic significance of erlotinib therapy.Results:A total 42lung adenocarcinoma patients were included in this study.Overall disease control rate was 59.5%(partial response [PR],2.4%;stable disease [SD],57.1%).Median overall survival was 7.1months,and median progression-free survival was 3.4months.The number of patients who achieved PR and non-PR (SD+progressive disease [PD])with gefitinib were 22(52%)and 20(48%),respectively.Patients with PR for gefitinib showed significantly longer survival times than those with non-PR (9.2vs.4.7months;p =0.014).In particular,among PR patients,those with TTP <12months on gefitinib showed significantly longer survival times than those with TTP ≥12months (10.3vs.6.4months;p =0.04).Conclusions:Erlotinib may exert survival benefit for lung adenocarcinoma patients with less than 12months of TTP of prior gefitinib who achieved PR for gefitinib.© 2011 Elsevier Ireland Ltd. All rights reserved.1.IntroductionGefitinib and erlotinib are oral epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs).Gefitinib has been reported to be effective in limited populations such as never smokers,Asians,and patients with adenocarcinoma,and is particularly effective in patients with EGFR mutations [1–3].Erlotinib,which has a sim-ilar quinazoline frame to gefitinib,is the first EGFR-TKI shown to provide survival benefit in patients with non-small cell lung cancer (NSCLC)[4]:the BR.21trial revealed significantly longer sur-vival times among patients who received erlotinib compared with a placebo group [4].In addition,these two EGFR-TKIs have been found to occasionally induce a particularly significant response in EGFR-mutant patients.However,despite this documented efficacy,most cancer clones acquire resistance to these particular com-pounds over time [5].Previous studies have demonstrated that amplified MET onco-gene and secondary EGFR T790M mutations are most commonly responsible for resistance to gefitinib and erlotinib [6,7].Indeed,several previous studies showed that secondary EGFR T790M muta-tion and MET amplification occurred in nearly half and 20%of lung∗Corresponding author.Tel.:+81722523021;fax:+81722511372.E-mail address:kazu.taizo@ (K.Asami).cancer specimens that had become resistant to EGFR-TKIs,respec-tively [8–11].In addition,the majority of patients who showed secondary resistance had EGFR mutations such as exon 19dele-tion mutations or L858R point mutation,which have been found to be sensitive to EGFR-TKIs [12,13].Several reports have demonstrated clinical benefits when administering erlotinib to NSCLC patients following failure of gefi-tinib [14–18];in contrast,one previous report has suggested that no erlotinib-derived clinical benefit can be expected in patients who failed gefitinib [19].However,reports thus far have all had small sample sizes,and clear findings regarding efficacy of erlotinib in patients who failed gefitinib have yet to be obtained.Consequently,whether or not erlotinib is useful in these patients remains contro-versial.We hypothesize that tumor clones may require exposure to gefi-tinib treatment with a positive response for a specific duration to acquire secondary common resistance to EGFR-TKIs.Even if a patient experiences tumor progression on gefitinib therapy,sub-sequent erlotinib therapy may nevertheless still be able to inhibit progression,provided the tumor clones did not acquire secondary resistance.As such,in positive-responder patients with confirmed progression within a specific duration of gefitinib treatment,some tumor clones may remain sensitive to erlotinib,and therefore these patients may still experience survival benefit with erlotinib treat-ment.0169-5002/$–see front matter © 2011 Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.lungcan.2010.12.014212K.Asami et al./Lung Cancer73 (2011) 211–216Here,we conducted a retrospective study primarily aimed at assessing overall survival(OS)of patients who received erlotinib therapy after failure with gefitinib.We also attempted to charac-terize the clinical features of patients who benefited from erlotinib treatment.2.Patients and methodsWe retrospectively reviewed records for patients with histopathologically diagnosed lung adenocarcinoma who received erlotinib after experiencing progression on gefitinib at Kinki-chuo Chest Medical Center between December2008and October2009. Responses were evaluated based on patient records and radio-graphic studies,such as chest roentgenograms and computed tomographic(CT)and magnetic resonance imaging(MRI)scans. We examined EGFR mutation status using the PCR-invader method with paraffin sections of biopsy specimens from patients.Time to progression(TTP)with gefitinib was defined as the period from initiation of gefitinib therapy to the date when dis-ease progression was confirmed.Overall survival was defined as the period from initiation of erlotinib therapy to the date of death or last follow-up.Disease control rate(DCR)was defined as com-plete response(CR)plus partial response(PR)plus stable disease (SD).Evaluation of response to gefitinib and erlotinib therapy by CT scan was performed according to the response evaluation crite-ria in solid tumors(RECIST).Stable disease plus progressive disease (PD)with prior gefitinib treatment was defined as“non-PR.”Categorical outcomes,including DCRs,were compared using the 2test,and survival distribution was estimating using the Kaplan–Meier method.Overall survival and progression-free sur-vival(PFS)were compared with regard to demographic factors such as gender,performance status,EGFR mutation status,response to gefitinib,TTP with gefitinib,and toxicity grade of skin rash,which may be associated with survival,using the log-rank test.Values were considered statistically significant for p<0.05.A multivariate Cox-proportional-hazards model was used to determine the clini-cal variables which influenced OS.Statistical analyses were carried out using SPSS software ver.11.0for Windows(IBM,Chicago,IL, USA).3.Results3.1.Patient characteristicsForty-two patients with lung adenocarcinoma were reviewed in the present study.All patients became refractory to gefitinib dur-ing the course of treatment and were subsequently switched to erlotinib therapy.Patient characteristics are described in detail in Table1.Thirty patients(71%)had received1or2regimens before Table1Patient characteristics.Number(%)Median age,years(range)65(31–85) SexMale13(31)Female29(69)Smoking historyNever28(67)Former/current14(33)ECOG score0–124(57)2–418(43)Cancer stageIIIB8(19)IV34(81) Number of previous treatments with erlotinib1–230a(71)3≤12(29) EGFR mutationExon19deletion mutation14(33)L858R14(33)Exon18point mutation1(2)Wild13(32)TTP with gefitinib treatment,months(range)8.1(0.9–40.7) <1229(69)≤1213(31)Response to gefitinibCR0(0)PR22(53)SD17(40)PD3(7)EGFR,epidermal growth factor receptor;TTP,time to progression;CR,complete response;PR,partial response;SD,stable disease;PD,progressive disease;ECOG, Eastern Cooperative Oncology Group.a Two patients received gefitinib asfirst-line treatment.initiation of erlotinib and2(7%)had received gefitinib asfirst-line treatment.EGFR mutations were detected in29(69%)patients:14(33%) had exon19deletions,14(33%)had L858R mutations,and1(2%) had an exon18point mutation.The median TTP with gefitinib treatment was8.1months.Thirteen(31%)patients had TTPs of12 months or more,while29(69%)had TTPs of less than12months. Twenty-two(53%)patients receiving gefitinib achieved PR,and17 (40%)achieved SD.None achieved CR while receiving gefitinib ther-apy.The response rate(RR)and DCR for gefitinib were53%(22of 42patients)and93%(39of42patients),respectively.Of the22 patients who achieved PR with gefitinib,19(86%)were found to have EGFR mutations.Of the20patients who had SD or PD(non-PR)Table2Response to erlotinib according to the response to prior gefitinib and EGFR mutation status.EGFR mutation Response to gefitinibPR(n=22)Non-PR a(n=20)SD(n=17)PD(n=3)Positive,n(%)Negative b,n(%)Positive,n(%)Negative,n(%)Positive,n(%)Negative,n(%) Response to erlotinib PR(N=1)1(4.5)0(0)0(0)0(0)0(0)0(0) SD(N=24)14(64)1(4.5)3(18)5(29)0(0)1(33)PD(N=17)4(18)2(9)6(35)3(18)1(33)1(33)EGFR,epidermal growth factor receptor;PR,partial response;SD,stable disease;PD,progressive disease.Overall disease control rate(PR+SD)was73%(EGFR mutation-positive:15/22[68%],EGFR mutation-negative:1/22[5%])among patients who achieved PR with gefitinib and45%(EGFR mutation-positive:3/20[15%],EGFR mutation-negative:6/20[30%])among patients with non-PR(SD+PD for gefitinib)with gefitinib treatment.Overall disease control rate was62%(PR for gefitinib:15/29[52%],non-PR for gefitinib:3/29[10%])in EGFR mutation-positive patients and54%(PR for gefitinib:1/13[8%],non-PR for gefitinib:6/13[46%])in EGFR mutation-negative patients.a Defined as SD plus PD with prior gefitinib therapy.b EGFR wild-type.K.Asami et al./Lung Cancer73 (2011) 211–216213 Table3Response to erlotinib stratified by TTP with prior gefitinib treatment.(A)TTP with gefitinib<12monthsResponse to gefitinib TTP with gefitinib(months)<12(n=29)PR(n=11)n(%)Non-PR(n=18)SD(n=15)n(%)PD(n=3)n(%) Response to erlotinib PR(n=1)1b(9)0(0)0(0)SD(n=17)8a,b(73)8a(44)1(6)PD(n=11)2(18)7(39)2(11)B.TTP with gefitinib≥12monthsResponse to gefitinib TTP with gefitinib(months)≥12(n=13)PR(n=11)n(%)Non-PR(n=2)SD(N=2)n(%)PD(N=0)n(%) Response to erlotinib PR(n=0)0(0)0(0)0(0)SD(n=7)7(64)0(0)0(0)PD(n=6)4(36)2(100)0(0)EGFR,epidermal growth factor receptor;PR,partial response;SD,stable disease;PD,progressive disease;TTP,time to progression.Overall disease control rate was62%(PR for gefitinib:9/29[31%],non-PR for gefitinib:9/29[31%])in patients with TTP of gefitinib<12months.Overall disease control rate was54%(PR for gefitinib:7/13[54%], non-PR for gefitinib:0/13[0%])in patients with TTP of gefitinib≥12months.a Ten patients showed improvement of target lesions,but not to PR standards.Seven and three patients achieved PR and SD,respectively,with gefitinib treatment.b A second biopsy from progression lesions was performed in three patients(one had PR and2had SD with erlotinib)who achieved PR with gefitinib.Exon19deletion mutations which were the same pattern as detected infirst biopsy specimen for primary diagnosis of NSCLC were identified,whereas EGFR T790M mutation,which endowed secondary common resistance to EGFR-TKIs,was not identified in those biopsy specimens.with gefitinib,EGFR mutations were detected in10(50%).Among patients with EGFR mutations,only one showed PD with gefitinib therapy,and RR and DCR in this group were66%(19of29)and97% (28of29),respectively.3.2.ResponseOn erlotinib therapy,1of42patients achieved PR,and24had SD.No patients achieved CR with erlotinib.Overall RR and DCR for erlotinib were2.4%(one of42)and59.5%(25of42),respectively.Response to erlotinib categorized by response to prior gefi-tinib duration and EGFR mutation status is described in Table2. Among patients who achieved PR with gefitinib,one achieved PR and15patients achieved SD with erlotinib therapy.Patients who achieved PR with gefitinib showed higher DCRs with erlotinib than patients who had non-PR with gefitinib(16[73%]of22vs.9[45%] of20),albeit without statistical significance(p=0.07).In addi-tion,EGFR mutation status was not found to be associated with response to erlotinib;in terms of DCR,no significant difference was noted between EGFR-mutant patients(18/29)and EGFR non-mutant patients group(7/13)(62%vs.54%,p=0.616).Time to progression with prior administration of gefitinib was not found to be associated with achieving a response with subse-quent erlotinib.Details regarding response to erlotinib categorized by TTP with gefitinib are shown in Table3.DCR among patients experiencing progression after less than12months of gefitinib therapy was18/29(62%).In contrast,DCR among patients with TTPs of12months or more was7/13(54%).No statistical significant difference in DCR was noted between these two groups accord-ing to TTP with prior administration of gefitinib(p=0.62).Of the 24patients who achieved SD with erlotinib therapy,10showed improvement in target lesions which had been exacerbated during gefitinib treatment;all10were EGFR-mutant patients(4L858R, 5exon19deletion mutations,and1exon18point mutation),and TTPs with gefitinib were all less than12months.Of the two patients who received gefitinib asfirst-line treatment,one had an EGFR L858R mutation and showed responses to gefitinib and subsequent erlotinib of PR and SD,respectively.While this particular patient showed a relatively long TTP(39.5months)with gefitinib,disease progression was confirmed4months after initiation of erlotinib therapy,and OS was58.6months.The other patient who received gefitinib asfirst-line treatment had EGFR-wild type,and responses to both gefitinib and subsequent erlotinib treatment were PD.TTP and OS in this patient were3and7.4months,respectively.A second biopsy of the progressed lesions was performed in three patients after gefitinib therapy failed.While exon19deletion mutations of the same pattern as noted in thefirst biopsy specimen for primary diagnosis were also detected on this second biopsy, we noted no EGFR T790M mutations.Of note,however,was the fact that imagingfindings for lesions after erlotinib therapy were improved on the second biopsy(Table3).3.3.SurvivalMedian OS and median progression-free survival(PFS)were 7.1months(95%confidence interval[CI]:4.4–9.8months)and3.4 months(95%CI:1.1–5.7months),respectively(Fig.1).Multivariate analysis of prognostic factors was performed using a Cox propor-tional hazards model to determine which clinical variables were most strongly associated with OS(Table4).Response to gefitinibTable4Multivariate analysis of prognostic variables for OS by use of a Cox proportional-hazards model.Multivariate analysisp a Hazard ratio95%CISex0.51 1.350.55–3.31 ECOG score0.190.580.25–1.31 EGFR mutation0.78 1.130.48–2.70 Response to gefitinib0.0050.230.80–0.64 TTP of gefitinib0.050.340.12–1.01 Grade of skin rash0.290.640.27–1.47EGFR,epidermal growth factor receptor;TTP,time to progression;CI,confidence interval;ECOG,Eastern Cooperative Oncology Group;PR,partial response.Response to gefitinib was the only independent prognostic factor.TTP with gefitinib showed borderline significance.Variables were compared as paired categories:sex(female vs.male),ECOG score(0–1vs.2–4),response to gefitinib(PR vs.non-PR),TTP of gefitinib(<12months vs.≥12months),grade of skin rash(3vs.1–2).a p<0.05was considered significant.214K.Asami et al./Lung Cancer73 (2011) 211–216Fig.1.Kaplan–Meier plot of survival time with erlotinib.(A)Overall survival rates and(B)progression-free survival rates of42patients.MST:median survival time; mPFS:median progression-free survival.was found to be the only independent prognostic factor(hazard ratio=0.23;95%CI:0.08–0.64,p=0.005),and time to progression with gefitinib showed borderline significance(hazard ratio=0.34; 95%CI:0.12–1.01,p=0.05).Kaplan–Meier curves of survival time according to response to prior gefitinib therapy are shown in Fig.2.Patients who achieved PR while receiving gefitinib therapy showed significantly longer OS(p=0.014).However,no significant difference was noted in PFS between patients with PR for gefitinib and those with non-PR(4.7 months[95%CI:2.9–6.5months]vs.1.8months[95%CI:1.4–2.2 months];p=0.122).Time to progression with gefitinib showed a borderline significant impact on survival with erlotinib therapy. However,among patients who achieved PR with gefitinib,TTP with gefitinib therapy was strongly correlated with survival time. Kaplan–Meier curves of survival time for patients who achieved PR with gefitinib stratified according to TTP are shown in Fig.3. Patients with TTPs of less than12months with gefitinib ther-apy were found to have significantly longer OS(10.3months[95% CI:7.0–13.6months]vs.6.4months[95%CI:2.6–10.2months]; p=0.04)and longer PFS(6.4months[95%CI:3.6–9.2months]vs.3.4 months[95%CI:1.2–5.6months];p=0.19)than patients with TTPs of12months or more.However,no statistically significant differ-ence was noted between the two groups in terms of PFS(p=0.19).Fig.2.Kaplan–Meier plot of survival time with erlotinib.(A)Overall survival rates and(B)progression-free survival rates stratified by response to prior gefitinib.Non-PR is defined as SD plus PD with gefitinib therapy.In addition,we found that skin rash was not predictive of sur-vival with erlotinib therapy.All patients in the present study were affected by rash of some grade while receiving erlotinib.The degree of skin rash toxicity due to erlotinib exceeded the grade noted dur-ing gefitinib treatment in32patients.Seven patients required dose reduction of erlotinib due to grade3skin ing a Cox propor-tional hazard model,we determined that skin rash grade had no impact on survival(hazard ratio=0.64[95%CI:0.27–1.47];p=0.29).4.DiscussionHere,we investigated survival potential in patients receiv-ing erlotinib after failure of gefitinib,focusing on response and TTP with gefitinib.Ourfindings suggest that administration of erlotinib subsequent to gefitinib may exert survival benefit in for-mer gefitinib-positive responders.Further,among those former responders,most with TTP<12months may not yet have sec-ondary resistance to EGFR-TKIs.Ourfindings suggest little chance for patients to achieve a high response with erlotinib therapy after experiencing progression with gefitinib therapy.This observation may be due to these two EGFR TKIs sharing the same mechanism of EGFR blockade or to cross resistance[5].Our retrospective study showed that response achieved with prior administration of gefitinib was the only prognostic factor for subsequent erlotinib therapy after experiencing progression on gefitinib therapy.In particular,among patients who achieved PR with gefitinib,patients with TTPs of less than12months with gefitinib therapy were found to have significantly longer OS thanK.Asami et al./Lung Cancer73 (2011) 211–216215Fig.3.Kaplan–Meier plot of survival time for patients who achieve PR with gefitinib.(A)Overall survival rates and(B)progression-free survival rates stratified by TTP with gefitinib.patients with TTPs of12months or more.In addition,most of these patients showed some degree of improvement in imagefind-ings after subsequent erlotinib therapy.We noted no EGFR T790M mutations in any of three patients who underwent a second biopsy of their progressed lesions after failure with gefitinib therapy.We therefore supposed that most patients with TTP<12months may have not yet acquired the EGFR T790M mutation.However,we only investigated the presence of a secondary EGFR T790M mutation in three patients in the present study.Validation of this hypothesis will require collection of more molecular information from patients who are no longer responsive to gefitinib in the future.Shepherd et al.demonstrated that TTP was2.6months in NSCLC patients who had previously been treated with docetaxel therapy [20].We observed that PFS was3.4months in patients with TTP≥12 months who achieved PR in our study,a duration which appears improved over that demonstrated by Shepherd et al.Given these findings,we posited that,regardless of duration of gefitinib ther-apy,subsequent erlotinib may be able to prolong PFS compared to chemotherapy with cytotoxic agent provided the patients demon-strated a positive response with gefitinib.However,given that our results were obtained in a retrospective study with an extremely small sample population,a prospective study is warranted to clar-ify whether or not erlotinib administered subsequent to gefitinib can elicit greater survival benefit in gefitinib-positive responders than chemotherapy with cytotoxic agents.We noted here that treatment with erlotinib following gefitinib resulted in more toxic grades of skin rash in patients,findings which suggest that erlotinib may have greater biological activity than gefitinib.Several other investigators have also suggested based on their ownfindings that erlotinib may have higher biological activity than gefitinib.Costa et al.showed that differing efficacy between gefitinib and erlotinib was due to differences in commonly admin-istered dosages between the two drugs[21].Gefitinib(250mg per day)is typically administered at one third of its maximum-tolerated dose,whereas erlotinib(150mg per day)is administered at its maximum tolerated dose.In vitro data showed that the mean concentration of gefitinib was0.24g/ml at the300-mg daily dose and1.1g/ml at1000mg/day.In contrast,median concentration of erlotinib at150mg/day was1.26g/ml.These previousfindings suggest that erlotinib(150mg/day)has a higher biological dose of EGFR inhibition than gefitinib(250mg/day).Recent studies have demonstrated that the increased biological activity of EGFR-TKIs is associated with control of tumor clones. Yoshimasu et al.reported observing a dose-response relationship between inhibition rates and gefitinib concentration[22].Clarke et al.reported that high-dose erlotinib was effective in controlling leptomeningeal metastases progression while receiving standard erlotinib therapy in EGFR-mutant patients[23].These authors demonstrated that a weekly1200-mg dose of erlotinib controlled leptomeningeal metastases in a patient who was no longer respon-sive to a standard daily dose of erlotinib(150mg).Ourfindings here suggest that a treatment duration of12 months of gefitinib therapy may be the borderline period for tumor clones to attain resistance to EGFR-TKIs.However,speculation as to whether or not previously EGFR-TKI-sensitive clones gradually grow resistant to EGFR-TKIs has not been resolved.Further studies are necessary to validate ourfindings.In conclusion,gefitinib responders may achieve survival ben-efits from erlotinib therapy after experiencing progression with gefitinib.Among patients who have been receiving gefitinib ther-apy for less than12months,tumor clones may not yet have acquired a secondary mutation.However,further studies are needed to clarify precisely how tumor clones attain such secondary resistance to EGFR-TKIs.Conflicts of interest statementNone declared.AcknowledgementsWe are grateful to the staff of Kinki-Chuo Chest Medical Cen-ter for their helpful comments.We are especially indebted to Dr. Masahiko Ando of Kyoto University for support on statistical anal-yses.References[1]Moscatello DK,Holgado-Madruga M,et al.Frequent expression of a mutantepidermal growth factor receptor in multiple human tumors.Cancer Res 1995;55(23):5536–9.[2]Janne PA,Engelman JA,et al.Epidermal growth factor receptor mutations innon-small-cell lung cancer:implications for treatment and tumor biology.J Clin Oncol2005;23(14):3227–34.[3]Baselga J,Arteaga CL.Critical update and emerging trends in epidermal growthfactor receptor targeting in cancer.J Clin Oncol2005;23(11):2445–59.[4]Pao W,Miller V,Zakowski M,et al.EGF receptor gene mutations are commonin lung cancers from“never smokers”and are associated with sensitivity of tumors to gefitinib and erlotinib.Proc Natl Acad Sci2004;101:13306–11. [5]Mitsudomi T,Kosaka T,Endoh H,et al.Mutations of the epidermal growth factorreceptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence.J Clin Oncol 2005;23(11):2513–20.[6]Mok TS,Wu Y-L,et al.Gefitinib or carboplatin–paclitaxel in pulmonary adeno-carcinoma.N Engl J Med2009;361(September(10)):947–57.[7]Shepherd FA,Rodrigues Pereira J,et al.Erlotinib in previously treated non-small-cell lung cancer.N Engl J Med2005;353(2):123–32.216K.Asami et al./Lung Cancer73 (2011) 211–216[8]Johnson JR,Cohen M,et al.Approval summary for erlotinib for treatmentof patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen.Clin Cancer Res 2005;11(18):6414–21.[9]Allan S,et al.Efficacy of erlotinib in patients with advanced non-small celllung cancer(NSCLC)relative to clinical characteristics:subset analysis from the TRUST study.In:Poster presented at ASCO.2008.[10]Baselga J,Rischin D,et al.Phase I safety,pharmacokinetic,and pharmaco-dynamic trial of ZD1839,a selective oral epidermal growth factor receptor tyrosine kinase inhibitor,in patients withfive selected solid tumor types.J Clin Oncol2002;20(November(21)):4292–302.[11]Hidalgo M,Siu LL,Nemunaitis J,Rizzo J,et al.Phase I and pharmacologic studyof OSI-774,an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies.J Clin Oncol2001;19(July(13)):3267–79.[12]Riely GJ,Pao W,et al.Clinical course of patients with non-small cell lung cancerand epidermal growth factor receptor exon19and exon21mutations treated with gefitinib or erlotinib.Clin Cancer Res2006;12(3(Pt1)):839–44.[13]Choong NW,Dietrich S,Seiwert TY,Tretiakova MS.Gefitinib response oferlotinib-refractory lung cancer involving meninges-role of EGFR mutation.Nat Clin Pract Oncol2006;3(January(1)):50–7[quiz1p following57].[14]Mitsudomi T,Kosaka T,Endoh H,Yoshida K.Mutational analysis of theEGFR gene in lung cancer with acquired resistance to gefitinib.J Clin Oncol 2006;24(18S):7074.[15]Pao W,Balak MN,Riely GJ,Li AR.Molecular analysis of NSCLC patients withacquired resistance to gefitinib or erlotinib.J Clin Oncol2006;24(18S):7078.[16]Bean J,Brennan C,Shih JY,Riely G,Viale A.MET amplification occurs with orwithout T790M mutations in EGFR mutant lung tumors with acquired resis-tance to gefitinib or erlotinib.Proc Natl Acad Sci USA2007;104(December(52)):20932–27.[17]Engelman JA,Zejnullahu K,Mitsudomi T.MET amplification leads to gefitinibresistance in lung cancer by activating ERBB3signaling.Science2007;316(May (5827)):1039–43.[18]Engelman JA,Zejnullahu K,Mitsudomi T,et al.MET amplification leads togefitinib resistance in lung cancer by activating ERBB3signaling.Science 2007;316(May(5827)):1039–43[Epub2007April26].[19]Balak MN,Gong Y,Riely GJ,et al.Novel D761Y and common secondary T790Mmutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors.Clin Cancer Res2006;12(Nov(21)):6494–501.[20]Shepherd FA,Dancey J,Ramlau R,et al.Prospective randomized trial ofdocetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy.J Clin Oncol 2000;18(10):2095–103.[21]Costa DB,Schumer ST,Tenen DG,et al.Differenctial responses to erlotinib inepidermal growth factor receptor(EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations.J Clin Oncol2008;26(7):1182–4.[22]Yoshimasu T,Ohta F,Oura S,et al.Histoculture drug response assay for gefi-tinib in non-small-cell lung cancer.Gen Thorac Cardiovasc Surg2009;57(March(3)):138–43[Epub2009March12].[23]Clarke JL,Pao W,Wu N,et al.High dose weekly erlotinib achieves thera-peutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer.J Neurooncol 2010;February.。
肿瘤动物模型和抗肿瘤药物的研究方法.

30
瘤株的组织学类型和生长特征趋于稳定,并 能在同系、同种或异种动物体内移植并连续传代。
生长特征,包括接种成活率、生长速度、自 动消退率、宿主寿命与宿主反应等。
细胞系:原代培养物经首次传代成功后即为细 胞系,由原先存在于原代培养物中的细胞世系 所组成。
40
(3)瘤细胞悬液接种法 如每次需要接种的动物数量较多时,可用瘤细胞 悬液接种:
取几个瘤块 --->除去坏死部分 ---> 混合瘤块 ---> 剪成小块 --->匀浆器单向研匀 ---> 放入无菌容器 ---> 生理盐水稀释成1:3~1:4悬液
每只动物接种0.2ml;整个操作应在30min内完成;每 次抽吸前应将细胞混匀。
8
[结果判定] 给药后每天观察肿瘤生长情况,记录发生时 间和位置。 肿瘤结节长到一定程度,每周用脚规测量皮下肿 瘤2次。测定肿块的最大径(a)和最小径(b)。 肿瘤体积(cm3) = ab2/2
9
将肿瘤体积变化对时间作出生长曲线,可由曲线 的斜率变化判断药物抑制肿瘤生长的作用,尤其 应观察肿瘤能否完全消退。
26
它比前述的自发性和诱发性动物肿瘤更易实施。 现有移植性肿瘤接种成功率可达100%, 可在同一 时间内获得大量(数十至百余只动物)、生长相当 均匀的肿瘤。
27
一、动物的选择
移植性肿瘤常用的动物为小鼠、大鼠和地鼠。 研究药物的抗肿瘤作用可选择三种或三种以上小 鼠、大鼠的移植性肿瘤进行实验治疗;抗肿瘤药 物筛选时,每批动物的来源应一致;
16
一、诱发性肿瘤动物模型的基本方法
降低氯化锂-匹罗卡品大鼠癫痫模型死亡率的实验研究

降低氯化锂-匹罗卡品大鼠癫痫模型死亡率的实验研究王冬梅;孙艺平;徐静;徐红;赵赫男;李骢;宫德正;田余祥【摘要】[目的]探讨有效控制氯化锂-匹罗卡品诱发癫痫持续状态,提高癫痫大鼠存活率的最佳条件.[方法]选用SD大鼠30只,腹腔注射氯化锂和匹罗卡品后,诱发大鼠癫痫持续状态1 h后,随机分为A组:只给东莨菪碱;B组:东莨菪碱+安定;C组:东莨菪碱+水合氯醛.比较3组大鼠间癫痫发作的持续状态、癫痫大鼠的死亡率、死亡发生的时间.[结果]A组大鼠癫痫发作的持续状态为16~20 h,死亡率为87.5%; B 组大鼠在用药后15 min内可控制癫痫发作的持续状态,死亡率为62.5%;C组大鼠在用药后3~10 min可完全控制大鼠的癫痫发作持续状态,大鼠全部存活.[结论]水合氯醛可有效控制氯化锂-匹罗卡品诱发大鼠的癫痫持续状态,提高癫痫大鼠的存活率.【期刊名称】《大连医科大学学报》【年(卷),期】2010(032)002【总页数】3页(P176-178)【关键词】氯化锂-匹罗卡品;癫痫;水合氯醛;死亡率【作者】王冬梅;孙艺平;徐静;徐红;赵赫男;李骢;宫德正;田余祥【作者单位】大连医科大学,机能实验室,辽宁,大连,116044;大连医科大学,机能实验室,辽宁,大连,116044;大连医科大学,机能实验室,辽宁,大连,116044;大连医科大学,机能实验室,辽宁,大连,116044;大连医科大学,病理生理教研室,辽宁,大连,116044;大连医科大学,病理生理教研室,辽宁,大连,116044;大连医科大学,机能实验室,辽宁,大连,116044;大连医科大学,生化教研室,辽宁,大连,116044【正文语种】中文【中图分类】R742.1氯化锂-匹罗卡品诱发大鼠癫痫持续状态(status epileptieus,SE)模型因与人类颞叶癫痫非常相似,已得到广泛应用。
国内外文献报道,一般在使用匹罗卡品30 min 内,大鼠即可出现全身阵挛,癫痫持续状态持续数小时而不能自止,但癫痫持续状态模型病死率较高,达到50%左右[1],限制了此模型的推广使用。
药物研发中常用的动物模型

一、常用肿瘤模型实验动物介绍:1、BALB/c 小鼠(近交系)特性与用途:◇其发病率低,但对致癌因子敏感。
乳腺肿瘤发生率约为10﹪~20﹪。
◇有一定数量的卵巢、肾上腺和肺部肿瘤的发生,对放射线极度敏感。
易患慢性肺炎。
◇多数个体于6月龄以后出现免疫球蛋白过多症。
主要是IgG1和IgA量的增加。
◇免疫球蛋白的绝对量依饲养条件而异。
腹腔注射矿物油后可引起浆细胞瘤。
◇广泛地应用于肿瘤学、生理学、免疫学、核医学研究,以及单克隆抗体研究和生产等。
2、DBA/2 小鼠(近交系)特征与用途:◇免疫:在普通饲养条件下三月龄鼠血清免疫球蛋白量为1000ug/ml左右,仅相当C57BL/6,C3H/He和BALB/c的1/2。
其中,IgM值较高,而IgG为低值。
在IgG各亚类中,IgG1最高,IgG2最低。
缺乏补体C5。
对鼠斑疹伤寒补体C5较敏感。
◇肿瘤:对DBA/1 的大部分移植瘤有抗性。
雌鼠白血病发病率为34%,雄鼠为18%,经产母鼠乳腺癌发生率为50- 60%,雌雄鼠中均有淋巴瘤生长。
◇微生物和寄生虫:对疟原虫、利什曼原虫有抗力。
对猫后睾吸虫、曼氏血吸虫较敏感。
对白色念球菌有抗力,由于具有Hc0等位基因,对新型隐球菌有抗力。
◇生理:红细胞多。
血压较低。
维生素K缺乏,氯仿和氧化乙烯引起的死亡率高。
肾上腺脂质贮存少,心脏有钙盐沉着。
具低嗜酒性及吗啡嗜好。
对百日咳组织胺易感因子敏感。
◇病理:听源性癫痫发作率在35日龄时为100%,55日龄时为5%,约一半动物肝可出现由巨噬细胞构成的蜡样质的肉芽肿。
3、ICR 小鼠(封闭群)特征与用途:◇适应性强,体格健壮,繁殖力强,生长速度快,实验重复性较好。
◇雌鼠自发性畸胎瘤和管状腺瘤发病率为0%~1%,用氨基甲酸乙酯诱发时,11~16天胚胎期畸胎瘤和管状腺瘤发病率为5.9%,离乳个体管状腺瘤和囊瘤发生率为30%,孕鼠为3%。
◇是国际通用的封闭群小鼠(封闭群又称远交群,是指以非近亲交配方式进行繁殖生产的一个实验动物种群,在不从其外部引入新个体的条件下,至少连续繁殖4代以上)◇是进行免疫药物筛选,复制病理模型较常用的实验动物。
动物模型-动物效应下考察药效的基本要素

动物模型-动物效应下考察药效的基本要素Models — Essential Elements to Address Efficacy Underthe Animal Rule2009年1月 美国FDA发布2010年3月 药审中心组织翻译博福-益普生(天津)制药有限公司翻译药审中心最终核准目录I.前言 (4)II.背景 (4)III.动物(效应)法规的考量 (5)IV.关于动物模型的基本数据要素的讨论 (7)A.影响疾病或状态的CBRN试剂的特性 (7)1、激发试剂 (7)2、致病性决定因子 (7)3、暴露途径 (8)4、暴露量 (8)B.宿主易感性和对病原性物质的响应 (8)C.疾病的自然病程:病理生理的可比性 (9)1、疾病/状态发作的时间 (10)2.疾病或状态进展的时间进程 (10)3.临床表现(症状和体征) (10)D.治疗干预的触发点 (11)E.医学治疗的特征 (11)1、产品类别 (12)2、作用机理 (12)3、体外活性 (12)4、在相似的病理生理学疾病/状态下的活性 (12)5、健康动物/人体的药物动力学(PK) (13)6、在受影响动物/人类中的PK/PD(药物动力学/药效学) (13)7、与可能合并使用的医药产品的PK相互作用 (13)8、与可能联合应用的医药产品的协同或拮抗作用 (14)F.动物药效研究实验设计上的考量 (14)1、终点 (15)2、治疗干预的时间选择 (15)3、给药途径 (15)4、给药方案 (16)V.人类安全性信息 (16)VI.动物模型的基本数据要素 (17)附件A:简称和缩写 (19)动物模型-动物效应下考察药效的基本要素I.前言本指南为潜在的申办方(工业、学术界和政府)提供关于开发疗效研究动物模型的信息。
本指南关注于在动物(效应)法规(the Animal Rule)下,开发药品或生物制品用于申请批准或许可时,需要着重强调的动物模型关键特征(基本数据要素)的确定(药品参考21CFR314.600,生物制品参考21CFR601.90)。
环磷酰胺对小鼠免疫抑制的动物模型建立及其免疫功能的测定

环磷酰胺对小鼠免疫抑制的动物模型建立摘要:[目的]建立环磷酰胺对小鼠的免疫抑制的动物模型,从天然免疫功能和获得免疫功能两方面检测对小鼠的影响。
[方法]初次免疫设立实验组给予OVA-AL(OH)3(0.5/只)和对照组给予NS(0.5/只);8d,10d,12d,14d分别给实验组和对照组注射NS和CTX(100mg/kg);与15d加强免疫分别注射NS和OVA-AL(OH)3(0.5/只);22d检测免疫功能[结果]与NS-NS组相比,CTX-NS组的SI值,双向免疫及ELISA的效价无明显差别,但吞噬细胞指数及吞噬细胞百分率明显降低;与NS-OVA相比CTX-OVA组的SI 明显降低,双向免疫剂ELISA效价都降低[结论]环磷酰胺对小鼠的T淋巴细胞,B淋巴细胞以及吞噬细胞有抑制作用Abstract:[aims] To establish animal model of cyclophosphamide in mice immunosuppression , test in mice from two aspects of natural immune function and immunity function [methods] Primary immune to set up the experimental group given OVA-AL(OH)3 and The control group given NS ; the 8th,10th,12th,14th day give the experimental group NS and the control group CTX ,and the 15th strengthen the immune;the 22th test the immune function [results] Compared with NS - NS group, the SI value of CTX - NS group, bidirectional immune potency and ELISA has no obvious difference, but the phagocyte index and phagocytes percentage decreased obviously; Compared with NS - OVA CTX - SI of OVA group was obviously lower, bidirectional immune agent ELISA titer were reduced [conclusion] Cyclophosphamide in mice of T lymphocyte, B lymphocyte and phagocytes have inhibition关键词:环磷酰胺 CTX 免疫抑制动物模型淋巴细胞建立一种环磷酰胺免疫抑制小鼠模型,并将其与正常小鼠进行对照,通过检测小鼠的天然及获得免疫功能对比其改变与不同,从而可知环磷酰胺对免疫抑制的作用,有利于进一步进行对临床应用抗肿瘤的研究。
Paclitaxel微管聚合物稳定剂CAS号33069-62-4生物活性数据
Clinical pharmacokinetics of paclitaxel. Sonnichsen DS, et al. Clin Pharmacokinet. 1994 Oct;27(4):256-69. PMID: 7834963.
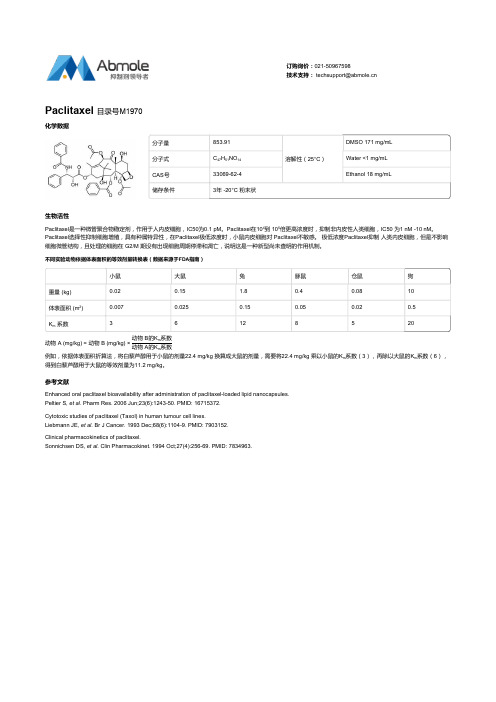
生物活性 Paclitaxel是一种微管聚合物稳定剂,作用于人内皮细胞,IC50为0.1 pM。Paclitaxel在104到 105倍更高浓度时,抑制非内皮性人类细胞,IC50 为1 nM -10 nM。 Paclitaxel选择性抑制细胞增殖,具有种属特异性,在Paclitaxel极低浓度时,小鼠内皮细胞对 Paclitaxel不敏感。 极低浓度Paclitaxel抑制 人类内皮细胞,但是不影响 细胞微管结构,且处理的细胞在 G2/M 期没有出现细胞周期停滞和凋亡,说明这是一种新型尚未查明的作用机制。
订购询价:021-50967598 技术支持: techsupport@
Paclitaxel 目录号M1970
化学数据 分子量 分子式 CAS号 储存条件
853.91 C47H51NO14 33069-62-4 3年 -20°C 粉末状
溶解性(25°C)
DMSO 171 mg/mL Water <1 mg/mL Ethanol 18 mg/mL
不同实验动物依据体表面积的等效剂量转换表(数据来源于FDA指南)
小鼠
大鼠
兔
豚鼠
1药理药效研究 动物模型

(一)生物大分子主要包括核酸、蛋白质、多糖等,其主要特征是由小分子的构件分子组成,具有复杂的空间结构,而且结构与生物活性密切相关。
生物小分子包括游离核苷酸、辅基辅酶、寡糖、寡肽、寡核苷酸、类脂、维生素、信号分子以及许多生物大分子的代谢产物等等。
乳糖操纵子包括:启动子;操纵基因;结构基因(Z, Y, A)(二)基因按其功能可分为:结构基因、调节基因、rRNA基因与tRNA基因。
常见的调节基因有microRNAs,siRNAs,piRNAs ……真核生物的RNA聚合酶(3种):RNA聚合酶I,II,III。
种类I II III分布核仁核基质核基质产物rRNA mRNA, microRNA 5S rRNA, tRNA, siRNA任一链上的可读框(ORF)方向总是由5,→3,。
原核生物转座因子主要有二类:插入序列(IS);复合型转座子(Tn)根据转座的的机制和结果,可将转座分为:复制型转座、保守型转座。
质粒DNA可以有共价闭合环状DNA、缺口的环状DNA、线性DNA三种结构状态。
琼脂糖凝胶电泳时涌动速度的快慢:超螺旋<线性<环状DNA(与分子量有关)按质粒复制的调控及其拷贝数可分两类:严紧控制型质粒、松弛控制型质粒。
影响质粒稳定性的因素:①宿主细胞分裂时质粒能否均衡地分配到子代细胞。
②质粒分子自身结构的稳定性。
真核生物基因组可分为细胞核基因组和细胞器基因组。
真核生物基因组存在大量的重复序列:单拷贝序列、中度重复序列(Alu家族)、高度重复序列。
DNA多态性主要包括单核苷酸多态性(SNP)和串联重复序列多态性。
真核基因组的另一特点是存在多基因家族与假基因真核生物有两类细胞器能携带遗传物质:线粒体和叶绿体。
高等动物线粒体基因组具有独特的特点:①母系遗传;②线粒体DNA损伤后不易修复;(三) ③遗传密码与通用遗传密码存在差别。
蛋白质组学研究需要两条互补的实验工作流程:基于凝胶的工作流程和基于液相色谱的工作流程。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
O RIGINAL A RTICLEPaclitaxel Poliglumex(PPX-Xyotax)and Concurrent Radiation for Esophageal and Gastric CancerA Phase I StudyTom Dipetrillo,MD,*Luka Milas,MD,†Devon Evans,MD,*Paul Akerman,MD,*Thomas Ng,MD,* Tom Miner,MD,*Dennis Cruff,MD,*Bharti Chauhan,MD,*David Iannitti,MD,*David Harrington,MD,*and Howard Safran,MD*Objectives:To determine the maximal tolerated dose(MTD)and dose limiting toxicities of poly(L-glutamic acid)-paclitaxel(PPX)and con-current radiation(PPX/RT)for patients with esophageal and gastric cancer.Methods:Patients with esophageal or gastric cancer receiving chemo-radiation for loco-regional,adjuvant,or palliative intent were eligible. The initial dose of PPX was40mg/m2/wk,for6weeks with50.4Gy radiation.Dose levels were increased in increments of10mg/m2/wk of PPX.Results:Twenty-one patients were enrolled over5dose levels.Sixteen patients had esophageal cancer and5had gastric cancer.Twelve patients received PPX/RT as definitive loco-regional therapy,4patients had undergone resection and received adjuvant PPX/RT,and5patients had metastatic disease and received PPX/RT for palliation of dyspha-gia.Dose limiting toxicities of gastritis,esophagitis,neutropenia,and dehydration developed in3of4patients treated at the80mg/m2dose level.Four of12patients(33%)with loco-regional disease had a complete clinical response.Conclusions:The maximally tolerated dose of PPX with concurrent radiotherapy is70mg/m2/wk for patients with esophageal and gastric cancer.Key Words:paclitaxel polyglumex,concurrent chemoradiation, esophageal cancer,gastric cancer,macromolecule drug conjugate (Am J Clin Oncol2006;29:376–379)P PX is a drug conjugate that links paclitaxel to a biodegrad-able polymer,poly-L-glutamic acid.1Preclinically,poly(L-glutamic acid)-paclitaxel(PPX)has demonstrated tumor tissue radiation enhancement factors from4.0to8.0compared with1.5 to 2.0for paclitaxel.2PPXs macromolecular structure may underlie its improved radiation enhancement.PPX has a molec-ular weight of40,000compared with854for paclitaxel.3Solid tumors are more permeable to macromolecules thannormal tissue because of altered capillary endothelium.4Further-more,macromolecules persist longer in tumor tissue because ofthe relative decrease in lymphatics compared with non-neoplas-tic tissue.5Radiation increases the vascular permeability of solidtumors.6,7Local tumor irradiation in tumor cell lines increasesvascular permeability and increases PPX uptake.2Tumor se-creted vascular endothelial growth factor also increases vascularpermeability.8The combination of PPX and radiation in theovarian Oca-1carcinoma implanted in C3Hf/Kam mice pro-duced a significantly greater tumor growth delay than treatmentwith radiation and paclitaxel when both drugs were given at anequivalent paclitaxel dose(enhancement factors of4.44versus1.50).2These preclinicalfindings support the hypothesis that thesupra-additive effect of combined PPX/RT is because of themodulation of the enhanced permeability and retention effect ofmacromolecules by radiation.Paclitaxel is an important radiation sensitizer in uppergastrointestinal malignancies.9–14We initiated a phase I trial ofPPX/RT for patients with esophageal and gastric cancer.This isthefirst report of a clinical study utilizing a poly L-glutamic acidmacromolecule as a radiation sensitizer in human malignancies.MATERIALS AND METHODS EligibiltyPatients with a histologically confirmed diagnosis of esoph-ageal adenocarcinoma or squamous cell carcinoma,or gastric ade-nocarcinoma were enrolled between January2004and May2005.PPX/RT could be administered as definitive loco-regional oradjuvant therapy.Patients with metastatic disease with dyspha-gia were also eligible.Required laboratory parameters includedabsolute neutrophil countϾ1,500L hemoglobinϾ10g/dL, plateletsϾ100,000L,creatinineϽ1.5mg/dL,bilirubinϽ1.5 mg/dL,alanine aminotransferase,and alkaline phosphatase Ͻ2.5ϫupper limit of normal,and calciumϽ12mg/dL.Patients with previous irradiation,Eastern Cooperative Oncology Group(ECOG)performance statusϾ1,life expectancyϽ12weeks,tracheobronchial invasion,tracheoesophagealfistula,unstableFrom The*Brown University Oncology Group,Providence,RI;and the †Department of Experimental Radiation Oncology,The University of Texas M.D.Anderson Cancer Center,Houston TX.Reprints:Devon Evans,MD,Brown University,Department of Hematology/ Oncology,164Summit Avenue,Providence,RI02906.E-mail:devans@ .Copyright©2006by Lippincott Williams&WilkinsISSN:0277-3732/06/2904-0376DOI:10.1097/01.coc.0000224494.07907.4ecardiac disease within6months,active infection,brain metas-tasis,laryngeal or phrenic nerve paralysis,unstable neurologic symptoms,sensory or motor neuropathy greater than grade1,or other concurrent malignancy,were excluded.A complete his-tory and physical examination were performed on all patients before treatment.Height,weight,performance status,and tumor stage were recorded.Required staging studies included a com-puted tomography(CT)scan of the chest,abdomen,and pelvis. Pre-and post-treatment endoscopies were required only in pa-tients with loco-regional disease.The study was approved by the institutional review boards of all participating hospitals and Brown University.All patients gave written informed consent according to federal and institutional guidelines. ChemotherapyPPX was administered as a10-minute infusion before radiotherapy on days1,8,15,22,29,36on an outpatient basis. Patients were treated with PPX according to a dose escalation scheme,with patients enrolled in cohorts of3at each dose level. The initial dose level was40mg/m2/wk in3patients,and was increased in increments of10mg/m2/wk in the next patient cohort if no patients had a dose limiting toxicity(DLT).All toxicities were graded according to the NCI Common Toxicity Criteria,Version3.Dose limiting toxicities were defined as grade3esophagitis,enteritis or gastritis,grade3nausea(despite maximal antinausea treatment)or dehydration,any other grade3 or4nonhematologic toxicity,and grade4neutropenia or throm-bocytopenia lasting more than1week.Toxicities were consid-ered dose limiting only if they resulted from toxicity of chemo-radiation,and not from disease progression.If one or2patients experienced a DLT,then3additional patients were enrolled at the same dose level.If no more than2patients total experienced a DLT at that dose level,than the dose was escalated for the next patient cohort.IfϾ2patients had a DLT,then the dose was de-escalated to the next dose level,and3additional patients enrolled.The MTD was defined as the dose at which no more than2of6patients have a DLT.RadiotherapyPatients received radiation concurrently with chemother-apy to a total dose of50.4Gy,administered at1.8Gy/d,for a total of28days(5days/wk)using aՆ10-MV linear accelerator. Three-dimensional conformal radiation was used to treat all patients.For esophageal and gastric cancers extending to the esophagus,the treatmentfields extended5cm beyond the superior and inferior borders,and2cm beyond the lateral margins.For gastric tumors confined to the stomach,the radia-tionfields encompassed the primary tumor and draining lymph nodes with a2to3cm margin in all directions.The right lateral margins included the portal,hepatic,and common bile duct as well as the head of the pancreas.Postsurgical patients receiveda radiation dose of45Gy delivered in25fractions(1.8Gy/d)for5.5weeks followed by a cone down after45Gy to encompass gross or microscopic disease with a margin of1to1.5cm,for a total dose of50.4Gy.ResponseResponse to treatment was assessed by pre-and posttreat-ment upper endoscopy.Only patients with loco-regional disease had disease assessable for response.A clinical complete re-sponse was defined as no tumor visible on follow-up endoscopy and no tumor on posttreatment biopsy.RESULTSPatient CharacteristicsTwenty-one patients(all Caucasian)were enrolled in this study.Patient characteristics are listed in Table1.Sixteen pa-tients had esophageal cancers,and5patients had gastric cancers extending to the gastroesophageal junction.Twenty patients had adenocarcinomas,and1patient had squamous cell carcinoma. Twelve patients received PPX/RT to treat loco-regional disease. Four patients received PPX/RT for adjuvant therapy after resec-tion,and5patients with metastatic disease at study entry were treated with PPX/RT for local control and symptom palliation. Seven patients had been treated previously with chemotherapy. ToxicityPatients received PPX/RT over5dose levels(see Table2). At the80mg/m2dose level,3of4patients had dose limiting toxicities including grade3esophagitis/gastritis(nϭ2),grade3 dehydration(nϭ1),and grade4neutropenia(nϭ1).One of6 patients at the70mg/m2dose level had grade3esophagitis. There were no grade3or4toxicities at dose levels below PPX 70mg/m2/wk.Dose limiting toxicities did not correlate with either disease site(gastric versus esophageal)or previous che-motherapy treatment.Except for the4patients who experienced a dose limiting toxicity,all patients completed the full6weeks of concurrent chemoradiation.Acute toxicities during chemoradiation are listed in Table3. Multiple toxicities in a single patient are scored as separate events.Esophagitis and dehydration were the most common grade3/4toxicities.One grade2hypersensitivity reaction(urti-caria)was noted among the21patients treated.The patient was TABLE1.Patient CharacteristicsNo.patients enrolled21 SexMale18 Female3 Tumor typeEsophageal16 Gastric5 PathologyAdenocarcinoma20 Squamous cell1 Treatment indicationLoco-regional12 Adjuvant4 Palliation(metastatic)5 Prior chemotherapy7 Median age in years(range)63(41–91)American Journal of Clinical Oncology•Volume29,Number4,August2006PPX-Xyotax and Radiation for Esophagogastric Cancersuccessfully retreated without further hypersensitivity reaction following dexamethasone and diphenhydramine premedication. No alopecia was observed in this study.ResponseTwelve patients had loco-regional disease and were as-sessable for response on posttreatment endoscopy.Four of12 patients(33%)had complete clinical response.An additional7 patients hadϾ50%tumor reduction as assessed by posttreat-ment endoscopy.Loco-regional activity was seen in patients who developed systemic metastases during treatment.DISCUSSIONPaclitaxel is a commonly used radiation sensitizer in multiple tumor types.9–18Paclitaxel synchronizes cells in G2/M, the most radiosensitive phase of the cell cycle,by enhancing microtubule assembly and preventing microtubule depolymer-ization.19–21Paclitaxel has the exceptional property of killing tumor cells in the absence of wild-type p53function in vitro.22 We demonstrated that p53gene mutations are not predictors of unfavorable response to paclitaxel/RT.23Paclitaxel radiosensi-tizes small bowel progenitor cells less than tumor cells poten-tially increasing the therapeutic index of paclitaxel radiosensi-tization in upper gastrointestinal cancers.24PPX is a drug conjugate that links paclitaxel to a biode-gradable polymer,poly-L-glutamic acid.The resulting macro-molecule has several beneficial properties.It is water-soluble, eliminating the need for Cremophor EL and thus reducing the risk of hypersensitivity reactions,and can be given as a10-minute infusion.The hyperpermeable angiogenic vasculature and suppressed lymphatic clearance of tumors facilitate PPX retention within the tumor space,resulting in higher intratumor drug concentrations over prolonged periods.This effect has been described as the enhanced permeability and retention effect(EPR).PPX is absorbed by tumor cells by endocytosis,bypassing the MDR pathway that commonly causes chemotherapy drug resistance.25,26PPX has been evaluated in a phase III trial in patients with performance status2advanced nonsmall cell lung cancer.This study demonstrated a similar response and sur-vival of PPX and carboplatin as compared with paclitaxel and carboplatin.27Radiation increases the vascular permeability of solid tumors.6,7The modulation of the EPR of macromolecules by radiation may underlie the striking preclinical enhancement of radiation and PPX.2This is thefirst clinical trial of PPX and concurrent RT.The maximum tolerated dose of PPX is70 mg/m2/wk with50.4Gy radiation for patients with esophageal and gastric cancer.The toxicities observed in this study of esophagitis,gastritis,and nausea were attributable to PPX in-duced RT enhancement.Substantial preliminary activity was demonstrated in this phase I study with one-third of patients undergoing a complete clinical response to PPX/RT alone.Loco-regional activity was demonstrated in some patients developing systemic progression illustrating that PPXs most important effect was as a radiation sensitizer.We have initiated a phase I study evaluating PPX, cisplatin,and concurrent radiation for patients with esophageal and gastric cancer.Based on the strong preclinical data and the promising preliminary results with PPX/RT,further evaluation of PPX radiosensitization in other human tumors is warranted.REFERENCES1.Singer JW,Baker B,de Vries P,et al.Poly-(L)-glutamic acid–paclitaxel(CT-2103)͓XYOTAXt͔,a biodegradable polymeric drug conjugate.Adv Exp Med Biol.2003;519:81–99.2.Chun Li,Shi Ke,Qing-Ping Wu,et al.Tumor irradiation enhances thetumor-specific distribution of poly(l-glutamic acid)-conjugated pacli-taxel and its antitumor efficacy.Clin Cancer Res.2000;6:2829–2834.3.Li C,Yu DF,Newman RA,et plete regression of well-estab-lished tumors using a novel water-soluble poly(l-glutamic acid)-pacli-taxel conjugate.Cancer Res.1988;58:2404–2409.4.Gerlowski LE,Jain RK.Microvascular permeability of normal andneoplastic tissue.Microvasc Res.1986;31:288–305.5.Maeda H,Matsumura Y.Tumoritropic and lymphotropic principles ofmacromolecular drugs.Crit Rev Ther Drug Carrier Syst.1989;6:193–210.6.Baker DG,Krochak RJ.The response of the microvascular system toradiation:a review.Cancer Invest.1989;7:287–294.7.Chohen FM,Kuwatsuru R,Shames DM,et al.Contrast-enhancedmagnetic resonance imaging estimation of altered capillary permeability in experimental mammary carcinomas after X-irradiation.Invest Radiol.1994;29:970–977.8.Dvorak HF,Brown LF,Detmar M,et al.Vascular permeability factor/vascular endothelial growth factor,microvascular hyperpermeability and angiogenesis.Am J Pathol.1995;146:1029–1039.9.Safran H,King T,Choy H,et al.Paclitaxel and concurrent radiation forlocally advanced pancreatic and gastric cancer:a phase I study.J Clin Oncol.1997;15:901–907.10.Safran H,Wanebo HJ,Hesketh PK,et al.Paclitaxel and concurrentradiation for gastric cancer.Int J Radiat Oncol Biol Phys.2000;46:889–894.TABLE2.Dose Limiting Toxicities(DLTs)PPX Dose (mg/m2/wk)No.PatientsToxicities(grade3–4)DehydrationGastritis/Esophagitis Weakness ANC40300005040000604000070601008042211TABLE3.ToxicityՆGrade2No.Patients(n؍21)Grade2Grade3Grade4NonhematologicalEsophagitis/gastritis530Nausea/vomiting310Anorexia400Dehydration720Fatigue/weakness610Infection200Hypersensitivity100HematologicalNeutropenia001Thrombocytopenia110Dipetrillo et al American Journal of Clinical Oncology•Volume29,Number4,August200611.Safran H,Gaissert H,Akerman P,et al.Paclitaxel,cisplatin andconcurrent radiation for esophageal cancer.Cancer Invest.2001;1:1–7.12.Safran H,Moore T,Iannitti D,et al.Paclitaxel and concurrent radiationfor locally advanced pancreatic cancer.Int J Radiat Oncol Biol Phys.2001;49:1275–1279.13.Ajani JA,Mansfield PF,Crane CH,et al.Paclitaxel-based chemoradio-therapy in localized gastric carcinoma:degree of pathologic response and not clinical parameters dictated patient outcome.J Clin Oncol.2005;23:1237–1244.14.Brenner B,Ilson DH,Minsky BD,et al.Phase I trial of combined-modality therapy for localized esophageal cancer:escalating doses of continuous-infusion paclitaxel with cisplatin and concurrent radiation therapy.J Clin Oncol.2004;22:45–52.15.Choy H,Akerley W,Safran H,et al.A phase I trial of concurrent weeklytaxol administered as a3hour infusion and radiation therapy for advanced non-small cell lung cancer.J Clin Oncol.1994;12:2682–2686.16.Choy H,Akerley W,Glantz M,et al.Concurrent paclitaxel and radiationtherapy for solid tumors.Cancer Control.1996;3:310–318.17.Choy H,Safran H,Akerley W,et al.Phase II trial of weekly paclitaxeland concurrent radiation therapy for locally advanced non-small cell lung cancer.Clin Cancer Res.1998;4:1931–1936.18.Choy H,Akerley W,Safran H,et al.Multi-institutional phase II trial ofpaclitaxel,carboplatin and concurrent radiation therapy for locally ad-vanced non-small cell lung cancer.J Clin Oncol.1998;16:3316–3322.19.Sinclair WK,Morton RA.X-ray sensitivity during the cell generationcycle of cultured Chinese hamster cells.Radiat Res.1996;29:450–474.20.Liebmann J,Cook JA,Fisher J,et al.In vitro studies of taxol as aradiation sensitizer in human tumor cells.J Natl Cancer Inst.1994;86: 441–446.21.Minarik L,Hall EJ.Taxol in combination with acute and low dose rateirradiation.Radiother Oncol.1994;32:124–128.22.Wahl AF,Donaldson KL,Fairchild C,et al.Loss of normal p53functionconfers sensitization to Taxol by increasing G2/M arrest and apoptosis.Nature Med.1996;2:72–79.23.King T,Akerley W,Fan A,et al.p53mutations do not predict responseto paclitaxel in metastatic non-small cell lung cancer.Cancer.2000;89: 769–773.24.Mason KA,Milas L,Peters LJ.Effect of paclitaxel(taxol)alone and incombination with radiation on the gastrointestinal mucosa.Int J Radiat Oncol Biol Phys.1997;38:623–631.25.Duncan R.The dawning era of polymer therapeutics.Nat Rev DrugDiscov.2003;2:347–360.26.McCormick-Thomson LA,Duncan R.Poly(amino acid)copolymers asa potential soluble drug delivery system,I:pinocytic uptake and lyso-somal degradation measured in vitro.J Bioact Compat Polym.1989;4: 242–251.nger CJ,Socinski MA,Ross H,et al.Paclitaxel poliglumex(PPX)/carboplatin vs paclitaxel/carboplatin for the treatment of PS2 patients with chemotherapy-naı¨ve advanced non-small cell lung can-cer(NSCLC):a phase III study.Proc Am Soc Clin Oncol.2005;LBA: 7011.American Journal of Clinical Oncology•Volume29,Number4,August2006PPX-Xyotax and Radiation for Esophagogastric Cancer。