生物信息学分析实例
生物信息学实验报告3(三)蛋白质序列分析

⽣物信息学实验报告3(三)蛋⽩质序列分析(三)蛋⽩质序列分析实验⽬的:掌握蛋⽩质序列检索的操作⽅法,熟悉蛋⽩质基本性质分析,了解蛋⽩质结构分析和预测。
实验内容:1、检索SOX-21蛋⽩质序列,利⽤ProParam⼯具进⾏蛋⽩质的氨基酸组成、分⼦质量、等电点、氨基酸组成、原⼦总数及疏⽔性(ProtScale⼯具)等理化性质的分析。
2、利⽤PredictProtein、PROF、HNN等软件预测分析蛋⽩质的⼆级结构;利⽤Scan Prosite软件对蛋⽩质进⾏结构域分析。
3、利⽤TMHMM、TMPRED、SOSUI等⼯具对蛋⽩质进⾏跨膜分析;采⽤PredictNLS进⾏核定位信号分析;利⽤PSORT进⾏蛋⽩质的亚细胞定位预测;利⽤CBS(http://www.cbs.dtu.dk/services/ProtFun/)⽹站⼯具预测蛋⽩的功能,将序列⽤Blocks、SMART、InterProScan、PFSCAN等搜索其保守序列的特征,进⾏motif 的结构分析。
4、利⽤Swiss-Model数据库软件预测该蛋⽩的三级结构,结果⽤蛋⽩质三维图象软件Jmol查看。
CPHmodels 也是利⽤神经⽹络进⾏同源模建预测蛋⽩质结构的⽅法和⽹络服务器I-TASSER预测所选蛋⽩质的空间结构。
5、分析蛋⽩质的翻译后修饰:分析信号肽及其剪切位点: SignalIP http://www.cbs.dtu.dk/services/SignalP/;分析糖链连接点:分析O-连接糖蛋⽩,NetOGlyc,http://www.cbs.dtu.dk/services/NetOGlyc/;分析N-连接糖蛋⽩,NetNGlyc,http://www.cbs.dtu.dk/services/NetNGlyc/。
6、利⽤检索的序列,进⾏同源⽐对,获得并分析⽐对结果。
实验步骤(⼀)1、在NCBI 蛋⽩质数据库中查找SOX-21蛋⽩质序列分别选择⽖蟾(Xenopus laevis)、⼩家⿏[Mus musculus]、猕猴[Macaca mulatt a]的SOX-21蛋⽩质序列,并保存其FASTA格式。
生物信息学在疾病检测方面的实例

生物信息学在疾病检测方面的实例引言:随着科技的发展,生物信息学在疾病检测领域发挥着越来越重要的作用。
通过对DNA、RNA和蛋白质等生物大分子的序列和结构信息进行分析,生物信息学可以帮助我们更加准确地诊断和预测疾病。
本文将以多个实例为例,介绍生物信息学在疾病检测方面的应用。
实例一:基因突变与遗传疾病生物信息学可以帮助我们分析基因突变与遗传疾病之间的关系。
例如,在遗传性疾病中,特定基因的突变可能导致蛋白质结构异常,进而引发疾病。
通过生物信息学的方法,我们可以对这些突变进行分析,了解突变对蛋白质功能的影响,从而预测疾病的发生和发展趋势。
实例二:基因表达与肿瘤疾病生物信息学可以帮助我们分析基因表达与肿瘤疾病之间的关系。
通过对肿瘤细胞中基因表达谱的分析,我们可以发现与肿瘤相关的基因。
这些基因的表达水平的异常可能与肿瘤的发生和发展相关。
生物信息学的方法可以帮助我们对大规模的基因表达数据进行挖掘和分析,以便发现新的肿瘤标志物和治疗靶点。
实例三:药物设计与疾病治疗生物信息学可以帮助我们设计和优化药物,从而提高疾病治疗效果。
通过对药物和靶标蛋白的结构信息进行分析,我们可以预测药物与靶标蛋白的结合模式,并设计出更加有效的药物。
此外,生物信息学还可以帮助我们筛选药物靶点,加快药物研发的速度和效率。
实例四:个体化医疗与疾病预测生物信息学可以帮助我们进行个体化医疗和疾病预测。
通过对个体基因组的测序和分析,我们可以了解个体的遗传变异情况,并预测个体患某种疾病的风险。
此外,生物信息学还可以结合临床数据和生活习惯等信息,从而为个体提供更加个性化的医疗和健康管理方案。
结论:生物信息学在疾病检测方面的应用不断发展,为我们提供了更加准确和个性化的疾病诊断和预测手段。
通过对基因突变、基因表达、药物设计和个体基因组等信息的分析,生物信息学可以帮助我们更好地理解疾病的发生和发展机制,为疾病的预防、诊断和治疗提供支持和指导。
随着生物信息学技术的不断进步,相信生物信息学在疾病检测方面的应用将会越来越广泛,并为人类的健康事业做出更大的贡献。
生物信息学(五篇范例)

生物信息学(五篇范例)第一篇:生物信息学生物信息学(Bioinformatics)是在生命科学的研究中,以计算机为工具对生物信息进行储存、检索和分析的科学。
它是当今生命科学和自然科学的重大前沿领域之一,同时也将是21世纪自然科学的核心领域之一。
其研究重点主要体现在基因组学(Genomics)和蛋白质组学(Proteomics)两方面,具体说就是从核酸和蛋白质序列出发,分析序列中表达的结构功能的生物信息。
具体而言,生物信息学作为一门新的学科领域,它是把基因组DNA序列信息分析作为源头,在获得蛋白质编码区的信息后进行蛋白质空间结构模拟和预测,然后依据特定蛋白质的功能进行必要的药物设计。
基因组信息学,蛋白质空间结构模拟以及药物设计构成了生物信息学的3个重要组成部分。
从生物信息学研究的具体内容上看,生物信息学应包括这3个主要部分:(1)新算法和统计学方法研究;(2)各类数据的分析和解释;(3)研制有效利用和管理数据新工具。
生物信息学是一门利用计算机技术研究生物系统之规律的学科。
目前的生物信息学基本上只是分子生物学与信息技术(尤其是因特网技术)的结合体。
生物信息学的研究材料和结果就是各种各样的生物学数据,其研究工具是计算机,研究方法包括对生物学数据的搜索(收集和筛选)、处理(编辑、整理、管理和显示)及利用(计算、模拟)。
1990年代以来,伴随着各种基因组测序计划的展开和分子结构测定技术的突破和Internet的普及,数以百计的生物学数据库如雨后春笋般迅速出现和成长。
对生物信息学工作者提出了严峻的挑战:数以亿计的ACGT序列中包涵着什么信息?基因组中的这些信息怎样控制有机体的发育?基因组本身又是怎样进化的?生物信息学的另一个挑战是从蛋白质的氨基酸序列预测蛋白质结构。
这个难题已困扰理论生物学家达半个多世纪,如今找到问题答案要求正变得日益迫切。
诺贝尔奖获得者W.Gilbert在1991年曾经指出:“传统生物学解决问题的方式是实验的。
举例说明生物信息学的主要应用

举例说明生物信息学的主要应用生物信息学是一个跨学科的领域,将计算机科学、统计学和生物学相结合,利用大数据和信息技术来分析生物学数据。
它在当前的科学研究和医学领域发挥着重要的作用。
下面将举几个生物信息学的主要应用例子,以展示它的广泛应用和重要性。
1. 基因组学研究:基因组学是生物信息学的重要应用之一。
通过对多种生物体基因组的测序和比较分析,可以揭示基因组结构和功能之间的关系,以及基因组变异对生物特征和疾病的影响。
例如,人类基因组计划就是通过生物信息学的方法完成了人类基因组的测序和分析,为后续疾病研究和医学个性化治疗提供了基础。
2. 蛋白质结构预测:蛋白质是生物体中功能最为重要的分子之一。
通过生物信息学的方法,可以根据蛋白质的序列信息来预测其三维结构,从而揭示其功能和相互作用。
这对于药物设计和疾病治疗非常重要。
例如,许多药物的研发过程中都会使用蛋白质结构预测来进行虚拟筛选,以提高筛选效率。
3. 转录组学研究:转录组学是研究生物体基因表达的一种方法,通过测定和分析特定时间和空间点上的RNA序列来揭示基因调控网络。
生物信息学的方法可以帮助我们挖掘转录组数据中隐藏的模式和规律,从而深入理解基因调控的机制。
这为研究生物体发育、疾病发生和治疗提供了重要的线索。
4. 肿瘤基因组学研究:肿瘤是生物信息学的一个重要应用领域。
通过分析肿瘤中的基因组信息,可以发现潜在的致癌基因、突变和其他遗传变异,为肿瘤的早期诊断和治疗选择提供依据。
例如,通过测序和比较正常细胞和癌细胞的基因组,可以发现癌症相关的驱动基因,从而为个性化治疗奠定基础。
5. 生物多样性研究:生物信息学在生物多样性研究中也发挥着重要作用。
通过对全球各地生物样本的DNA测序和分析,可以揭示物种的遗传多样性和进化关系。
这对于保护生物多样性、发现新的物种和了解生态系统的功能具有重要意义。
综上所述,生物信息学在基因组学、蛋白质结构预测、转录组学、肿瘤基因组学和生物多样性研究等领域发挥着重要的作用。
生物信息学分析实例

Bioinformatics
第4步:提交计算脚本
[zouly@big mauve-test]$ qsub mauve-test.qsub 注意:非并行程序不需要指定CPU数量 记录比对信息的文件:/disk1/zouly/ec_sf1_sf2.mauve 记录比对结果的文件:/disk1/zouly/ec_sf1_sf2.alignment
Bioinformatics
第3步:编写计算脚本文件mauve-test.sge
[zouly@big mauve-test]$ vi mauve-test.qsub #!/bin/bash #$ -cwd #$ -j y #$ -S /bin/bash # /disk1/biosoft/mauve_2.3.1/linux-x64/mauveAligner --output =ec_sf1_sf2.mauve --output-alignment=ec_sf1_sf2.alignment NC_000913.gbk NC_000913.gbk .sml NC_011353.gbk NC_011353.gbk .sml NC_004337.gbk NC_004337.gbk .sml NC_004741.gbk NC_004741.gbk .sml NC_011283.gbk NC_011283.gbk .sml NC_012731.gbk NC_012731.gbk.sml
Bioinformatics
生物信息学高性能计算平台 应用实例分析
Bioinformatics Center Lingyun Zou
生物信息学在转录组富集分析中的应用

生物信息学在转录组富集分析中的应用一、生物信息学概述生物信息学是一门交叉学科,它结合了生物学、计算机科学、数学和统计学等多学科知识,以研究生物数据的获取、存储、分析和解释。
随着高通量测序技术的发展,生物信息学在转录组学研究中扮演着越来越重要的角色。
转录组富集分析是生物信息学中的一项关键技术,它可以帮助研究者识别和量化基因表达的变化,从而揭示生物体在不同状态下的分子机制。
1.1 生物信息学的核心领域生物信息学的核心领域包括基因组学、转录组学、蛋白质组学和代谢组学等。
这些领域通过分析生物体的遗传信息、基因表达模式、蛋白质结构与功能以及代谢途径,为理解生命过程提供了重要视角。
1.2 生物信息学的应用场景生物信息学的应用场景非常广泛,包括但不限于以下几个方面:- 疾病机理研究:通过分析疾病状态下的基因表达变化,揭示疾病发生的分子机制。
- 药物靶点发现:利用生物信息学方法预测药物作用的分子靶点,加速新药研发。
- 个体化医疗:根据个体的基因组信息,为患者提供个性化的治疗方案。
二、转录组富集分析的基本原理转录组富集分析是一种定量分析基因表达水平的方法,它通过比较不同样本或条件下的基因表达差异,识别出表达量显著变化的基因。
这一过程通常涉及以下几个步骤:2.1 数据获取首先,需要通过高通量测序技术,如RNA测序(RNA-Seq),获取样本的转录组数据。
这些数据包含了样本中所有RNA分子的序列信息。
2.2 数据处理获取的原始测序数据需要经过质量控制、序列比对、转录本组装等步骤,以确保数据的准确性和可靠性。
2.3 表达量定量利用生物信息学工具,如Cufflinks、eXpress等,对转录本的表达量进行定量分析,计算每个基因的表达水平。
2.4 差异表达分析通过比较不同样本或条件下的基因表达水平,使用统计学方法,如DESeq2、edgeR等,识别出差异表达的基因。
2.5 结果解释与验证对差异表达的基因进行功能注释和富集分析,以理解其生物学意义。
Python数据分析实战之生物信息学数据分析案例

Python数据分析实战之生物信息学数据分析案例生物信息学是生命科学与信息科学相结合的交叉学科,它通过对生物数据的收集、处理和分析,揭示生物学中的规律和机制。
Python作为一种强大的编程语言,在生物信息学领域也得到了广泛的应用。
本文将介绍Python在生物信息学数据分析方面的实战案例,带您领略Python在解决生物学问题上的威力。
1. 数据获取与预处理在生物信息学数据分析中,数据的获取和预处理是至关重要的步骤。
我们常常需要从公共数据库如NCBI、Ensembl等下载生物数据,并对其进行清洗和格式转换以便后续分析。
使用Python的`Biopython`库可以方便地实现这一步骤,例如:```pythonfrom Bio import SeqIO# 从GenBank下载序列数据seq_record = SeqIO.read("sequence.gb", "genbank")# 清洗数据,去除无用信息clean_seq = clean_data(seq_record.seq)# 将序列保存为FASTA格式文件SeqIO.write(clean_seq, "clean_sequence.fasta", "fasta")```2. 序列分析与比对生物信息学中常见的任务之一是对生物序列进行分析和比对,以寻找序列之间的相似性和差异性。
Python提供了丰富的工具和库来实现这些功能,例如`Biopython`中的`Seq`和`Align`模块:```pythonfrom Bio.Seq import Seqfrom Bio.Align import pairwise2# 创建序列对象seq1 = Seq("ATCGATCG")seq2 = Seq("ATGGATCG")# 序列比对alignments = pairwise2.align.globalxx(seq1, seq2)```3. 基因组学数据分析基因组学数据分析是生物信息学中的重要分支,涉及到对基因组序列、基因结构和基因组功能的研究。
生物信息学技术在医学研究中的应用案例分析

生物信息学技术在医学研究中的应用案例分析概述:生物信息学是利用计算机科学和信息学原理来解决生物学问题的一门学科。
随着技术的快速发展和数据量的急剧增加,生物信息学在医学研究中的应用变得越来越重要。
本文将通过分析几个具体的案例,探讨生物信息学技术在医学研究中的应用及其优势。
1. 基因组学和转录组学基因组学和转录组学是生物信息学在医学研究中最常应用的技术之一。
通过对基因组和转录组的研究,可以揭示基因和基因表达与疾病之间的关联。
例如,在癌症研究中,研究人员可以通过测序和分析癌细胞和正常细胞的基因组和转录组数据,识别突变和差异表达的基因,并从中发现与癌症发展相关的重要调控网络和信号通路。
这些发现有助于揭示癌症发生的机制,为精准医学和个体化治疗提供基础。
2. 蛋白质组学蛋白质组学研究的是细胞或生物体内所有蛋白质的组成、结构、功能和相互作用。
生物信息学技术在蛋白质组学中的应用主要包括蛋白质结构预测和蛋白质-蛋白质相互作用的预测。
通过预测蛋白质结构和相互作用,可以加速药物研发过程。
例如,通过计算蛋白质的结构,可以预测药物与蛋白质结合的方式和位置,提高药物的设计和筛选效率。
此外,还可以通过预测蛋白质-蛋白质相互作用来探索疾病内部的信号通路,并发现新的药物靶点。
3. 肿瘤基因组学肿瘤基因组学是生物信息学在肿瘤研究中的一项重要应用。
通过对肿瘤样本中的基因组数据进行分析,可以发现与肿瘤发生、发展和治疗相关的重要基因和变异。
例如,在肿瘤突变分析中,研究人员通过对肿瘤样本的全外显子组测序,可以发现存在的突变,这些突变可能是驱动肿瘤生长和扩散的关键因素。
这些发现可以帮助医生选择合适的治疗策略,并为个体化治疗提供指导。
4. 药物设计和筛选对药物的设计和筛选一直是医学研究中的难题之一。
生物信息学技术在药物设计和筛选中的应用可以大大缩短研发周期和降低研发成本。
例如,通过利用计算机模型和模拟技术,可以预测药物与靶点的亲和力和选择性,进而指导合理的药物设计和优化。
生信分析实例[G]
![生信分析实例[G]](https://img.taocdn.com/s3/m/44d167e8102de2bd960588ed.png)
抗原决定簇预测 抗原决定簇是指能与抗原相应抗体结合的抗原上有限部位的特殊分子结构,也称为表位 (epitope)。 哈佛大学的 Predicting Antigenic Peptides 预测软件是使用 Kolaskar 和 Tongaonkar(1990)方法 预测氨基酸序列内可能引起抗体反应的抗原片段。 以生物信息学为基础的蛋白质抗原决定簇的预测对于分子生物学实验,如诊断试剂的制备、 抗体制备筛选等,都是必不可少的工具,减少了了实验研究的盲目性(万涛等,1997)。 据报道,运用 Predicting Antigenic Peptides 预测软件并结合多种方法综合分析预测的成功率 可达 86%(孙沫逸等,2003)。 /Tools/antigenic.pl 实例:RGDV Pns9 基因 RGDV S9 片段可能有两个开放阅读框(ORF),另一阅读框位于主阅读框下游,但目前尚未获 得该 ORF 表达的实验证据。 主 ORF 转入表达载体 pGEX-4T-1 或 pET29-a 均无法得到成功表达,故目前无法获取完整 Pns9 蛋白的抗血清。 原因分析:使用生物学软件 RNAstructure 4.2 采用最小自由能法(总能量值=283.3kcal/mol)折 叠 S9 片段主 ORF 对应的核苷酸序列,发现核苷酸序列开始位置的第 10-36 碱基间形成强势 的发夹状结构。
选择强抗原性肽段的原则 • 肽段长度12-15个氨基酸; • 肽段内无4个以上连续相邻的疏水性残基,疏水性残基数目<6,带正电荷氨基酸越 多越好; • 亲水性及可及性参数均较高; • 肽段位于转角附近,易形成无规则卷曲。 • 蛋白质的羧基端非常理想(灵活性、暴露性)
生信分析实例[G]
![生信分析实例[G]](https://img.taocdn.com/s3/m/e01d75c45fbfc77da269b1f2.png)
Blast result of amino acid sequence for Major ORF of RGDV Seg8
核苷酸序列=>氨基酸序列
• ExPASy 上的Translate tool /tools/dna.html
• 生物学软件BioEdit a) 查看密码子用法(Codon Usage Database ) b) 整理制作密码子使用频率表 c) 翻译成氨基酸
或Oligonucleotide Properties Calculator /biotools/oligocalc.html
序列同源分析 (By BLAST 2 SEQUENCES)
序列矩阵图示意
序列同源 序列易位 序列交换 序列插入
蛋白功能分析
理化性质
理论pI、亲/疏水性、 不稳定系数等
功能域
跨膜螺旋、信号肽、 结构功能域、亚细胞 定位等
M
Motif 搜索
-PORSITE数据库、 -ProfileScan数据库
三维结构模拟
-同源模建 -折叠识别
二级结构预测
PHD 神经网络
蛋白质理化性质分析
• 在线分析 • ExPasy服务器上的ProtParam • /tools/protparam.html
• 核苷酸序列的基本分析 • 分子量、碱基组成 • 序列变换-反向、反向互补、转换为RNA序列等 • 限制性酶切分析
• 基因结构分析: • 启动子及转录因子结合位点 • 重复序列 • CpG Island (HTF Island) • ORF分析(内含子/外显子分析)
• 电子克隆(e-PCR) • 利用EST数据库的重叠序列克隆新基因
>20h >10h 37.11 85.34 -0.243
生物信息学在疾病检测方面的实例

生物信息学在疾病检测方面的实例随着科技的不断进步,生物信息学作为一门新兴的交叉学科,已经在疾病检测方面发挥着越来越重要的作用。
通过对基因组、蛋白质组和代谢组的研究,生物信息学可以帮助科学家发现疾病的潜在机制,提供新的治疗策略,并为个体化医疗提供支持。
以下将以几个实际例子来说明生物信息学在疾病检测方面的应用。
例子1:基因组学在肿瘤研究中的应用肿瘤是世界范围内的一种主要疾病,生物信息学在肿瘤研究中扮演着重要的角色。
通过对大规模癌症基因组数据的分析,科学家可以发现不同癌症类型之间的遗传变异,从而识别出致病基因。
例如,研究人员通过对乳腺癌患者基因组数据的分析,发现了BRCA1和BRCA2基因的突变与乳腺癌的高风险相关。
这些发现为乳腺癌的早期检测和个体化治疗提供了依据。
例子2:蛋白质组学在糖尿病研究中的应用糖尿病是一种常见的慢性代谢性疾病,生物信息学在糖尿病研究中起到了重要的作用。
通过对大规模蛋白质组数据的分析,科学家可以发现与糖尿病相关的蛋白质标志物,并进一步研究其功能和调控机制。
例如,在一项研究中,科学家通过对糖尿病患者和健康人群的蛋白质组数据进行比较,发现了一种与糖尿病相关的蛋白质标志物,并验证了其在糖尿病发生发展过程中的重要作用。
这些发现为糖尿病的早期诊断和治疗提供了新的思路。
例子3:代谢组学在心血管疾病研究中的应用心血管疾病是全球范围内的主要死因之一,生物信息学在心血管疾病研究中也发挥着重要作用。
通过对大规模代谢组数据的分析,科学家可以发现与心血管疾病相关的代谢物,并进一步研究其调控机制和生物学功能。
例如,在一项研究中,科学家通过对心血管疾病患者和健康人群的代谢组数据进行比较,发现了一种与心血管疾病风险相关的代谢物,并验证了其在心血管疾病发生发展过程中的重要作用。
这些发现为心血管疾病的早期预测和治疗提供了新的线索。
生物信息学在疾病检测方面的应用已经取得了显著的进展。
通过对基因组、蛋白质组和代谢组的研究,生物信息学可以帮助科学家发现疾病的潜在机制,提供新的治疗策略,并为个体化医疗提供支持。
生物信息学分析在肿瘤诊疗中的应用研究

生物信息学分析在肿瘤诊疗中的应用研究随着科技的发展,生物信息学分析在肿瘤诊疗中的应用已成为当今医学领域的一个热点研究方向。
生物信息学,指的是运用计算机技术对生命科学中的大量数据进行整合、分析和解读的一门综合性学科。
而在肿瘤诊疗方面,生物信息学则是指利用现代计算技术、生物技术和统计学方法对肿瘤相关基因、蛋白质、信号通路等多种生物信息进行挖掘和分析,以实现对肿瘤的更准确、更个性化的诊断和治疗。
一、生物信息学分析在肿瘤诊疗中的应用生物信息学分析在肿瘤诊疗中的应用主要体现在以下几个方面:1. 肿瘤的基因检测和基因分型现代生物学研究发现,几乎所有的肿瘤都是由基因突变导致的,而基因突变又是导致肿瘤发生的主要因素。
因此,对肿瘤的基因检测和基因分型已经成为肿瘤诊疗中的重要环节。
随着高通量测序技术的发展,现在可以快速检测出数千种基因的突变情况,并帮助医生更好地诊断肿瘤的类型、预测肿瘤的进展和治疗效果。
这些基因检测和基因分型的结果可以为肿瘤诊疗提供有力的依据,帮助医生制定更合理的治疗方案。
2. 肿瘤基因组学分析生物信息学分析可以帮助科学家和医生研究肿瘤的基因组学特征,包括基因变异、基因表达、RNA处理、蛋白质结构和功能等,从而深入了解肿瘤细胞生长、分化和转移的机制。
基因组学分析还可以揭示肿瘤发展的分子机制,探究肿瘤的起源和发展过程,为肿瘤诊断和治疗提供更准确的依据。
3. 肿瘤免疫治疗随着肿瘤免疫治疗的发展,生物信息学分析也为肿瘤免疫治疗提供了有力的支持。
肿瘤免疫治疗利用免疫系统的天然防御机制,激发机体的自身免疫反应来杀灭肿瘤细胞。
生物信息学分析可以帮助医生筛选出具有潜在免疫治疗反应的患者,并确定最佳的免疫治疗方案,从而提高免疫治疗的疗效和安全性。
4. 肿瘤筛查和早期诊断生物信息学分析还可以帮助实现肿瘤的早期筛查和诊断。
通过对大规模的肿瘤数据进行挖掘和分析,可以找到与肿瘤发展相关的生物标志物和代谢物,从而研制出更精准的肿瘤筛查和早期诊断试剂。
高通量测序的生物信息学分析报告
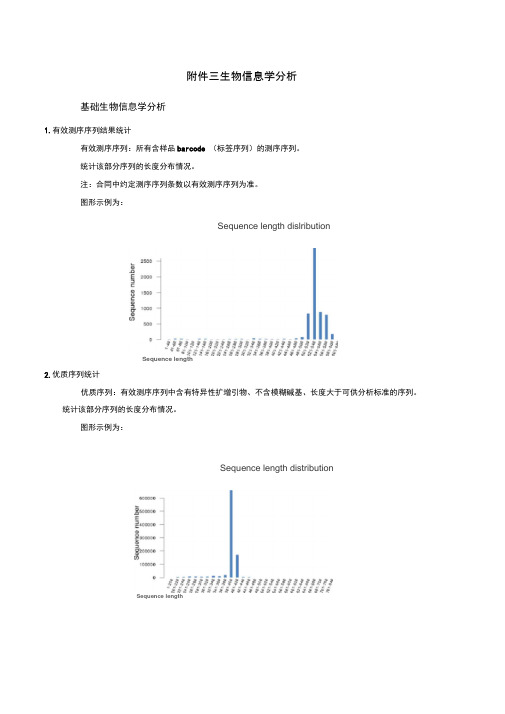
附件三生物信息学分析基础生物信息学分析1.有效测序序列结果统计有效测序序列:所有含样品barcode (标签序列)的测序序列。
统计该部分序列的长度分布情况。
注:合同中约定测序序列条数以有效测序序列为准。
图形示例为:Sequence length dislributionSequence length2.优质序列统计优质序列:有效测序序列中含有特异性扩增引物、不含模糊碱基、长度大于可供分析标准的序列。
统计该部分序列的长度分布情况。
图形示例为:Sequence length distributionSequence length3.各样本序列数目统计:统计各个样本所含有效测序序列和优质序列数目。
结果示例为:4. OTU生成:根据序列的相似性,将序列归为多个OTU(操作分类单元),以便后续分析。
5. 稀释曲线(rarefaction 分析)根据第4条中获得的OTU数据,做出每个样品的Rarefaction 曲线。
本合同默认生成OTU相似水平为0.03 的rarefaction 曲线。
rarefaction 曲线结果示例:Number of R«ads SampladM; 0.036. 指数分析计算各个样品的相关分析指数,包括:丰度指数:ace'chao多样性指数: sha nnon\simps on本合同默认生成OTU 相似水平为0.03的上述指数值。
多样性指数分析结果示例:IDR HM 4H机MOTU■chM|A2W0MO ICNMtlOOO.iOM}S(KS) J O L WCO,UM亠帼血期’ th注:默认分析以上所列指数,如有特殊需要请说明7. Shannon-Wiener 曲线利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线, 反映各样本在不同测序数量 时的微生物多样性。
当曲线趋向平坦时, 说明测序数据量足够大, 可以反映样品中绝大多数的微生物信 息。
绘制默认水平为:0.03。
基于生物信息学的胰腺癌预后关键基因及通路分析

基于生物信息学的胰腺癌预后关键基因及通路分析基于生物信息学的胰腺癌预后关键基因及通路分析胰腺癌是一种高度侵袭性的肿瘤,其发病率和死亡率均居世界癌症统计前列。
胰腺癌的预后与许多因素密切相关,其中最重要的是基因及其通路的异常激活。
生物信息学技术的应用使得我们能够在基因组水平上更深入地理解胰腺癌的预后。
本文将基于生物信息学的方法,以胰腺癌的预后为中心,探讨关键基因及其通路的分析。
胰腺癌预后关键基因的发现是基于大规模基因表达数据的分析。
我们将胰腺癌病人的基因表达数据与他们的生存信息进行比较,筛选出与预后相关的基因。
通过差异表达分析,我们发现了一些关键基因,如KRAS、TP53和CDKN2A等。
这些基因的异常表达与胰腺癌的预后密切相关。
进一步地,我们对这些关键基因进行功能富集分析,从而获得它们参与的关键通路。
通过生物信息学数据库的查询,我们发现胰腺癌预后关键基因参与了多种重要的通路,如细胞周期、MAPK信号通路和PI3K信号通路等。
这些通路的异常激活会促进胰腺癌的恶性发展和预后的恶化。
此外,我们还通过构建基因共表达网络,探索这些关键基因之间的相互作用。
基于共表达网络的分析,我们可以发现一些关键的基因模块,这些模块在胰腺癌预后中发挥重要作用。
通过重新分析这些模块,我们发现了新的关键基因,如BIRC5和EGFR等。
这些基因可能会进一步影响预后相关的通路,从而影响胰腺癌病人的生存。
综上所述,基于生物信息学的方法为胰腺癌预后关键基因及其通路的分析提供了重要的工具。
通过分析基因表达数据、功能富集分析以及基因共表达网络的构建,我们能够深入了解胰腺癌的预后机制。
这些研究结果为胰腺癌的预后评估提供了新的理论和实践基础,为胰腺癌的精准治疗和个体化医疗提供了新的思路。
然而,需要指出的是,本文仅仅是基于生物信息学的分析结果,仍然需要进一步的实验验证来验证这些关键基因及其通路的预后效果。
此外,胰腺癌是一种高度复杂的疾病,还有许多其他因素可能影响其预后,这需要在未来的研究中进一步探索综合以上研究结果,我们发现胰腺癌预后关键基因参与了多种重要的通路,如细胞周期、MAPK信号通路和PI3K信号通路等。
生物信息学分析范文

生物信息学分析范文生物信息学的应用非常广泛,主要包括基因组学、蛋白质组学和转录组学等方面。
在基因组学中,生物信息学可以用于预测基因的结构和功能,鉴定基因的变异和突变,以及研究基因的演化和分布。
在蛋白质组学中,生物信息学可以用于预测蛋白质的结构和功能,鉴定蛋白质的修饰和相互作用,以及研究蛋白质的表达和调控。
在转录组学中,生物信息学可以用于分析基因的转录和表达,鉴定基因的调控元件和信号通路,以及研究基因的功能和调控网络。
生物信息学的方法主要包括序列比对、结构预测和功能注释等方面。
序列比对是生物信息学中最常用的方法之一,它可以用于比较不同物种或样本的基因组、蛋白质或RNA序列,以及分析它们的相似性和差异性。
结构预测是生物信息学中另一个重要的方法,它可以用于预测蛋白质的三维结构,以及分析蛋白质的稳定性和功能。
功能注释是生物信息学中最有挑战性的方法之一,它可以用于预测基因或蛋白质的功能、鉴定代谢途径和信号通路,以及研究基因或蛋白质的功能调控网络。
生物信息学在基因组学中的应用非常广泛。
通过分析基因组的序列和结构,生物信息学可以用于预测基因的结构和功能,鉴定基因的变异和突变,以及研究基因的演化和分布。
例如,通过比对不同物种或样本的基因组序列,可以鉴定基因的保守区域和编码区域,以及分析它们的相似性和差异性。
同时,生物信息学还可以预测基因的启动子、转录因子结合位点和DNA甲基化位点,以及研究基因的调控网络和表达模式。
生物信息学在蛋白质组学中的应用也非常重要。
通过分析蛋白质的序列和结构,生物信息学可以用于预测蛋白质的结构和功能,鉴定蛋白质的修饰和相互作用,以及研究蛋白质的表达和调控。
例如,通过比对不同物种或样本的蛋白质序列,可以鉴定蛋白质的保守区域和功能域,以及分析它们的相似性和差异性。
同时,生物信息学还可以预测蛋白质的二级结构、三级结构和配体结合位点,以及研究蛋白质的功能调控网络和信号通路。
生物信息学在转录组学中的应用也越来越重要。
生物信息学的应用举例

生物信息学的应用举例生物信息学是一门整合生物学、计算机科学和统计学的交叉学科,它的应用范围越来越广泛,下面列举一些生物信息学的应用。
1. 基因组学研究生物信息学在基因组学研究中起着重要作用,可以通过测序、比对、注释等方法对基因组进行全面的分析。
比如,人类基因组计划就是利用生物信息学技术完成的。
2. 蛋白质组学研究生物信息学在蛋白质组学研究中也有广泛的应用,可以通过蛋白质质量谱、蛋白质结构预测等方法对蛋白质进行研究。
3. 基因功能预测生物信息学可以通过分析基因序列、比对已知基因和蛋白质序列等方法预测基因的功能。
这对基因治疗、疾病诊断等方面具有重要意义。
4. 基因表达谱分析生物信息学可以对基因表达谱进行分析,研究在不同生理状态下基因的表达情况。
这对了解基因调控机制、疾病发生机制等方面具有重要意义。
5. 药物研发生物信息学可以通过分析蛋白质结构、药物分子与蛋白质相互作用等方法来辅助药物研发,并且可以通过药物代谢途径分析、药物副作用预测等方法来提高药物研发的效率和成功率。
6. 食品安全监测生物信息学可以通过对食品样本进行基因分析、毒素检测等方法来保证食品安全。
比如,通过对食品中病原菌基因的检测,可以及时发现并防止食品污染事件的发生。
7. 生物多样性研究生物信息学可以对不同物种的基因组进行比对和分析,从而研究物种间的进化关系、生态环境和群体遗传学等方面的问题,对生物多样性的研究具有重要意义。
8. 个性化医疗生物信息学可以通过对个体基因组的分析来实现个性化医疗。
比如,通过对癌症病人的基因组分析,可以确定最佳治疗方案,提高治疗效果。
9. 植物基因改良生物信息学可以对植物基因组进行分析,发现有利基因,优化植物品种,提高农作物的产量和质量。
10. 生物信息学教育生物信息学已成为生命科学中不可或缺的领域,生物信息学教育也越来越重要。
通过生物信息学教育,可以培养生命科学领域的专业人才,推动生物信息学的发展和应用。
生物信息学在新药研发中的应用案例研究

生物信息学在新药研发中的应用案例研究引言:在当今的药物研发领域中,生物信息学成为了不可或缺的工具。
生物信息学是将计算机科学、统计学和生物学相结合的交叉学科,通过对生物数据的分析和解释,可以为药物研发提供重要的支持和指导。
本文将介绍几个生物信息学在新药研发中的成功应用案例,展示了其在发现靶点、模拟药物相互作用和优化药物设计等方面的重要作用。
案例一:生物信息学在靶点发现中的应用生物信息学在靶点发现中发挥着重要的作用。
通过对已知基因组和蛋白质组的分析,可以找到与特定疾病相关的靶点。
例如,在乳腺癌的研究中,研究人员使用生物信息学的方法,分析了乳腺癌组织和正常组织的基因表达谱,发现了多个与乳腺癌发生相关的基因。
通过进一步的挖掘和验证,他们最终确定了一个新的乳腺癌靶点,并成功开发出针对该靶点的药物。
案例二:生物信息学在药物相互作用模拟中的应用药物的研发需要了解药物与靶点之间的相互作用,而生物信息学可以提供模拟药物和靶点结合的重要信息。
例如,在抗HIV药物研发中,研究人员使用生物信息学的工具,模拟了药物与HIV核心蛋白的相互作用过程,并预测了药物与蛋白之间的键合模式和亲和性。
这些模拟结果有助于研究人员深入了解药物与靶点之间的相互作用机制,并引导后续的药物设计工作。
案例三:生物信息学在药物设计中的应用生物信息学在药物设计中可以帮助研究人员优化和改良药物的结构。
例如,在抗癌药物的研发中,研究人员通过生物信息学的计算方法,对已有的抗癌药物进行结构分析和比较,找到药物与靶点结合的关键位点。
然后,他们利用这些信息,通过设计合成一系列的类似化合物,进一步优化药物的活性和选择性。
案例四:生物信息学在药物剂量个体化中的应用生物信息学在药物剂量个体化方面也发挥着重要的作用。
通过分析基因组学数据,研究人员可以预测个体对特定药物的代谢能力和药效反应。
这对于优化药物的剂量方案、减少药物不良反应以及提高药物治疗效果具有重要意义。
例如,在心血管疾病的治疗中,研究人员通过基因组学数据分析,预测患者对抗凝血药物的剂量需求,并制定了个体化的用药方案,取得了良好的治疗效果。
生物信息学在疾病检测方面的实例

生物信息学在疾病检测方面的实例随着科学技术的不断进步,生物信息学作为一门新兴的学科,已经在许多领域发挥了重要的作用。
其中,生物信息学在疾病检测方面的应用尤为突出。
本文将通过几个具体的实例,来展示生物信息学在疾病检测方面的重要性和潜力。
一、癌症基因检测癌症是当今社会中最常见的疾病之一,而基因突变是导致癌症发生的主要原因之一。
通过生物信息学技术,科学家们可以对人类基因组进行全面的测序,并分析其中的突变情况。
通过比较癌症患者和正常人的基因组,可以发现与癌症相关的突变基因,从而提前进行预测和诊断。
这种基因检测技术不仅可以帮助人们及早发现患病风险,还可以为个性化治疗提供依据,提高治疗效果。
二、遗传疾病筛查遗传疾病是由基因突变引起的疾病,通常会在家族中遗传。
通过生物信息学技术,科学家们可以对家族中患有遗传疾病的患者进行基因测序,发现与疾病相关的突变基因,并对家族成员进行筛查。
这样,可以及早发现患病的风险,并采取相应的预防措施,减少疾病的发生和传播。
三、微生物感染检测微生物感染是导致许多疾病的重要原因之一,如肺炎、尿路感染等。
通过生物信息学技术,科学家们可以对微生物的基因组进行测序,发现与感染相关的基因,并通过基因表达谱分析来确定感染的类型和程度。
这种微生物感染检测技术可以帮助医生更准确地诊断和治疗微生物感染,减少误诊和滥用抗生素的情况发生。
四、药物反应预测药物反应的差异是导致许多药物治疗失败的主要原因之一。
通过生物信息学技术,科学家们可以对个体基因组进行测序,并通过比较不同个体的基因组差异,预测药物的疗效和副作用。
这种药物反应预测技术可以帮助医生选择最合适的药物和药物剂量,提高治疗效果,减少药物不良反应的发生。
通过以上几个实例,我们可以看出生物信息学在疾病检测方面的重要性和潜力。
生物信息学技术的发展,为疾病的预防、诊断和治疗提供了新的思路和方法。
相信随着技术的不断进步,生物信息学在疾病检测方面的应用还会有更大的突破和发展。
生物信息学在疾病检测方面的实例

生物信息学在疾病检测方面的实例生物信息学在疾病检测方面的应用非常广泛,以下是两个例子:
1. 癌症的基因变异检测
癌症是由基因变异引起的一类疾病。
生物信息学可以通过测定癌
症患者的基因组序列,找出基因序列和正常组织中存在差异的位置,
然后对这些变异进行分析,确定哪些变异与癌症相关。
这种方法可以
用于早期癌症的检测,同时为治疗提供了一些针对性的药物解决方案。
例如,一些基因阵列分析技术可以检测出乳腺癌中的HER2基因扩增,
这种扩增需要使用一种特殊的抗癌药物进行治疗。
2. 个人基因组分析
个人基因组分析是基于人类基因组序列编码中的单个核苷酸多态
性(SNP)来预测一些疾病。
这种检测可以使用基因芯片或测序技术进行。
通过分析个人基因组中的SNP,可以预测患病的风险,例如乳腺癌、阿尔茨海默病等。
基于个人基因组分析得到的结果,可以指导个体在
日常生活中采取一些预防措施,减少疾病的发病率。
例如,一些基因
检测服务机构可以对卡方检验进行基因分析,同时提供许多与遗传疾
病相关的健康信息,并根据个人情况提供一些预防和干预措施。
总的来说,生物信息学在疾病检测中有着很广泛的应用,以其准
确性和实用性而受到广泛的关注。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ORF预测的可靠性检验
设计引物:Primer Premier 5.0
评估引物质量:Oligo 6.65 或Oligonucleotide Properties Calculator
NCBI的BLAST 2 SEQUENCES程序
/blast/bl2seq/wblast2.cgi
核苷酸序列=>氨基酸序列
制作密码子用法表
蛋白质理化性质分析
在线分析
ExPasy服务器上的ProtParam
/tools/protparam.html
生物学软件
BioEdit-氨基酸成分
Seqtools-亲、疏水性残基,蛋白溶解度
蛋白质功能性区域分析
疏水性分析
在线的ProtScale 程序
/cgi-bin/protscale.pl
使用生物学软件BioEdit7.05
采用Kyte-Doolittle的TGRESE算法
调整计算窗口大小n=9
附:该参数用于估计每种氨基酸残基的平均显示尺度,有助于对数据进行平滑。
跨膜区分析
在线分析
TMHMM Server v. 2.0
http://www.cbs.dtu.dk/services/TMHMM/
TMpred
/software/TMPRED_form.html
TMP
http://www.mbb.ki.se/tmap/
信号肽预测
SignalP 3.0 Server
几种人工神经网络法的组合
G+、G-、真核生物为训练集
http://www.cbs.dtu.dk/services/SignalP/
卷曲螺旋是控制蛋白质寡聚化的元件
/software/COILS_form.html
亮氨酸拉链结构:亲脂性的α螺旋,包含有许多集中在螺旋一边的疏水氨基酸,两条多肽链以此形成二聚体。
每隔6个残基出现一个亮氨酸。
由赖氨酸(Lys)和精氨酸(Arg)组成DNA结合区。
Domain分析
结构域是蛋白序列的功能、结构和进化单元,由50-300个氨基酸组成,有独特的空间构象。
类型:全平行结构域、反平行结构域、α+β结构域、α/β结构域及他折叠类型
EMBL的SMART服务器
http://smart.embl-heidelberg.de/
提交序列后=>系统每隔10秒刷新一次=>结果
模体(Motif)搜索
PROSITE数据库
确定新的蛋白质序列是否属于已知家族
N-糖基化位点的模式(Pattern):N[^P][ST][^P]
其中^P表示除Pro外的任意氨基酸
缺点:数量与质量上存在问题
/prosite/
Profile数据库
基于最佳的多重比对质量(包括人工校正)
优点:确保重要信息不被遗漏
http://myhits.isb-sib.ch/cgi-bin/motif_scan
蛋白质二级结构预测
蛋白质二级结构是指α螺旋、β折叠、无规则卷曲(Coils)等元件
预测方法:
基于统计的预测方法,如Chou-Fasman法、人工神经网络法等
基于知识的预测方法:Lim方法、Cohen方法
混合方法:选择性合并以上提到的各种方法
预测准确率:>70%,其中PHD神经网络预测的平均准确度及最佳残基的准确率分别高达72%和90%
二级结构预测的标准:PHD
/
同源模建
原理:比较模建,利用已知结构的同源蛋白建立目的蛋白的结构模型,再用理论计算方法进化优化,最终得到合理的3D模型。
关键:模板的选择
适用:同源性>30%的同源蛋白质
步骤:(6步曲)
目的序列与模板序列的匹配;
根据多重比对结果确定同源蛋白质的保守区及相应的框架结构;
目的蛋白质结构保守区的主链模建;
目标蛋白质结构变异区的主链模建;
侧链的安装和优化;
优化和评估模建的结构
系统发育分析
NJ法-邻接法:
特点:NJ法是基于最小进化原理经常被使用的一种算法,它构建的树相对准确,假设少,计算速度快,只得一颗树。
缺点:序列上的所有位点等同对待,且所分析的序列的进化距离不能太大
适用:进化距离不大,信息位点少的短序列
MP法-最大简约法
特点:基于进化过程中碱基替代数目最少这一假说
缺点:推测的树不是唯一的,变异大的序列会出现长枝吸引而导致建树错误。
适用:序列残基差别小,具有近似变异率,包含信息位点比较多的长序列
ML法-最大似然法
原理:考虑到每个位点出现的残基的似然值,将每个位置所有可能出现的残基替换概率进行累加,产生特定位点的似然值。
ML法对所有可能的系统发育树都计算似然函数,似然函数值最大的那颗树即最可能的系统发育树
优点:在进化模型确定的情况下,ML法是与进化事实吻合最好的建树算法
缺点:计算强大非常大,极为耗时
建树相关软件:
PAUP-/
PHLIP-/phylip.html
MEGA-
TreePuzzle-http://www.nsc.liu.se/software/biology/puzzle5/
TreeView-/rod/treeview.html
MEGA用法: [生信相关]
PHYLIP3.65界面
PHYLIP建树的子程序:
Dnapars-核苷酸序列最大简约法
Protpars-蛋白质序列最大简约法
Dnaml-核苷酸序列最大似然法
Dnamlk-核苷酸序列最大似然法(分子钟假说)
Proml-蛋白质序列最大似然法
Promlk-蛋白质序列最大似然法(分子钟假说)
Dnadist-核苷酸序列距离法->距离矩阵
Prodist-蛋白质序列距离法->距离矩阵
Seqboot-重复抽样检验
Consense-构建严格的一致树
抗原决定簇预测
抗原决定簇是指能与抗原相应抗体结合的抗原上有限部位的特殊分子结构,也称为表位(epitope)。
哈佛大学的Predicting Antigenic Peptides 预测软件是使用Kolaskar 和Tongaonkar(1990)方法预测氨基酸序列内可能引起抗体反应的抗原片段。
以生物信息学为基础的蛋白质抗原决定簇的预测对于分子生物学实验,如诊断试剂的制备、抗体制备筛选等,都是必不可少的工具,减少了了实验研究的盲目性(万涛等,1997)。
据报道,运用Predicting Antigenic Peptides 预测软件并结合多种方法综合分析预测的成功率可达86%(孙沫逸等,2003)。
/Tools/antigenic.pl
实例:RGDV Pns9基因
RGDV S9片段可能有两个开放阅读框(ORF),另一阅读框位于主阅读框下游,但目前尚未获得该ORF表达的实验证据。
主ORF转入表达载体pGEX-4T-1或pET29-a均无法得到成功表达,故目前无法获取完整Pns9蛋白的抗血清。
原因分析:使用生物学软件RNAstructure 4.2采用最小自由能法(总能量值=283.3kcal/mol)折叠S9片段主ORF对应的核苷酸序列,发现核苷酸序列开始位置的第10-36碱基间形成强势的发夹状结构。
选择强抗原性肽段的原则
∙肽段长度12-15个氨基酸;
∙肽段内无4个以上连续相邻的疏水性残基,疏水性残基数目<6,带正电荷氨基酸越多越好;
∙亲水性及可及性参数均较高;
∙肽段位于转角附近,易形成无规则卷曲。
∙蛋白质的羧基端非常理想(灵活性、暴露性)。