高通量测序入门
高通量测序,名词解释

高通量测序基础知识汇总一代测序技术:即传统的Sanger测序法,Sanger法是根据核苷酸在待定序列模板上的引物点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以A、T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,通过检测得到DNA碱基序列。
二代测序技术:next generation sequencing(NGS)又称为高通量测序技术,与传统测序相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定,从而使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序(Deep sequencing)。
NGS主要的平台有Roche(454 &454+),Illumina(HiSeq 2000/2500、GA IIx、MiSeq),ABI SOLiD等。
基因:Gene,是遗传的物质基础,是DNA或RNA分子上具有遗传信息的特定核苷酸序列。
基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状。
DNA:Deoxyribonucleic acid,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱基、脱氧核糖、磷酸。
脱氧核糖核酸通过3',5'-磷酸二酯键按一定的顺序彼此相连构成长链,即DNA链,DNA链上特定的核苷酸序列包含有生物的遗传信息,是绝大部分生物遗传信息的载体。
RNA:Ribonucleic Acid,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成。
高通量测序生物信息学分析 内部极品资料 初学者必看

略。由于物种基因组的大小相差比较大,如细菌、真菌等微生物,其基因组一般比较小,可以单 独采用 Roche 454(20-30x)或 Solexa 采用高覆盖率(60×左右)的策略进行测序。而对于一些基因 组比较大(100M 以上)的物种(如植物),会采用一些技术平台组合的方法进行测序。考虑到平台
进行深度测序,完成基因组拼接。 采用 De Novo 测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部
的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及 进行比较基因组学研究,为后续的相关研究奠定基础。
实验流程:
公司服务内容
1.基本服务:DNA 样品检测;测序构建;高通量测序;数据基本分析(Base calling,去接头, 去污染);序列组装达到精细图标准
库中发现有 5%的插入片段在 0~500bp 的读段,将有可能增加 De Novo基因组信息,需要调查近缘物种的重复序列分布,能够帮助实验设计。详情
见问题 4
-4-
6.基因组 De Novo 需要多大的覆盖率?
基因组的覆盖率是指测序得到的碱基总量(bp)与基因组大小(Genome)的比值,它是评价测序量 的指标之一。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结片段包含基因组中较大跨度(2-10 kb) 片段两端的序列,更具体地说:首先将基因组 DNA 随机打断到特定大小(2-10 kb 范围可选); 然后经末端修复,生物素标记和环化等实验步骤后,再把环化后的 DNA 分子打断成 400-600 bp 的片段并通过带有链亲和霉素的磁珠把那些带有生物素标记的片段捕获。这些捕获的片段再经末 端修饰和 测序量与测序覆盖度的关系
高通量测序 名词解释

高通量测序基础知识汇总一代测序技术:即传统的Sanger测序法,Sanger法是根据核苷酸在待定序列模板上的引物点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以A、T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,通过检测得到DNA碱基序列。
二代测序技术:next generation sequencing(NGS)又称为高通量测序技术,与传统测序相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定,从而使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序(Deep sequencing)。
NGS主要的平台有Roche(454 & 454+),Illumina(HiSeq 2000/2500、GA IIx、MiSeq),ABI SOLiD等。
基因:Gene,是遗传的物质基础,是DNA或RNA分子上具有遗传信息的特定核苷酸序列。
基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状。
DNA:Deoxyribonucleic acid,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱基、脱氧核糖、磷酸。
脱氧核糖核酸通过3',5'-磷酸二酯键按一定的顺序彼此相连构成长链,即DNA链,DNA链上特定的核苷酸序列包含有生物的遗传信息,是绝大部分生物遗传信息的载体。
RNA:Ribonucleic Acid,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成。
2022理论培训课第1讲高通量测序基础知识和原理简介

2022理论培训课第1讲高通量测序基础知识和原理简介
理论课程第一讲,让我们从测序的基础知识和原理开始~你想知道什么是高通量测序吗?
你想了解测序仪是通过什么原理获取信号的吗?
你想看看多种测序仪可以各自应用到哪些工作场景中吗?
让我们一起来探讨一下~讲师介绍:
强裕俊,中国疾病预防控制中心传染病预防控制所测序工程师,研究方向病原微生物和宏基因组学,长期从事于病原微生物高通量测序相关工作,拥有多年3730、454、Miseq、BGIseq、Nonapore等不同型号测序仪相关工作经验及处理各类样本量近万份,参与发表SCI 文章数篇。
本系列所有资料,仅用于内部学习交流,未经授权,不可他用。
长按关注
公众号名称:微微悦明
科学的乐趣是获得新知识的喜悦~
高通量测序、大数据病原微生物检测和监测健康大数据行业资讯记录与分享
我有一壶酒
独酌无相亲
倾尽江湖里
共饮天下人。
高通量测序技术简介

高通量测序技术简介近年来,随着生物技术的发展,高通量测序技术在生物学研究、临床医学、农业科技等众多领域中发挥着越来越重要的作用。
本文将为读者简单介绍高通量测序技术的基本原理、应用及未来发展方向。
一、高通量测序技术基本原理高通量测序技术(High-Throughput Sequencing,简称HTS)是指通过同时测序数以亿计上万条DNA片段的方法,快速准确地得出基因信息。
其核心技术包括样品制备、DNA片段库构建和测序。
样品制备主要包括DNA抽提、纯化和切割等步骤。
DNA片段库构建通常分为两种方式:文库构建(Library Preparation)和逆相PCR法(Inverse PCR)构建。
其中文库构建方法包括Genomic DNA文库构建、cDNA文库构建和ChIP-seq文库构建等。
测序分为Sanger测序和第二代/第三代测序两种。
目前,Illumina、Ion Torrent、PacBio和Nanopore等公司的测序技术已开始广泛应用。
二、高通量测序技术的应用高通量测序技术在生物领域中的应用越来越广泛。
具体应用包括以下几个方面:1、基因组学:基因组学是高通量测序技术最早应用的领域之一。
通过对整个基因组进行测序,可以深入研究基因的结构、组织与表达等方面的信息,促进基因组学的发展。
2、转录组学:高通量测序技术在转录组学中的应用主要为RNA测序,可以发现RNA剪切变异、可变外显子和SNPs (Single Nucleotide Polymorphisms)等。
3、表观基因组学:表观基因组学是研究基因组DNA序列和其组杂化状况的学科。
高通量测序技术可以对DNA甲基化、组蛋白修饰、染色质状态等进行充分研究。
4、单细胞测序技术:在原有的基础上,在单细胞尺度上进行分析,可以识别不同类型的单细胞和细胞异质性在不同生理状态下的基因表达差异。
5、临床医学:高通量测序技术在临床上可以进行新生儿常染色体脆性综合征、癌症个性化治疗、基因疾病等多方面的风险评估。
高通量测序技术(NGS)

高通量测序技术(NGS)学习感悟:近来,看到了《高通量测序揭秘中药如何杀死癌细胞》的文章,什么是高通量测序?教材中只有PCR技术扩增技术知识,查找了一些资料,获得了肤浅的理论知识。
一、高通量测序技术简介高通量测序技术(High-throughput sequencing)又称“下一代”测序技术,以能一次并行对几十万到几百万条DNA分子进行序列测定和一般读长较短等为标志。
实验过程:样本准备,文库构建,测序反应,数据分析。
(1)将目标DNA剪切为小片段(2)单个小片段DNA分子结合到固相表面(3)单分子独立扩增(4)每次只复制一个碱基(A,C,T,G)并检测信号(5)高分辨率的成像系统高通量测序以其高输出量与高解析度的特性,不仅为我们提供了丰富的遗传学信息,而且使得测序的费用和时间大大缩短。
在高通量测序发展的过程中,也有很多的问题需要我们去解决:数据在临床诊断上的作用,测序数据的储存和分析,数据的安全和信息隐私等。
二、测序行业技术发展概况自FrederickSanger提出双脱氧核苷酸末端终止法以来,测序技术已经历了近40年的发展,根据核心技术的区别与进步,可以分为三代:第一代测序技术——始于19771977年,Sanger提出了双脱氧核苷酸末端终止法,同年A.M.Maxam和W.Gilber也提出了化学酶解法,两者的提出标志着第一代测序技术的诞生。
第二代测序技术(NGS)——始于20052005年,开发出全球第一台商业化的第二代DNA测序仪GS20,拉开了基因产业发展的序幕。
之后数年NGS行业内经历了激烈的竞争,逐步形成较为稳定的格局:(1)LifeTechnologies于2013年被著名科研服务供应商ThermoFisher收购,SOLiD平台逐步淡出市场,主推2011和2012年陆续发布的Ion PGM和IonProton两款测序设备。
(2)Illumina则在全面接收Solexa的研发平台之后,开发出了著名的HiSeq平台系列。
高通量测序技术的数据分析方法教程

高通量测序技术的数据分析方法教程随着生物技术的发展,高通量测序技术(high-throughput sequencing technology)已成为生物学、医学和生物信息学研究中的重要工具。
高通量测序技术可以快速而准确地测定DNA或RNA序列,透过大量的数据来揭示生物体的基因组、转录组以及其他生物学过程中的变化。
然而,正确且高效地分析测序数据是高通量测序技术应用的关键一步。
本文将介绍高通量测序技术的数据分析方法教程。
首先,分析高通量测序数据前,我们需要了解常见的测序平台和数据格式。
当前常用的高通量测序平台包括Illumina、ABI SOLiD、Ion Torrent等,而测序数据通常以FASTQ、SAM/BAM和VCF等格式存储。
FASTQ格式用于存储原始测序数据,其中包含了每个测序读段的序列信息及其对应的质量分数。
而SAM/BAM格式则是将测序读段比对到参考基因组之后的结果,其中SAM是比对结果的文本格式,而BAM则是对应的二进制格式。
VCF(Variant Call Format)格式则用于存储基因型变异信息。
接下来,我们将介绍高通量测序数据的基本分析流程。
通常,测序数据分析可以分为质控、比对、变异检测和功能注释几个主要步骤。
在质控步骤中,我们需要对测序数据进行质量评估和过滤。
质量评估可以通过查看测序数据的质量分数、GC含量、碱基分布和测序错误率等指标来判断测序数据的质量。
使用质量评估工具如FastQC和NGS QC Toolkit可以帮助我们快速准确地评估测序数据的质量,并进行相应的过滤工作,去除低质量的测序读段。
接下来,我们需要将测序读段比对到参考基因组上。
比对工作可以通过软件如Bowtie、BWA和HISAT等进行。
比对结果通常以SAM格式存储,然后可以进行排序、去重和索引等处理,生成最终的BAM格式文件。
在变异检测步骤中,我们需要从比对后的BAM文件中检测样本中存在的变异信息。
变异检测可以通过多种工具来实现,如GATK、Samtools和VarScan等。
测序 基础知识

转录组高通量测序中,reads、contigs、scaffold、unigene、singleton高通量测序时,在芯片上的每个反应,会读出一条序列,是比较短的,叫read,它们是原始数据;有很多reads通过片段重叠,能够组装成一个更大的片段,称为contig(克隆群);多个contigs通过片段重叠,组成一个更长的scaffold;一个contig被组成出来之后,鉴定发现它是编码蛋白质的基因,就叫singleton;多个contigs组装成scaffold之后,鉴定发现它编码蛋白质的基因,叫unigene。
基因组测序方法:链中止法测序:通过合成与单链DNA互补的多核甘酸链,由于合成的互补链可在不同位置随机终止反应,产生只差一个核苷酸的DNA分子,从而来读取待测DNA分子的顺序。
化学降解法测序:在待定的核苷酸碱基中引入化学集团,再用化合物处理,使DNA分子在被修饰的位置降解。
自动化测序:与链终止测序原理相同,这姿势用不同的荧光色彩标记ddNTP,如ddA TP 标记红色荧光,ddCTP标记蓝色荧光,ddGTP标记黄色荧光,ddTTP标记绿色荧光。
由于每种ddNTP带有各自待定的荧光颜色,二简化为由1个泳道同时判读4种碱基。
非常规DNA测序毛细管电泳、光点测序、DNA芯片测序、随机的组装(鸟枪法)鸟枪法:就有可能出现错装。
鸟枪法策略指导测序策略不需要背景信息构建克隆群时间短需要几年时间需要大型计算机得到的是草图(Draft)得到的是精细图谱EST (Expressed sequence tag)测序EST是一种重要的基因组图分子标记,以EST为探针很容易从cDNA文库中筛选全基因,又可从BAC克隆中找到其基因组的基因序列。
优点:mRNA可直接反转录成cDNA,而且cDNA文库也可比较容易构建。
对cDNA文库大量测序,即可获得大量的EST序列EST为基因的编码区,不包括内含子和基因间区域,一次测序的结果足以鉴定所代表的基因。
高通量测序技术的基本原理及其应用

高通量测序技术的基本原理及其应用高通量测序技术是一种用于分析DNA或RNA序列的先进工具。
自2005年首次商业化以来,高通量测序技术已经成为生物医学研究领域中最受欢迎的技术之一。
本文将介绍高通量测序技术的基本原理以及其在各种生物研究中的应用。
一、高通量测序的基本原理高通量测序技术通过对DNA或RNA序列进行多轮扩增和差异式回收来实现序列的读取。
这些扩增和回收过程通过从核酸库中选取并扩增特定区域的DNA或RNA序列并将这些序列与标志物添加到瓶底上的方法来实现。
在扩增过程中,DNA序列被切成小碎片,并与适配器连接。
这些适配器具有序列信息,以帮助下一阶段将它们区分开来。
然后,这些DNA片段被反复复制和放大,以产生大量的DNA片段。
这些片段被装入流式细胞仪等设备中,以便单个分子可以被读取。
在差异式回收的过程中,将标记DNA(即在扩增过程中附加的标签)与扩增的DNA片段分离。
这是通过在特定区域上捕获(将标记DNA与其匹配的DNA区域连接)完成的。
这些DNA片段然后被读取并映射到基因组或转录组上,以详细分析其序列。
二、高通量测序技术的应用高通量测序技术可以用于许多应用领域,如基因组学,转录组学,表观遗传学和元基因组学。
以下是一些例子:1.基因组学高通量测序技术被广泛用于研究基因组结构和功能。
它可以识别基因组中的单核苷酸多态性(SNP),从而对个体或种群中的基因组变异进行研究。
此外,它也可以用于构建DNA序列库,用于组装参考基因组和研究基因组进化。
2.转录组学高通量测序技术可以用于分析特定细胞中的基因表达模式和代谢途径。
这些信息可以帮助生物学家理解细胞的生长和分化,并对某些疾病的发生有所帮助。
此外,通过将RNA序列映射到基因组上,可以有效地注释基因组,并识别各种转录本和剪切变异。
3.表观遗传学高通量测序技术可以用于研究表观遗传学变异,如DNA甲基化和组蛋白修饰。
通过研究这些变异,生物学家可以了解这些变异是如何影响细胞表达模式的。
高通量测序技术的使用教程

高通量测序技术的使用教程引言:高通量测序技术是一种重要的基因组学研究手段,它能够迅速、可靠地获取大量的DNA或RNA序列信息。
本文将介绍高通量测序技术的使用教程,包括样品准备、文库构建、测序平台选择和数据分析等方面的内容。
一、样品准备:在进行高通量测序前,样品的准备非常关键。
首先,需要根据研究目的选择合适的样品类型,如组织、细胞、血液等。
其次,对于DNA样品,需要通过核酸提取方法将DNA纯化。
对于RNA样品,需要注意样品的采集、保存和提取过程中避免RNA的降解。
另外,样品的数量和质量也对高通量测序结果有影响,因此要确保样品含量充足且质量良好。
二、文库构建:文库构建是高通量测序的关键步骤,它决定了待测DNA或RNA的插入片段、测序标签和文库构建方法。
常见的文库构建方法包括PCR扩增文库、tagmentation文库和mate-pair文库等。
在文库构建的过程中,需要选择适当的酶切位点、测序引物和适配体,确保文库构建的高效和准确。
三、测序平台选择:目前市面上有多种不同的高通量测序平台可供选择,如Illumina HiSeq、Ion Torrent PGM等。
选择合适的测序平台需要考虑测序通量、测序质量、运行成本和处理时间等因素。
一般来说,Illumina HiSeq是最常用的高通量测序平台,它具有较高的通量和测序质量,适用于大多数研究项目。
四、测序实验操作:根据测序平台和文库构建方法的要求,进行相应的实验操作。
首先,需要将文库片段与测序引物结合,然后进行PCR扩增、片段选择和质检等步骤。
在实验过程中,应注意反应体系的准备、温度控制和反应时间等细节,确保实验操作的准确性和稳定性。
五、数据分析:测序完成后,得到的数据需要进行后续的数据分析。
常见的数据分析包括序列比对、变异分析、基因表达分析和基因组装等。
这些分析通常需要通过专业的测序分析软件(如BWA、SAMtools、GATK等)进行。
对于初学者来说,可以借助一些在线的分析平台(如Galaxy、BaseSpace等)进行数据分析,减轻分析的难度。
高通量DNA测序技术

高通量DNA测序技术随着科技的发展和进步,越来越多的科研领域需要对DNA进行测序。
而高通量DNA测序技术的出现,使得DNA测序工作更加快捷、精准和高效。
本文将从以下几个方面介绍高通量DNA测序技术的相关内容。
一、高通量DNA测序技术的定义和原理高通量DNA测序技术是指利用生物学和计算机学方法,对DNA序列进行高速、高效的测序技术。
其核心原理是将DNA分割成较短的片段,使用不同的方法来扩增和测序这些片段,最后通过计算机处理和分析这些片段的碱基序列并将其组装起来,得到完整的DNA序列。
常用的高通量测序技术包括Illumina、Ion Torrent、Roche 454和Pacific Biosciences等,其中Illumina是最为常用的技术之一。
其工作原理是将待测DNA片段嵌入在底物上,然后循环地进行扩增、预处理和测序过程,每一个循环产生的信号都代表了DNA片段的一个碱基,最终得到高精度的DNA序列。
二、高通量DNA测序技术的优势和应用场景相比传统的Sanger测序技术,高通量DNA测序技术有以下几个显著的优势:1.高通量:能够同时测序大量DNA样本,提高测序效率和速度;2.高准确性:能够对多个DNA片段进行测序并进行校准,提高测序准确性;3.高可靠性:不受DNA质量和样本数量等因素的影响,能够在最短时间内快速得到结果;4.低成本:相对于传统测序技术,高通量DNA测序技术的成本更低,更加适合大规模测序。
高通量DNA测序技术适用于生命科学的多个领域,如基因功能研究、病原体检测、基因突变研究等。
例如,应用高通量DNA测序技术可以对基因的表达进行分析和量化,从而探究不同基因在不同条件下的调控机制;同时,也可以对病原体的基因组进行测序,更好地了解其特性和病理机制。
三、高通量DNA测序技术的未来发展方向随着科技的不断发展和进步,高通量DNA测序技术也将会不断完善和升级。
目前,一些新技术已经在高通量DNA测序技术领域中得到了应用,例如长读段技术、基于纳米孔的测序技术等,这些新技术能够更加准确地测序DNA,并且可以降低测序的误差和偏差。
高通量测序基础学习培训

富集的外泌体
• 干冰运输即可。也可以送4ml左右血浆或血清, 我们来富集。
1. 一般每组至少3个样本
2. 取样的代表性及准确性
• 取材时间、部位、处理条件等方面尽可能保持一致 • 准确记录,并按要求(低温、迅速)采集、制备、贮存、运输进行实验处
理
3. 样本编号
• 比如:癌症病人组可用编号 C_01; C_02; C_03……;正常病人组可用 编号 N_01; N_02
2) 2100 bioanalyzer检测片段大小:其峰值大小应该是目的片段大 小加上接头的序列长度。
示例1 2100 bioana分析
• 数据质控和筛选过滤,得到高质量数据 • 根据不同的测序类型进行差异基因分析、功能注
释、预测新的基因等
• 判断是否存在降解应以2100 bioanalyzer模拟电泳图为准 • 植物样品检测结果为 25S/18S ,具有相同的参考意义
RIN值 浓度、总量
• RNA Intergrity Number,RNA完整性指数,满分为10分 • 建议使用符合RIN值≥ 7且rRNA 28S/18S≥ 0.7
• 起始总RNA浓度≥ 100 ng/µl • 起始总RNA量 ≥ 2 µg/样品(如小RNA测序) • 或起始总RNA量 ≥ 20 µg/样品(如转录组测序)
示例1 2100 bioanalyzer峰图和模拟电泳图
示例2
(报告一)
样序列打 碎成200-500bp的
小片采用特异性染料结合的方式特异性的检测样本浓度 来实现精确定量。
主要测序平台
Illumina 高通量测序平台,主要由HiSeq测序仪和MiSeq测序仪组成。
HiSeq 2500平台
• 转录组测序、小RNA测序
高通量测序的流程

高通量测序的流程高通量测序技术作为现代生物学和医学研究中不可或缺的重要工具,已经在基因组学、转录组学和生物信息学等领域广泛应用。
本文将深入探讨高通量测序的流程及其技术要点,以及在不同应用场景下的具体应用。
一、高通量测序技术是一种通过并行处理大量DNA或RNA分子的方法,能够快速、准确地测定样本中的基因组序列信息。
其广泛应用于基因变异分析、群体遗传学研究、肿瘤基因组学以及微生物群落结构分析等多个领域。
二、高通量测序的主要步骤1. 样本准备与DNA/RNA提取高通量测序的第一步是样本的准备和核酸的提取。
样本可以是来自生物体的任何组织或细胞,提取得到的DNA或RNA质量和纯度直接影响后续测序结果的可靠性。
常用的提取方法包括酚/氯仿法、商业提取试剂盒以及磁珠法,选择合适的方法取决于样本类型和实验室设施的情况。
2. 文库构建DNA或RNA提取后,需要将目标核酸转化为可用于高通量测序的文库。
文库构建的关键步骤包括断裂、末端修复、连接连接子、文库扩增和文库纯化等。
每个步骤都需要精确控制反应条件和使用高质量的试剂,以避免污染和损伤目标DNA/RNA。
3. 测序平台选择与测序类型确定在文库构建完成后,需要根据具体实验设计选择合适的测序平台和测序类型。
目前常用的高通量测序平台包括Illumina、Ion Torrent、PacBio和Oxford Nanopore等,每种平台都有其特定的优缺点和适用场景。
测序类型主要分为全基因组测序(WGS)、全外显子组测序(WES)、RNA测序和甲基化测序等,根据研究问题的不同选择合适的测序类型至关重要。
4. 数据与质控测序平台的数据需要经过严格的质量控制(QC)流程,包括去除低质量序列、去除接头序列、去除PCR重复序列和去除污染序列等步骤。
质控后的数据才能用于后续的生物信息学分析。
5. 数据分析与解释质控通过的数据将进行生物信息学分析,这包括序列比对、变异检测、表达定量、功能注释和数据可视化等步骤。
如何使用高通量技术对基因进行筛查

如何使用高通量技术对基因进行筛查随着现代生物技术的发展,DNA测序技术不断推陈出新。
现在,高通量测序技术已经成为了基因测序的主流。
高通量测序技术可以高效、精准、快速地进行基因测序,大大提高了我们对基因的认识和研究。
在本文中,我们将探讨如何使用高通量技术对基因进行筛查。
一、高通量测序技术的基本原理高通量测序技术是指在同一时间内进行大量的DNA分析,通过扩大分析范围,来加速DNA序列的测定速度和降低成本。
根据测序原理的不同,高通量测序技术有以下几种:1.链终止法:该方法是1986年由斯里兰卡的谢科尔斯基组研发出来的。
该方法需要使用标记有不同荧光染料的4种dNTP(即ATCG),在DNA合成过程中加入。
DNA链延伸到有某个dNTP的位置后,该荧光染料就会释放出荧光信号标记位置的碱基。
通过监测这些荧光信号,就可以突破测序速度和通量的瓶颈。
2.桥接PCR法:该方法是将DNA片段接到测序板上,然后通过PCR反应来产生许多DNA链,最后进行荧光检测。
该方法能在不断增加测序片段的同时,也能提高质量、稳定性和重复性。
3.扩增桥接法:该方法是将DNA片段连接成桥,并逐一地进行扩增。
可形成数百万个DNA片段,然后通过荧光标记的方法检测DNA片段测序。
4.纳米孔法:该方法是利用原子层沉积技术在纳米空间中制备出纳米孔。
DNA分子通过这些孔,来逐个被检测和测序。
五、如何使用高通量技术对基因进行筛查1.基因组测序:基因组测序是高通量测序技术的主要应用之一。
该技术可以对整个基因进行测序,包括所有编码和非编码区域。
通过比较两个不同人的基因组序列,我们可以找到其差异,从而帮助我们理解人类基因的多样性和基因在足迹中的重要性。
2.转录组测序:转录组是指在一个特定细胞或组织的基因表达水平。
通过利用转录组测序技术,我们可以得到组织中所有表达的核糖核酸序列,从而研究基因的表达模式和功能。
3.外显子组测序:外显子是基因组中的编码蛋白序列的区域。
高通量测序科研入门常用名词意义整理
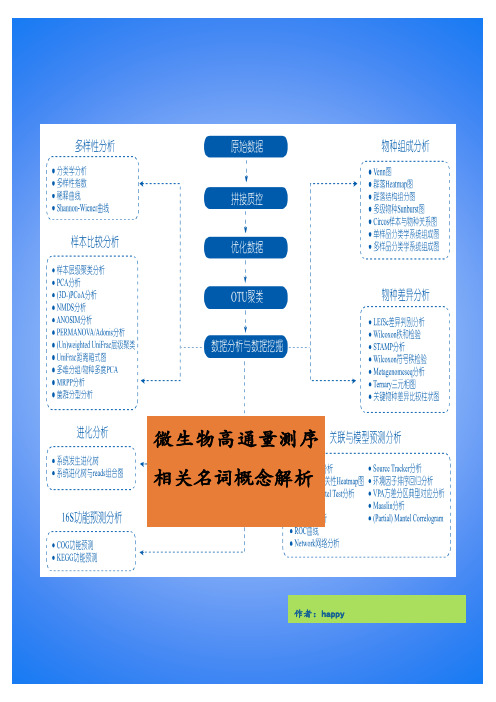
微生物高通量测序相关名词概念解析作者:happy目录一、OTU分类和统计 (2)二、生物信息分析 (2)三、16SrRNA (3)四、Alpha多样性 (4)五、稀疏性分析(rarefaction analysis)和稀疏性曲线(rarefaction curve) (7)六、Shannon-Weiner指数 (8)七、Rank Abundance 曲线 (9)八、微生物种属鉴定及相关分析 (10)九、OTU群落聚类及相关分析 (14)十、Rank Abundance 曲线 (15)十一、韦恩图(Venn) (16)一、OTU分类和统计OTU(operationaltaxonomicunits)是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。
通常按照97%的相似性阈值将序列划分为不同的OTU,每一个OTU通常被视为一个微生物物种。
相似性小于97%就可以认为属于不同的种,相似性小于93%-95%,可以认为属于不同的属。
样品中的微生物多样性和不同微生物的丰度都是基于对OTU的分析。
Coverage是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低。
该指数实际反映了本次测序结果是否代表样本的真实情况。
计算公式为:C=1-n1/N其中n1=只含有一条序列的OTU的数目;N=抽样中出现的总的序列数目。
分类水平统计表主要是对每个样本在分类学水平上的数量进行统计,并且在表格中列出了在每个分类学水平上的物种数目(只显示前10个样本,如果样本超过10个,请查看结果中taxon_all.txt文件)其中SampleName表示样本名称;Phylum表示分类到门的OTU数量;Class表示分类到纲的OTU数量;Order表示分类到目的OTU数量;Family表示分类到科的OTU数量;Genus表示分类到属的OTU数量;Species表示分类到种的OTU数量。
高通量测序流程和原理

高通量测序流程和原理高通量测序技术是一种快速、准确地测定DNA序列的方法,它在基因组学、转录组学和生物信息学等领域有着广泛的应用。
本文将介绍高通量测序的流程和原理,帮助读者更好地了解这一重要的生物技术。
首先,高通量测序的流程可以分为样品准备、文库构建、测序和数据分析四个主要步骤。
在样品准备阶段,需要从生物样品中提取DNA或RNA,并进行质量检测和浓度测定。
接下来是文库构建,这一步骤包括DNA片段的末端修复、连接接头、文库扩增等操作,最终得到适合测序的文库。
然后是测序阶段,高通量测序技术包括Illumina测序、Ion Torrent测序、PacBio测序等多种方法,每种方法都有其特定的原理和应用范围。
最后是数据分析,通过生物信息学软件对测序数据进行处理、比对、拼接和注释,最终得到样品的基因组或转录组信息。
其次,高通量测序的原理主要包括DNA片段化、文库构建、测序、数据分析等几个方面。
首先是DNA片段化,将DNA样品通过超声波、酶切或化学方法打断成数百到数千碱基对的片段。
接着是文库构建,将DNA片段末端修复、连接接头、文库扩增,构建成适合测序的文库。
然后是测序,根据不同的测序平台和技术,可以实现单端测序、双端测序、长读长测序等多种模式。
最后是数据分析,通过生物信息学软件对测序数据进行处理,包括去除低质量序列、比对到参考基因组、拼接成序列等步骤,最终得到样品的基因组或转录组信息。
总之,高通量测序技术在生命科学研究、临床诊断和个性化医疗等领域有着重要的应用前景。
通过了解高通量测序的流程和原理,可以更好地理解其在生物学研究中的作用,促进相关技术的发展和创新。
希望本文能够对读者有所帮助,谢谢阅读!。
高通量测序流程和原理

高通量测序流程和原理
高通量测序(High-throughput sequencing)是一种快速、高效的DNA测序技术,也被称为第二代测序技术。
它的出现极大地推动了基因组学和生物信息学的发展,为基因组变异、表达调控、蛋白质组学等研究领域提供了强大的支持。
高通量测序的流程可以简单概括为DNA提取、文库构建、测序仪测序和数据分析四个步骤。
首先是DNA提取,从样本中提取出所需的DNA,可以是基因组DNA、表达物的cDNA等。
接下来是文库构建,将提取的DNA片段连接到测序引物上,形成文库。
然后是测序仪测序,将文库中的DNA片段进行高通量测序,得到大量的原始测序数据。
最后是数据分析,对原始数据进行质控、比对、组装和功能注释等一系列分析,最终得到所需的生物信息学结果。
高通量测序的原理主要基于测序引物的引导下,通过不断地合成和检测新的核苷酸碱基,从而逐渐构建起整个DNA片段的序列。
常见的高通量测序技术包括Illumina测序、Ion Torrent测序、PacBio测序等,它们各自采用不同的原理和方法,但都能实现高通量的DNA测序。
在实际应用中,高通量测序技术被广泛应用于基因组测序、转录组测序、表观基因组测序等领域。
它不仅在科学研究中发挥着重要作用,还在临床诊断、生物工程、农业育种等领域有着广阔的应用前景。
总之,高通量测序技术以其快速、高效、准确的特点,成为现代生物学研究中不可或缺的重要工具,为我们深入了解生命的奥秘提供了有力支持。
随着技术的不断进步和应用的不断拓展,相信高通量测序技术将为生命科学领域带来更多的惊喜和突破。
高通量测序入门

高通量测序入门本人方向也仅限在rna-seq领域,所以其他领域的问题可能不太了解,只能按照自己的背景知识和请教别人解答,请大家慢拍砖!此外,由于实验室项目繁忙,我们可能无法及时发布或回复您。
请原谅我。
既然是入门专题,那就先简单说一下,要分析高通量测序数据的配置要求吧:声明:该配置不适用与从华大拿回分析结果直接写paper的同学。
我认识的一位同学一点生物信息背景也没有,直接用华大返回分析结果发了很好的文章,如果想这样的同学可直接跳过这篇,等待以后的专题。
言归正传:1.软配置:生物学理论知识:熟悉生命活动的基本过程,对复制、转录、翻译和转录后修饰有清晰的理解。
最好了解顺式元件和反式因子之间的区别。
推荐朱玉贤的分子生物学,掌握60%就差不多了(这是对希望通过测序数据进行生物分析的学生的要求。
他们是否在做软件开发并不重要。
例如,中国一些表现良好的实验室是主修数学或自动化的牛人,以下配置不适用于这些牛人)实验理论知识:不一定要做过实验,但至少要知道实验的过程,比如测序前样本的处理过程,序列片段化、加接头、pcr扩增等。
也许没有用,但将来出了问题,你可以很容易知道问题出在哪里编程知识:要求不必太高。
只需学习一些Perl。
对于主修生物学(我主修生物学)的学生,强烈建议使用Perl。
它似乎已经在第五版中出版了。
这本书很有趣。
当时我读了一个星期。
幽默的语言让我经常大笑,这让实验室里的同学觉得他们很紧张。
对于C语言基础的学生来说,这只是一道菜,可以在两天内通过。
此外,有空闲能力的学生可以学习一些R、python或Java,因为很多软件都是用R或python编写的。
如果你很懒或很忙,你就不能抽出时间。
只要学习Perl。
需要记住的一点是:除了基本知识之外,您还必须了解Perl学习过程中的哈希和模块。
当然,如果你的导师允许你对数据进行半个月的冗余,你只能学习周期。
统计学知识:只要大学上过生物统计也就差不多了(遇到二百五的老师你就比较悲剧了),最基本的知道什么是标准化,正态分布,pvalue以及卡方检验或fisher精确检验,多重检验,,fdr这些概念和计算方法也就差不多了。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
很高兴成为论坛特邀专家,鄙人会接下来的一段时间内写一些高通量测序数据方面的帖子,由浅入深,可能刚开始会比较简单一些,后面会有一些针对性的专题,也欢迎各位大侠或小菜提出建议或问题大家一起探讨。
为了活跃论坛建议大家直接跟帖或发新帖,我会尽快回复大家。
本人方向也仅限在RNA-seq 领域,所以其他领域的问题可能不太了解,只能按照自己的背景知识和请教别人解答,请大家慢拍砖!另外,由于实验室课题比较忙,所以可能不能及时发帖或回复大家,也请见谅。
既然是入门专题,那就先简单说一下,要分析高通量测序数据的配置要求吧:声明:该配置不适用与从华大拿回分析结果直接写paper 的同学。
我认识的一位同学一点生物信息背景也没有,直接用华大返回分析结果发了很好的文章,如果想这样的同学可直接跳过这篇,等待以后的专题。
言归正传:1. 软配置:生物理论知识:熟悉生命活动的基本过程,对复制、转录、翻译、转录后修饰有较清晰的认识,如果知道cis-element 和trans-factor 的区别就更好了。
推荐朱玉贤的分子生物学,能够掌握60% 就差不多了(这是对想通过测序数据进行生物分析同学的要求,如果是做软件开发等就无所谓了,比如国内做的很好的一些实验室,都是数学或自动化专业的牛人,以下一些配置也不适用这些牛人)实验理论知识:不一定要做过实验,但至少要知道实验的过程,比如测序前样本的处理过程,序列片段化、加接头、PCR 扩增等。
也许没有用,但将来出了问题,你可以很容易知道问题出在哪里编程知识:要求不用太高,学一些perl 就可以了,对于生物专业的同学(本人就是生物专业),强烈推荐perl 语言入门,好像现在已经出到第五版了。
此书极为搞笑,本人当时看了一个星期,其中幽默的语言导致本人经常笑出声音引得实验室同学以为神经了。
对于有C 语言基础的同学来说简直就是菜,两天就可以通了。
另外,学有余力的同学可以学一些R 以及python 或java. 因为好多软件都是用R 或python 写的,如果要是比较懒或三国杀很忙抽不出空就算了,学学perl 就好了。
切记一点:perl 的学习过程中除了基础知识,一定要看一下哈希和模块这两部分。
当然如果你们导师允许你对数据去个冗余也要半个月的话,你只学到循环就可以了。
统计学知识:只要大学上过生物统计也就差不多了(遇到二百五的老师你就比较悲剧了),最基本的知道什么是标准化,正态分布,p value 以及卡方检验或Fisher 精确检验,多重检验,,FDR 这些概念和计算方法也就差不多了。
推荐从以下统计软件中择一精通之:SAS(比较变态,硕士期间学了,后来就还给老师了)excel(入手比较容易,好好学学,功能比较全,我学的差)matlab(本人认为最牛的统计软件,有专门的论坛,有兴趣的同学可以google 一下) SPSS(上手比较容易,而且很多汉化的非常好,新手同学比较推荐,但是精通比较难)R (最好能学这个,我觉着学R 太必要了)perl (指CPAN 中的统计模块,不过需要一点技术)常见数据库:这个根据自己所做的方向,需要具体问题具体分析,常见的NCBI 以及EBI 和UCSC 还是需要了解的。
计算机操作要求:推荐linux 系统,掌握最基本的命令就可以了,还有一些shell 命令,建议买一本linux 入门的书看看;对于习惯windows 的同学,强烈建议学linux,开始的时候也许你觉得好多软件都有windows 版本的,但是早晚你会发现有很多软件没有,所以必须要学2. 硬件要求:计算机要求:现在电脑快跟白菜一个价了,所以建议个人电脑配置的好一点(如果有服务器就算了),推荐配置:64位系统(32 系统的话,内存受限,最多识别3G 多),redhat 或ubuntu 都可以,推荐ubuntu,它的apt-get 功能还是比较神器的,4个CPU 差不多,本人极力推荐装8G 内存,如果你不能忍受经常内存溢出的话。
当然如果有服务器,这些都不是问题。
至于显卡什么的,就算了,如果要是你想魔兽一下的话,可以跟你老板申请一下。
对了硬盘大点,因为测序数据一般比较大。
网络要求:这个好像你也管不了,一般实验室都已经固定了带宽。
遇有经常在数据库上下一些基因组或其他注释信息,所以还是进你所能的争取一下。
本人文字表达能力比较差,就唠唠叨叨先说这些,下次我会简单介绍一些高通量测序的基本知识和发展过程。
对于已经掌握这些入门知识(一般也是生物信息的入门知识)的同学可以飞过,如果你还有哪些不了解,可以简单的复习一下了!!高通量测序入门第二帖/bbs/thread-370713-1-1.html很高兴贴完第一帖得到那么多回复,本来这一帖早就该写的,因为最近课题比较紧而且遇到很多问题,所以拖到现在,向大家致歉!———————————————————扯淡分割线——————————————————————————正式开始之前,还是扯点八卦。
在第一帖之后,有个朋友给我发邮件问我华大的评价。
我也觉着华大是一个好有争议的话题。
我仔细想了一下那些质疑华大的人无非有两种理由:1. 华大太能忽悠2. 对于他们取得的成绩,很多人都说如果我有那么多钱我也能做。
我跟华大接触不是很多,而且我读博之前也那么觉得,可是我现在觉得我们应该好好的去阅读一下华大。
首先,现在的科研有几个不在忽悠(此忽悠不是贬义,试想,我们做的工作在发paper 时总是要写的意义重要一些,去让reviewer 觉得有发表的必要,这是不是忽悠;你去申请基金的时候,总是要把课题意义拔高再拔高,这是不是忽悠),大家都是在忽悠,何必五十步笑百步呢。
2. 给你那么多钱,你也不一定能有他们那么多成果。
华大到底拿了多少钱,我不知道,但是我知道拿他们那么多钱,没做出东西的人有的是。
我知道某个单位,要测某个微生物的基因组(为了影响就不说是什么物种了,说了物种就很容易知道哪个单位了),当时Roche 454 刚刚出来,该单位将测序意义定义为打破国外高科技技术垄断,人工与高通量测序技术赛跑。
人才啊,最后的结果是什么,在徘徊了两年,花费数十万(或上百万后),还是送到了华大,倒是真的没用454,因为已经出了通量更高的Illumina GA,最后文章发表在某杂志上,篇幅不到一页,亮点就是作者奇多,估算一下,每个作者不到十个单词。
当然这么极品的人还是比较少,我只是想说给你钱,你真的不知道怎么花。
————————————————扯淡完分割线————————————————————————————扯淡完,进正题,这一贴,主要简单介绍一些,测序数据分析的基本知识,心急的同学,不要着急,俗话说心急吃不了臭豆腐。
首先,介绍一下测序技术的发展过程和一些标志事件;说道测序,可能最先想到的是Sanger 和Maxam-Gilbert 这两个人,至于这两个人干了什么,就不用太清楚了,只要知道没有这两个人就不会有测序技术的今天.......就像没有GCD 就没有XZG 一样,自从有了这两个人就迎来了分子生物学的春天,自从有了这两个人分子生物学事业焕然一新.......事物的发展总是从量变到质变,在这个量变过程中,我们完成伟大的人类基因组计划还有很多的模式生物的基因组,那些鄙视华大的同学这里要记住这个过程中,华大是有贡献的。
质变来临: 忽如一夜春风来,ABI 3730 型测序仪渐行渐远,NGS (Next Generation sequence) 在哪里?马上就有答案。
Roche 454、Illumina GA、ABI SOLiD伴着春姑娘的脚步出现了。
这三种测序平台的原理、优缺点、发展历程估计大家已经听的很多了,如果想复习一下的同学可以google一下(俗话说,知之为知之,不知google 知)。
找不到?不能吧,两个检索方法:1. google 中输入: "filetype:ppt Next Generation sequence" .2. 直接pubmed 检索综述,找稍微好点的杂志,好好复习一下就好了。
由于本人用到的数据多是Illumina GA 平台,所以我后面的内容可能更倾向于这个平台。
先说几个概念:1. fasta 格式:其实我也不知道,为什么叫这个名字,其实也不用知道,你只要这是一种序列存储格式就好了,大概分为两行,第一行以> 开头,表明注释信息,第二行及往后均为序列信息。
2. fastq 格式:这个同样是序列存储格式,共分四行,前两行与fasta 一致,第三行一般是一个“+”字符,第四行就是序列质量分数,这个分数看起来有点奇怪,实际在对测序错误率进行log 变换后取整用ASCII 码的表述形式。
但是不同的测序仪换算方法稍有不同,这个换算过程,大家有兴趣可以看一下,针对自己用的平台要仔细看一下。
3. 序列比对:alignment, 好像没有什么好解释的,最简单的BLAST、BLAT 到后面的Seqmap/Bowtie/SOAP 等都是干这个用的,虽然我在工作中从来没有用过华大的SOAP, 但是某天无聊我测试了下,其性能绝对算不上差,而且protocol 竟有中文版,所以还值得试试。
现在出了N多的软件,反正原理就是两个,要么把基因组做索引,要么把测序的片段做索引.4. 好像知道这么多久可以进行数据分析了,可是我特别想写第四条,就把Illumina GA 测转录组样本提取流程说一下吧,测基因组的就更简单一些。
第一步:提取总的RNA,具体怎么做大家都比别人清楚,我说了你也不会听我的,不会的话就请你师姐/师兄教教你吧。
一般他们都比较热心,爱国爱家爱师妹嘛!第二步:纯化一下,一般真核都用Oligo(dT)纯化,原核好像直接去除rRNA(不知道什么原理,没做过)。
第三步:片段化,有的用酶,有的用超声破碎片段(听讲座的时候,无数次听到有人提问这个问题,无聊死了)第四步:反转录成cDNA然后PCR 扩增。
第五步:送个公司测序,然后攒人品等数据,好像现在北京有好多公司都能测,听老板说现在非常便宜,没有问过都少钱。
下一帖我会从测序数据回来后的分析开始讲,谢谢大家!。