欧洲药典附录313大全
欧洲药典7.4索引(现行版)

EUROPEAN PHARMACOPOEIA7.4INDEXTo aid users the index includes a reference to the supplement in which the latest version of a text can be found.For example:Amikacin sulfate...............................................7.1-3377means the monograph Amikacin sulfate can be found on page 3377of Supplement 7.1.Note that where no reference to a supplement is made,the text can be found in the principal volume.English index ........................................................................4401Latin index .................................................................................4433/forum.php?mod=viewthread&tid=21456&fromuid=1023/forum.php?mod=viewthread&tid=21456&fromuid=1023EUROPEAN PHARMACOPOEIA7.4EUROPEAN PHARMACOPOEIA 7.4Index Numerics 1.General parative table of porosity of sintered-glass filters...152.1.3.Ultraviolet ray lamps for analytical purposes...................152.1.4.Sieves.........................................................................................162.1.5.Tubes for comparative tests..................................................172.1.6.Gas detector tubes...................................................................172.1.Apparatus....................................................................................152.2.10.Viscosity -Rotatingviscometer method..........................282.2.11.Distillation range (30)2.2.12.Boiling point..........................................................................312.2.13.Determination of water by distillation (31)2.2.14.Melting point -capillary method........................................312.2.15.Melting point -open capillary method............................322.2.16.Meltingpoint -instantaneous method (32)2.2.17.Drop point..............................................................................322.2.18.Freezing point.......................................................................342.2.19.Amperometrictitration (34)2.2.1.Clarity and degree of opalescence of liquids.....................212.2.20.Potentiometric titration......................................................342.2.21.Fluorimetry. (35)2.2.22.Atomic emission spectrometry..........................................352.2.23.Atomic absorption spectrometry......................................362.2.24.Absorption spectrophotometry,infrared........................382.2.25.Absorption spectrophotometry,ultraviolet and visible..................................................................................................402.2.26.Paper chromatography.. (41)2.2.27.Thin-layer chromatography................................................422.2.28.Gas chromatography...........................................................432.2.29.Liquidchromatography......................................................452.2.2.Degree of coloration of liquids............................................222.2.30.Size-exclusion chromatography........................................462.2.31.Electrophoresis (46)2.2.31.Electrophoresis (5.8.)................................................7.1-33452.2.32.Loss on drying.......................................................................512.2.33.Nuclear magnetic resonance spectrometry.. (52)2.2.34.Thermal analysis...................................................................542.2.35.Osmolality...................................................................7.3-37772.2.36.Potentiometric determination of ionic concentration using ion-selective electrodes.............................572.2.37.X-ray fluorescence spectrometry.......................................582.2.38.Conductivity.. (59)2.2.39.Molecular mass distribution in dextrans........................602.2.3.Potentiometric determination of pH..................................242.2.40.Near-infrared spectrophotometry.. (61)2.2.41.Circular dichroism................................................................652.2.42.Density of solids...................................................................662.2.43.Mass spectrometry (67)2.2.44.Total organic carbon in water for pharmaceutical use.......................................................................................................692.2.45.Supercritical fluid chromatography.................................702.2.46.Chromatographic separation techniques........................702.2.47.Capillary electrophoresis (5.8.)...............................7.1-33452.2.47.Capillary electrophoresis.........................................7.1-33132.2.48.Raman spectrometry...........................................................822.2.49.Falling ball viscometer method.........................................832.2.4.Relationship between reaction of solution,approximate pH and colour of certain indicators...................................................252.2.54.Isoelectric focusing..............................................................832.2.54.Isoelectric focusing (5.8.)........................................7.1-33452.2.55.Peptide mapping...................................................................852.2.55.Peptide mapping (5.8.).............................................7.1-33452.2.56.Amino acidanalysis (88)2.2.56.Amino acid analysis (5.8.)........................................7.1-33452.2.57.Inductively coupled plasma-atomic emission spectrometry.....................................................................................942.2.58.Inductively coupled plasma-mass spectrometry............962.2.59.Glycan analysis of glycoproteins.......................................972.2.5.Relative density.......................................................................252.2.60.Melting point -instrumental method.............................102 2.2.64.Peptide identification by nuclear magnetic resonance spectrometry..........................................................................7.2-35312.2.6.Refractiveindex......................................................................262.2.7.Optical rotation.......................................................................262.2.8.Viscosity (27)2.2.9.Capillaryviscometer method (27)2.2.Physical and physicochemical methods.................................212.3.1.Identification reactions of ions and functional groups..1072.3.2.Identification of fatty oils by thin-layer chromato-graphy (110)2.3.3.Identification of phenothiazines by thin-layer chromatography (110)2.3.4.Odour......................................................................................1102.3.Identification.. (107)2.4.10.Lead in sugars (117)2.4.11.Phosphates............................................................................1172.4.12.Potassium (117)2.4.13.Sulfates........................................................................7.3-37812.4.14.Sulfated ash (118)2.4.14.Sulfated ash (5.8.).....................................................7.1-33462.4.15.Nickel in polyols (118)2.4.16.Total ash...............................................................................1182.4.17.Aluminium. (118)2.4.18.Free formaldehyde (118)2.4.19.Alkaline impurities in fatty oils.......................................1192.4.1.Ammonium.. (113)2.4.21.Foreign oils in fatty oils bythin-layer chromato-position of fatty acids by gas chromatography (119)2.4.23.Sterols in fatty oils....................................................7.2-35352.4.24.Identification and control of residual solvents...7.2-35372.4.25.Ethylene oxide and dioxan.. (127)2.4.26.N,N -Dimethylaniline (128)2.4.27.Heavy metals in herbal drugs and fatty oils.................1292.4.28.2-Ethylhexanoic acid (130)position of fatty acids in oils rich in omega-3acids..................................................................................................1302.4.2.Arsenic (113)2.4.30.Ethylene glycol and diethylene glycol inethoxylatedsubstances.......................................................................................1322.4.31.Nickel in hydrogenated vegetable oils (132)2.4.32.Total cholesterol in oils rich in omega-3acids (133)2.4.3.Calcium...................................................................................1132.4.4.Chlorides.. (113)2.4.5.Fluorides (114)2.4.6.Magnesium..............................................................................1142.4.7.Magnesium and alkaline-earth metals (114)2.4.8.Heavy metals (114)2.4.9.Iron...........................................................................................1172.4.Limit tests. (113)2.5.10.Oxygen-flaskmethod (139)plexometric titrations................................................1402.5.12.Water:semi-micro determination.. (140)2.5.13.Aluminium in adsorbed vaccines (141)2.5.14.Calcium in adsorbed vaccines..........................................1412.5.15.Phenol in immunosera and vaccines (141)2.5.16.Protein in polysaccharide vaccines (141)2.5.17.Nucleic acids in polysaccharide vaccines......................1422.5.18.Phosphorus in polysaccharide vaccines (142)2.5.19.O -Acetylin polysaccharide vaccines (142)2.5.1.Acid value...............................................................................1372.5.20.Hexosamines in polysaccharide vaccines. (142)2.5.21.Methylpentoses in polysaccharide vaccines.................1432.5.22.Uronic acids in polysaccharide vaccines.......................1432.5.23.Sialic acid in polysaccharide vaccines (143)2.5.24.Carbondioxide in gases (143)2.5.25.Carbon monoxide in gases...............................................1442.5.26.Nitrogen monoxide and nitrogen dioxide in gases.. (145)2.5.27.Oxygeningases..................................................................1452.5.28.Water in gases.....................................................................1452.5.29.Sulfur dioxide.....................................................................1452.5.2.Ester value.............................................................................1372.5.30.Oxidising substances (146)/forum.php?mod=viewthread&tid=21456&fromuid=1023Index EUROPEAN PHARMACOPOEIA7.42.5.31.Ribose in polysaccharide vaccines (146)2.5.32.Water:micro determination (146)2.5.33.Total protein (147)2.5.34.Acetic acid in synthetic peptides (150)2.5.35.Nitrous oxide in gases (150)2.5.36.Anisidine value (150)2.5.37.Methyl,ethyl and isopropyl methanesulfonate in methanesulfonic acid...........................................................7.1-3321 2.5.38.Methyl,ethyl and isopropyl methanesulfonate in active substances..............................................................................7.3-3785 2.5.39.Methanesulfonyl chloride in methanesulfonic acid...........................................................................................7.4-4093 2.5.3.Hydroxyl value. (137)2.5.4.Iodine value (137)2.5.5.Peroxide value (138)2.5.6.Saponification value (139)2.5.7.Unsaponifiable matter (139)2.5.8.Determination of primary aromatic amino-nitrogen (139)2.5.9.Determination of nitrogen by sulfuric acid digestion..139 2.5.Assays (137)2.6.10.Histamine (162)2.6.11.Depressor substances (162)2.6.12.Microbiological examination of non-sterile products: microbial enumeration tests (163)2.6.12.Microbiological examination of non-sterile products: microbial enumeration tests(5.8.)....................................7.1-3346 2.6.13.Microbiological examination of non-sterile products:test for specified micro-organisms.. (167)2.6.13.Microbiological examination of non-sterile products:test for specified micro-organisms(5.8.)..................................7.1-3346 2.6.14.Bacterial endotoxins. (171)2.6.15.Prekallikrein activator (175)2.6.16.Tests for extraneous agents in viral vaccines for human use (176)2.6.17.Test for anticomplementary activity of immunoglobulin (177)2.6.18.Test for neurovirulence of live virus vaccines (179)2.6.19.Test for neurovirulence of poliomyelitis vaccine (oral) (179)2.6.1.Sterility(5.8.)................................................................7.1-3346 2.6.1.Sterility..........................................................................7.1-3325 2.6.20.Anti-A and anti-B haemagglutinins.......................7.2-3545 2.6.21.Nucleic acid amplification techniques (181)2.6.22.Activated coagulation factors (185)2.6.24.Avian viral vaccines:tests for extraneous agents in seed lots (185)2.6.25.Avian live virus vaccines:tests for extraneous agents in batches of finished product (188)2.6.26.Test for anti-D antibodies in human immunoglobu-lin.............................................................................................7.2-3546 2.6.27.Microbiological control of cellular products. (191)2.6.2.Mycobacteria (156)2.6.30.Monocyte-activation test (192)2.6.31.Microbiological examination of herbal medicinal products for oral use (197)2.6.7.Mycoplasmas (156)2.6.8.Pyrogens (161)2.6.9.Abnormal toxicity (162)2.6.Biological tests (153)2.7.10.Assay of human coagulation factor VII (219)2.7.11.Assay of human coagulation factor IX (219)2.7.12.Assay of heparin in coagulation factors (220)2.7.13.Assay of human anti-D immunoglobulin (220)2.7.14.Assay of hepatitis A vaccine (222)2.7.15.Assay of hepatitis B vaccine(rDNA)......................7.3-3794 2.7.16.Assay of pertussis vaccine(acellular).. (223)2.7.17.Assay of human antithrombin III (224)2.7.18.Assay of human coagulation factor II (224)2.7.19.Assay of human coagulation factor X (225)2.7.19.Assay of human coagulation factor X(2.7.19.) (225)2.7.1.Immunochemical methods (201)2.7.20.In vivo assay of poliomyelitis vaccine(inactivated) (225)2.7.21.Assay of human von Willebrand factor..........................2262.7.22.Assay of human coagulation factor XI (227)2.7.23.Numeration of CD34/CD45+cells in haemato-poietic products (228)2.7.24.Flow cytometry (229)2.7.25.Assay of human plasmin inhibitor (230)2.7.27.Flocculation value(Lf)of diphtheria and tetanus toxins and toxoids(Ramon assay) (231)2.7.28.Colony-forming cell assay for human haematopoietic pro-genitor cells (232)2.7.29.Nucleated cell count and viability (233)2.7.2.Microbiological assay of antibiotics.........................7.3-3789 2.7.30.Assay of human protein C (234)2.7.31.Assay of human protein S (235)2.7.32.Assay of humanα-1-proteinase inhibitor (236)2.7.4.Assay of human coagulation factor VIII (207)2.7.5.Assay of heparin (208)2.7.6.Assay of diphtheria vaccine(adsorbed) (209)2.7.7.Assay of pertussis vaccine(whole cell)...................7.2-3549 2.7.8.Assay of tetanus vaccine(adsorbed) (214)2.7.9.Test for Fc function of immunoglobulin (217)2.7.Biological assays (201)2.8.10.Solubility in alcohol of essential oils (240)2.8.11.Assay of1,8-cineole in essential oils (240)2.8.12.Determination of essential oils in herbal drugs (241)2.8.13.Pesticide residues (242)2.8.14.Determination of tannins in herbal drugs (243)2.8.15.Bitterness value (244)2.8.16.Dry residue of extracts (244)2.8.17.Loss on drying of extracts (244)2.8.18.Determination of aflatoxin B1in herbal drugs (244)2.8.1.Ash insoluble in hydrochloric acid (239)2.8.20.Herbal drugs:sampling and sample preparation (246)2.8.21.Test for aristolochic acids in herbal drugs (247)2.8.22.Determination of ochratoxin A in herbal drugs (249)2.8.23.Microscopic examination of herbal drugs (250)2.8.2.Foreign matter (239)2.8.3.Stomata and stomatal index (239)2.8.4.Swelling index (239)2.8.5.Water in essential oils (239)2.8.6.Foreign esters in essential oils (239)2.8.7.Fatty oils and resinified essential oils in essential oils..239 2.8.8.Odour and taste of essential oils (240)2.8.9.Residue on evaporation of essential oils (240)2.8.Methods in pharmacognosy (239)2.9.10.Ethanol content and alcoholimetric tables (268)2.9.11.Test for methanol and2-propanol (270)2.9.12.Sieve test (270)2.9.14.Specific surface area by air permeability (271)2.9.16.Flowability (272)2.9.17.Test for extractable volume of parenteral preparations (273)2.9.17.Test for extractable volume of parenteral preparations (5.8.).........................................................................................7.1-3346 2.9.18.Preparations for inhalation:aerodynamic assessment of fine particles.. (274)2.9.19.Particulate contamination:sub-visible particles(5.8.).........................................................................................7.1-3347 2.9.19.Particulate contamination:sub-visible particles..................................................................................7.1-3333 2.9.1.Disintegration of tablets and capsules(5.8.).........7.1-3346 2.9.1.Disintegration of tablets and capsules....................7.1-3331 2.9.20.Particulate contamination:visible particles. (287)2.9.22.Softening time determination of lipophilic suppositories (288)2.9.23.Gas pycnometric density of solids (288)2.9.25.Dissolution test for medicated chewing gums....7.4-4097 2.9.26.Specific surface area by gas adsorption (291)2.9.26.Specific surface area by gas adsorption(5.8.)....7.1-3347 2.9.27.Uniformity of mass of delivered doses from multidose containers. (294)2.9.29.Intrinsic dissolution (294)2.9.2.Disintegration of suppositories and pessaries (255)2.9.31.Particle size analysis by laser light diffraction (295)/forum.php?mod=viewthread&tid=21456&fromuid=1023EUROPEAN PHARMACOPOEIA 7.4Index2.9.32.Porosity and pore-size distribution of solids by mercury porosimetry.....................................................................................2992.9.33.Characterisation of crystalline and partially crystallinesolids by X-ray powder diffraction (XRPD)...............................3012.9.34.Bulk density and tapped density of powders...............3052.9.35.Powder fineness.................................................................3082.9.36.Powder flow.........................................................................3082.9.36.Powder flow (5.8.).....................................................7.1-33472.9.37.Optical microscopy.. (311)2.9.37.Optical microscopy (5.8.).........................................7.1-33472.9.38.Particle-size distribution estimation by analytical sieving. (313)2.9.38.Particle-size distribution estimation by analytical sieving (5.8.).........................................................................................7.1-33472.9.39.Water-solid interactions:determination of sorption-desorption isotherms and of water activity....7.1-33352.9.3.Dissolution test for solid dosage forms..................7.3-37972.9.40.Uniformity of dosage units......................................7.4-41012.9.41.Friability of granules and spheroids.....................7.4-41032.9.42.Dissolution test for lipophilic solid dosage forms.......3192.9.43.Apparent dissolution.........................................................3202.9.44.Preparations for nebulisation:characterisation.........7.3-38032.9.45.Wettability of porous solids including powders..........3212.9.4.Dissolution test for transdermal patches........................2632.9.5.Uniformity of mass of single-dose preparations.............2652.9.6.Uniformity of content of single-dose preparations........2662.9.7.Friability of uncoated tablets.............................................2662.9.7.Friability of uncoated tablets (5.8.)..........................7.1-33462.9.8.Resistance to crushing of tablets......................................2672.9.9.Measurement of consistency by penetrometry.. (267)2.9.Pharmaceutical technical procedures.................................2533.1.10.Materials based on non-plasticised poly(vinyl chloride)for containers for non-injectable,aqueous solutions............3493.1.11.Materials based on non-plasticised poly(vinyl chloride)for containers for dry dosage forms for oral administration (350)3.1.1.1.Materials based on plasticised poly(vinyl chloride)for containers for human blood and blood components.............3293.1.1.2.Materials based on plasticised poly(vinyl chloride)for tubing used in sets for the transfusion of blood and blood components.....................................................................................3323.1.13.Plastic additives (352)3.1.14.Materials based on plasticised poly(vinyl chloride)for containers for aqueous solutions for intravenous infusion..3553.1.15.Polyethylene terephthalate for containers forpreparations not for parenteral use...........................................3573.1.1.Materials for containers for human blood and blood components.....................................................................................3293.1.3.Polyolefines............................................................................3343.1.4.Polyethylene without additives for containers for parenteralpreparationsand for ophthalmic preparations (337)3.1.5.Polyethylene with additives for containers for parenteral preparations and for ophthalmic preparations.......................3383.1.6.Polypropylene for containers and closures for parenteralpreparations and ophthalmic preparations..............................3423.1.7.Poly(ethylene -vinyl acetate)for containers and tubing for totalparenteralnutritionpreparations (345)3.1.8.Silicone oil used as a lubricant.........................................3473.1.9.Silicone elastomer for closures and tubing....................3473.1.Materials used forthe manufacture of containers (329)3.2.1.Glass containers for pharmaceutical use........................3633.2.2.1.Plastic containers for aqueous solutions for infusion............................................................................................3683.2.2.Plastic containers and closures for pharmaceutical use.....................................................................................................3683.2.3.Sterile plastic containers for human blood andblood components.........................................................................3693.2.4.Empty sterile containers of plasticised poly(vinyl chloride)for human blood and blood components.................................3703.2.5.Sterile containers of plasticised poly(vinyl chloride)for human blood containing anticoagulant solution...................3713.2.6.Sets for the transfusion of blood and blood components.....................................................................................371 3.2.8.Sterile single-use plastic syringes.....................................3733.2.9.Rubber closures for containers for aqueous parenteralpreparations,for powders and for freeze-dried powders (374)3.2.Containers.................................................................................3633-O -Desacyl-4′-monophosphoryl lipid A.............................7.2-36394.1.1.Reagents........................................................................7.4-41094.1.2.Standard solutions for limit tests.............................7.4-42164.1.3.Buffersolutions...........................................................7.4-42204.1.Reagents,standard solutions,buffer solutions........7.4-41094.2.1.Primary standards for volumetric solutions..........7.4-42254.2.2.Volumetric solutions...................................................7.4-42254.2.Volumetric analysis.........................................................7.4-42254-Aminobenzoic acid.....................................................................13714.Reagents...............................................................................7.4-41095.10.Control of impurities in substances for pharmaceutical use............................................................................................7.4-42475.1.10.Guidelines for using the test forbacterial endotoxins..5205.11.Characters section in monographs....................................6375.1.1.Methods of preparation of sterile products....................5035.1.2.Biological indicators of sterilisation (504)5.12.Reference standards (641)5.1.3.Efficacy of antimicrobial preservation.............................5055.14.Genetransfer medicinalproducts for human use (647)5.1.4.Microbiological quality of non-sterile pharmaceutical preparations and substances for pharmaceutical use...........5075.1.4.Microbiological quality ofnon-sterile pharmaceutical preparations and substances for pharmaceutical use (5.8.).........................................................................................7.1-33475.1.5.Application of the F 0concept to steamsterilisation ofaqueous preparations...................................................................5085.15.Functionality-related characteristics of excipients.........6615.1.6.Alternative methods for control ofmicrobiologicalquality...............................................................................................5085.16.Crystallinity....................................................................7.4-42535.17.1.Recommendations on dissolution testing.....................6655.17.Recommendations on methods for dosage forms testing...............................................................................................6655.1.7.Viralsafety (518)5.1.8.Microbiological quality of herbal medicinal products for oral use (519)5.1.9.Guidelines for using the test for sterility (519)5.1.General texts on microbiology..............................................5035.2.1.Terminology used in monographs on biologicalproducts (527)5.2.2.Chicken flocks free from specified pathogens for theproduction and quality control of vaccines.............................5275.2.3.Cell substrates for the production of vaccines for humanuse.....................................................................................................5305.2.4.Cell cultures for the production of veterinaryvaccines (533)5.2.5.Substances of animal origin for the production ofimmunological veterinary medicinal products........................5355.2.6.Evaluation of safety of veterinary vaccines andimmunosera ...................................................................................5365.2.7.Evaluation of efficacy of veterinary vaccines andimmunosera (538)5.2.8.Minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinalproducts..................................................................................7.4-42335.2.9.Evaluation of safety of each batch of veterinary vaccines and immunosera............................................................................5475.2.Generaltexts on biological products...................................5275.3.Statistical analysis of results of biological assays and tests...................................................................................................5515.4.Residual solvents (583)5.5.Alcoholimetric tables (593)5.6.Assay of interferons.................................................................6075.7.Table of physical characteristics of radionuclidesmentionedin the European Pharmacopoeia (611)5.8.Pharmacopoeial harmonisation..................................7.1-33455.9.Polymorphism...........................................................................627/forum.php?mod=viewthread&tid=21456&fromuid=1023。
欧洲药典附录

第二部分、附录附录1 溶液的澄清度 .................................附录2 溶液颜色检查 .................................附录3 旋光度 .......................................附录4 铵盐检查法 ...................................附录5 氯化物检查法 .................................附录6 硫酸盐灰分 ...................................附录7 铁 ...........................................附录8 重金属 .......................................附录9 干燥失重 .....................................附录10 硫酸盐检查法 ................ 错误!未定义书签。
附录11 红外吸收分光光度法............................附录12 pH测定.......................................附录13 滴定 .........................................附录14 氯化物鉴别反应 ...............................附录15 指示剂颜色与溶液pH 的关系....................附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
国外药典介绍

欧洲药典增补版
• 欧洲药典第8版包括两个基本卷,于2013年7月出版发行,以后在每次欧洲
药典委员会全会做出决定后,通过非累积增补本更新,每年出3个增补本。第 8版累计共有8个非累积增补本(8.1~8.8)。 各增补版的出版日期及执行的日期。·
欧洲各册内容介绍
第一卷
各论举例、前言、介绍、总目录、第8版内容简介(包括新增内容、 修订内容和更正内容)
欧洲各册内容介绍第一卷各论举例前言介绍总目录第8版内容简介包括新增内容修订内容和更正内容版内容简介包括新增内容修订内容和更正内容genaralnotice凡例分析方法通论包装材料及包装试剂genaraltext通则各论通则剂型各论人用疫苗各论兽用疫苗各论人用免疫血通则各论通则剂型各论人用疫苗各论兽用疫苗各论人用免疫血清各论兽用免疫血清各论放射制剂及放射制剂起始物各论人20151215清各论兽用免疫血清各论放射制剂及放射制剂起始物各论人用手术缝合线各论兽用手术缝合线各论草药及草药制剂各论顺势疗法制剂各论用手术缝合线各论兽用手术缝合线各论草药及草药制剂各论顺势疗法制剂各论第二卷各论举例各论按字母的索引页总索引每卷均有侧面黑色索引标示各论举例各论按字母的索引页总索引每卷均有侧面黑色索引标示欧洲各册内容介绍增补举例
USP–NF 的不断 修订
修订公告 IRA拟议的修订 说明
勘误表
2019/12/16
美国药典的修订
USP–NF 不断进行修订。 修订包括 USP–NF 年度修订和每年两 次增补,以及 USP 网站上的加速修订。 USP 使用加速修订过程 加快修订美国药典–国家处方集 (USP–NF)。加速修订包括修订公 告、临时修订声明 (IRA) 和勘误表。 是 USP 最快的修订途径,可取代在 USP–NF 及其增补(印刷版 和在线版)中发布的标准。 在 USP 网站上发布的修订公告指示 其正式日期和纳入正式出版物中的日期。 IRA 在 PF 中发布,征求公众意见期为 90 天。 在意见(如果有) 通过审查并且 IRA 得到相关专家委员会的批准后,最终 IRA 将发 布在 USP 网站上。 与修订公告一样,IRA 可取代在印刷版和在线 版的 USP–NF 及其增补中发布的标准。 IRA 被纳入下一个可用的 USP–NF 或增补中。 是指在 USP–NF 或其增补中发布的文字有误,不能准确地反映专 家委员会批准的预期要求。 勘误表发布在网站上,并立即成为正 式版本。 勘误表被纳入下一个可用的正式出版物中。
欧洲药典8.0版附录2.9.40具体内容

欧洲药典8.0版附录2.9.40是一项具有重要意义的内容,它包含了关于卫生产品和医药制剂的质量要求和标准。
在这篇文章中,我们将深入探讨这一主题,从基础概念到具体内容,帮助你更好地理解欧洲药典8.0版附录2.9.40。
1. 了解欧洲药典8.0版附录2.9.40欧洲药典是欧洲药典委员会制定的标准规范,旨在保障卫生产品和医药制剂的质量、安全和有效性。
附录2.9.40则是其中的重要内容之一,它详细规定了一系列的质量要求,涵盖了原材料的选择、生产过程的控制、成品的质量检验等方方面面。
这些要求旨在确保药品的质量稳定、安全性高、有效性强。
2. 欧洲药典8.0版附录2.9.40的具体内容欧洲药典8.0版附录2.9.40的具体内容主要包括以下几个方面:- 原材料的要求:包括对原材料的来源、制备、存储和使用的规定,确保原材料的质量稳定和可追溯性。
- 生产过程的控制:包括药品的生产工艺、设备、人员培训等方面的要求,确保生产过程稳定可控、符合GMP要求。
- 药品的质量检验:包括对成品药品的各项质量指标和检测方法的规定,确保药品符合质量标准。
3. 个人观点和理解欧洲药典8.0版附录2.9.40的具体内容凸显了对药品质量和安全的高标准要求,这种规范的制定对保障患者用药安全、促进药品质量提升有着重要的意义。
而且,这种规范也对制药企业的生产经营提出了更高的要求,能够推动行业向着更加规范、科学的方向发展。
总结回顾通过本文的阐述,相信你对欧洲药典8.0版附录2.9.40有了更深入的了解。
这一规范的制定和实施,促进了药品质量的提升、促进了医药行业的良性发展,对患者和企业来说都具有重要意义。
希望你能在日常工作和学习中,更加关注和重视这一规范,促进药品质量和安全的保障。
4. 药品质量和安全的重要性药品质量和安全对于患者的健康和生命安全具有重要的意义。
优质的药品能够有效治疗疾病,保障患者的健康。
然而,如果药品质量不达标或者存在安全隐患,可能会导致患者用药失败或者出现严重的副作用,危及患者的生命安全。
eugmp附录1解读 -回复

eugmp附录1解读-回复“eugmp附录1解读”首先,需要明确eugmp是什么意思。
eugmp是欧洲药典协会(European Union Good Manufacturing Practices)的缩写,该协会制定了一系列药品生产管理的规范和指南,旨在确保药品的质量和安全性。
而“eugmp 附录1”则是eugmp的第一个附录,其中详细陈述了药品生产过程中的规定和要求。
eugmp附录1主要涵盖了药品生产过程中的设备、人员、文档等各个环节的规定。
下面将按照这些环节一步一步进行解读和回答。
1. 设备:根据eugmp附录1的规定,药品生产过程中所使用的设备必须符合一定的标准,例如必须具备适当的清洁性能、可靠性能和操作性能等。
此外,还要求对设备进行定期的维护和保养,并保证其符合相应的验证要求。
同时,还要求设备的使用必须经过适当的培训和授权,并进行记录。
2. 人员:eugmp附录1对药品生产过程中的人员要求也十分严格。
首先,要求药品生产厂家必须拥有足够的员工,以保证药品生产过程的顺利进行。
其次,要求严格培训和授权所有参与药品生产的人员,确保其具备足够的专业知识和技能。
此外,还要求人员要定期接受必要的培训和教育,以保证其在生产过程中的操作符合相应的标准。
3. 文档:eugmp附录1还对药品生产过程中的文档管理提出了要求。
具体来说,要求药品生产厂家必须确保所有的生产文档都要进行准确和完整的记录,包括原料的采购记录、生产过程中的记录以及产品的出货记录等。
此外,还要求对这些记录进行适当的存档和管理,以供后续的审查和追溯。
综上所述,eugmp附录1对药品生产过程中的设备、人员和文档等环节都提出了明确的要求。
这些要求旨在确保药品的质量和安全性,防止药品生产过程中出现的差错和问题。
只有严格遵守eugmp附录1的规定,药品生产厂家才能生产出高质量、安全可靠的药品,以满足患者和社会的需求。
然而,光有规定还不够,必须要有监督和执行的机制来确保这些规定得到遵守。
欧洲药典附录中文翻译

附录1溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静臵4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g 乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静臵24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容1000.0ml。
欧洲药典附录中文版.

第二部分、附录附录1 溶液的澄清度 (2)附录2 溶液颜色检查 (3)附录3 旋光度 (7)附录4 铵盐检查法 (9)附录5 氯化物检查法 (11)附录6 硫酸盐灰分 (13)附录7 铁 (14)附录8 重金属 (16)附录9 干燥失重 (21)附录10 硫酸盐检查法 (23)附录11 红外吸收分光光度法 (25)附录12 pH测定 (29)附录13 滴定 (34)附录14 氯化物鉴别反应 (37)附录15 指示剂颜色与溶液pH 的关系 (38)附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
欧洲药典7.0-凡例(全)

欧洲药典7.0-凡例(全)07/2010:10000 1. 凡例1.1. 概述凡例的内容适⽤于各论和欧洲药典中的其它章节。
欧洲药典以英语和法语形式发⾏,欧洲药典委员会的签署国可将药典内容译成其它语⾔,但若发⽣争议,应以英语和法语版为权威。
在欧洲药典中,如⽆特殊规定,“药典”是指欧洲药典,官⽅缩写Ph. Eur.也指欧洲药典。
⽂章中如果引⽤了各论中的标题和副标题意味着⽂章内容符合相关各论的要求。
⽂章参考药典中各论内容时,以斜体的各论题⽬或相关数字表⽰。
制剂在有效期内必须性质稳定,明确的有效期或说明书应由权⼒机构批准。
任何各论的物质也必须服从其使⽤期限。
任何药品的有效期和有效期的计算由权⼒机构经稳定性研究的试验结果决定。
除凡例和各论中另有说明,各论中的说明为强制要求;除了特定的引⽤信息,如果各论引⽤总论中内容时,该总论要求为法定要求。
各论中描述的活性物质,赋形剂,药物制剂和其它项⽬都是⼈⽤和兽⽤的(除⾮明确限制不可使⽤)。
药品项⽬必须符合各论的要求,否则不符合药典质量。
但并不要求产品放⾏前,⽣产商要做各论中的每项试验以满⾜药典要求。
⽣产商可通过原始数据,例如⽣产过程验证,和中间体控制,确保药品是否符合药典要求。
公布的环境参数,权⼒机构可适当采信,但不排除故意满⾜药典要求的可能。
检测和试验⽅法应基于药典标准的官⽅⽅法。
经权利机构允许可采⽤其它替代的分析⽅法以达到控制⽬的,并证明该⽅法是否能达到各论各标准。
若出现争论或异议,应以药典⽅法为准。
药典各论中的某些物质有多个等级可满⾜各种需要,除各论中另有说明,要求适⽤于各等级。
在⼀些各论中,特别是赋形剂,⼀系列相关的功能特性都有介绍,其中给出了⼀些特性的检测⽅法。
质量体系:在适宜的质量体系架构下,产⽣有疑问的项⽬时,应以各论中的质量标准为法定标准。
通则:各论中介绍的药物和制剂也应符合通则中的相关要求。
交叉引⽤的通则在各论中不特别指出。
除⾮限定了适⽤条件,如规定适⽤于药典各论中的物质,通则的内容适⽤于各论定义范围内的所有药物和制剂。
欧洲药典7.0-凡例(全)

07/2010:10000 1. 凡例1.1. 概述凡例的内容适用于各论和欧洲药典中的其它章节。
欧洲药典以英语和法语形式发行,欧洲药典委员会的签署国可将药典内容译成其它语言,但若发生争议,应以英语和法语版为权威。
在欧洲药典中,如无特殊规定,“药典”是指欧洲药典,官方缩写Ph. Eur.也指欧洲药典。
文章中如果引用了各论中的标题和副标题意味着文章内容符合相关各论的要求。
文章参考药典中各论内容时,以斜体的各论题目或相关数字表示。
制剂在有效期内必须性质稳定,明确的有效期或说明书应由权力机构批准。
任何各论的物质也必须服从其使用期限。
任何药品的有效期和有效期的计算由权力机构经稳定性研究的试验结果决定。
除凡例和各论中另有说明,各论中的说明为强制要求;除了特定的引用信息,如果各论引用总论中内容时,该总论要求为法定要求。
各论中描述的活性物质,赋形剂,药物制剂和其它项目都是人用和兽用的(除非明确限制不可使用)。
药品项目必须符合各论的要求,否则不符合药典质量。
但并不要求产品放行前,生产商要做各论中的每项试验以满足药典要求。
生产商可通过原始数据,例如生产过程验证,和中间体控制,确保药品是否符合药典要求。
公布的环境参数,权力机构可适当采信,但不排除故意满足药典要求的可能。
检测和试验方法应基于药典标准的官方方法。
经权利机构允许可采用其它替代的分析方法以达到控制目的,并证明该方法是否能达到各论各标准。
若出现争论或异议,应以药典方法为准。
药典各论中的某些物质有多个等级可满足各种需要,除各论中另有说明,要求适用于各等级。
在一些各论中,特别是赋形剂,一系列相关的功能特性都有介绍,其中给出了一些特性的检测方法。
质量体系:在适宜的质量体系架构下,产生有疑问的项目时,应以各论中的质量标准为法定标准。
通则:各论中介绍的药物和制剂也应符合通则中的相关要求。
交叉引用的通则在各论中不特别指出。
除非限定了适用条件,如规定适用于药典各论中的物质,通则的内容适用于各论定义范围内的所有药物和制剂。
欧洲药典在线查询全文
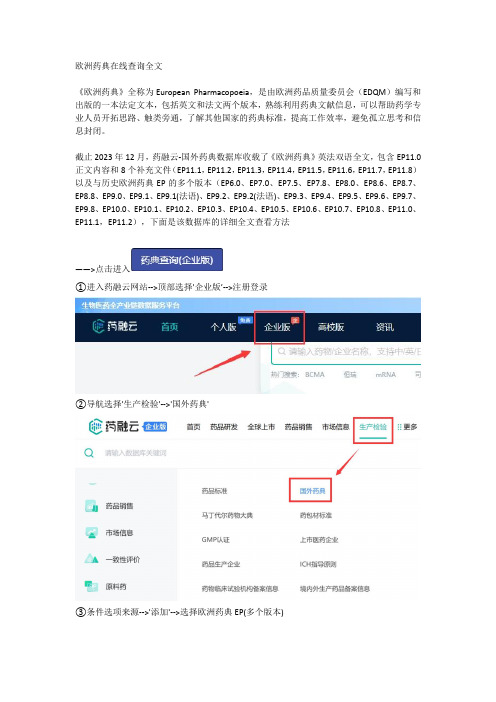
欧洲药典在线查询全文《欧洲药典》全称为European Pharmacopoeia,是由欧洲药品质量委员会(EDQM)编写和出版的一本法定文本,包括英文和法文两个版本,熟练利用药典文献信息,可以帮助药学专业人员开拓思路、触类旁通,了解其他国家的药典标准,提高工作效率,避免孤立思考和信息封闭。
截止2023年12月,药融云-国外药典数据库收载了《欧洲药典》英法双语全文,包含EP11.0正文内容和8个补充文件(EP11.1,EP11.2,EP11.3,EP11.4,EP11.5,EP11.6,EP11.7,EP11.8)以及与历史欧洲药典EP的多个版本(EP6.0、EP7.0、EP7.5、EP7.8、EP8.0、EP8.6、EP8.7、EP8.8、EP9.0、EP9.1、EP9.1(法语)、EP9.2、EP9.2(法语)、EP9.3、EP9.4、EP9.5、EP9.6、EP9.7、EP9.8、EP10.0、EP10.1、EP10.2、EP10.3、EP10.4、EP10.5、EP10.6、EP10.7、EP10.8、EP11.0、EP11.1,EP11.2),下面是该数据库的详细全文查看方法——>点击进入①进入药融云网站-->顶部选择'企业版’-->注册登录②导航选择'生产检验'-->'国外药典'③条件选项来源-->'添加'-->选择欧洲药典EP(多个版本)-----最后说下关于欧洲药典新版本EP11---《欧洲药典》EP11.0于2023年1月1日开始实施,是欧洲药品行业的权威参考书,EP11.0版本对之前版本的内容进行了全面更新和重新组织,新增和修改了600多个单体和试剂。
因此欧洲药典2024版将涵盖超过4000个全新的认证试剂,并提供7000多个化学物质的简要概述。
EP11.0版本的欧洲药典引入了一些重要的改进。
世界各国药典大汇总

世界各国药典大汇总中国药典(CHP):介绍就省了,大家都比较熟悉。
美国药典/国家处方集(简称USP/NF)U.S. Pharmacopeia / National Formulary:由美国政府所属的美国药典委员会(The United States Pharmacopeial Convention)编辑出版。
USP于1820年出第一版,1950年以后每5年出一次修订版,到2005年已出至第28版。
NF1883年第一版, 1980年15版起并入USP,但仍分两部分,前面为USP,后面为NF。
美国药典是美国政府对药品质量标准和检定方法作出的技术规定,也是药品生产、使用、管理、检验的法律依据。
NF收载了美国药典(USP)尚未收入的新药和新制剂。
美国药典正文药品名录分别按法定药名字母顺序排列,各药品条目大都列有药名、结构式、分子式、CAS登记号、成分和含量说明、包装和贮藏规格、鉴定方法、干燥失重、炽灼残渣、检测方法等常规项目,正文之后还有对各种药品进行测试的方法和要求的通用章节及对各种药物的一般要求的通则。
可根据书后所附的USP 和NF的联合索引查阅本书。
英国药典(BP):/《英国药典》是英国药品委员会(British Pharmacopoeia Commission)的正式出版物,是英国制药标准的重要来源。
英国药典不仅为读者提供了药用和成药配方标准以及公式配药标准,而且也向读者展示了许多明确分类并可参照的欧洲药典专著。
英国药典出版周期不定。
BP2004该药典由三卷本组成。
其中两卷为英国药典、一卷为英国兽药典(兽医药品部分)。
各条目均以药品名称字母顺序排列,内容包括药品性质、制法、血产品、免疫产品、电磁药品制法及外科材料等部分。
英国药典书后附有全部内容关键词索引。
欧洲药典(EP):/欧洲药典委员会1964年成立。
1977年出版第一版《欧洲药典》。
从1980年到1996年期间,每年将增修订的项目与新增品种出一本活页本,汇集为第二版《欧洲药典》各分册,未经修订的仍按照第一版执行。
欧洲药典7.0-凡例(全)
07/2010:10000 1. 凡例1.1. 概述凡例的内容适用于各论和欧洲药典中的其它章节。
欧洲药典以英语和法语形式发行,欧洲药典委员会的签署国可将药典内容译成其它语言,但若发生争议,应以英语和法语版为权威。
在欧洲药典中,如无特殊规定,“药典”是指欧洲药典,官方缩写Ph. Eur.也指欧洲药典。
文章中如果引用了各论中的标题和副标题意味着文章内容符合相关各论的要求。
文章参考药典中各论内容时,以斜体的各论题目或相关数字表示。
制剂在有效期内必须性质稳定,明确的有效期或说明书应由权力机构批准。
任何各论的物质也必须服从其使用期限。
任何药品的有效期和有效期的计算由权力机构经稳定性研究的试验结果决定。
除凡例和各论中另有说明,各论中的说明为强制要求;除了特定的引用信息,如果各论引用总论中内容时,该总论要求为法定要求。
各论中描述的活性物质,赋形剂,药物制剂和其它项目都是人用和兽用的(除非明确限制不可使用)。
药品项目必须符合各论的要求,否则不符合药典质量。
但并不要求产品放行前,生产商要做各论中的每项试验以满足药典要求。
生产商可通过原始数据,例如生产过程验证,和中间体控制,确保药品是否符合药典要求。
公布的环境参数,权力机构可适当采信,但不排除故意满足药典要求的可能。
检测和试验方法应基于药典标准的官方方法。
经权利机构允许可采用其它替代的分析方法以达到控制目的,并证明该方法是否能达到各论各标准。
若出现争论或异议,应以药典方法为准。
药典各论中的某些物质有多个等级可满足各种需要,除各论中另有说明,要求适用于各等级。
在一些各论中,特别是赋形剂,一系列相关的功能特性都有介绍,其中给出了一些特性的检测方法。
质量体系:在适宜的质量体系架构下,产生有疑问的项目时,应以各论中的质量标准为法定标准。
通则:各论中介绍的药物和制剂也应符合通则中的相关要求。
交叉引用的通则在各论中不特别指出。
除非限定了适用条件,如规定适用于药典各论中的物质,通则的内容适用于各论定义范围内的所有药物和制剂。
欧洲药典附录

第二部分、附录附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取硫酸肼溶于水,加水稀释至,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液?:在100ml容量平中,以水溶解乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1ⅠⅡⅢⅣ浊度标准液水附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B,则可9判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml 水混和)溶解,继续添加,并定容。
欧洲药典附录3.1.3.大全

3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3% –1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3% –二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
欧洲药典附录中文翻译
附录1溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静臵4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g 乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静臵24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容1000.0ml。
欧洲药典7.0-凡例(全)
07/2010:10000 1. 凡例1.1. 概述凡例的内容适用于各论和欧洲药典中的其它章节。
欧洲药典以英语和法语形式发行,欧洲药典委员会的签署国可将药典内容译成其它语言,但若发生争议,应以英语和法语版为权威。
在欧洲药典中,如无特殊规定,“药典”是指欧洲药典,官方缩写Ph. Eur.也指欧洲药典。
文章中如果引用了各论中的标题和副标题意味着文章内容符合相关各论的要求。
文章参考药典中各论内容时,以斜体的各论题目或相关数字表示。
制剂在有效期内必须性质稳定,明确的有效期或说明书应由权力机构批准。
任何各论的物质也必须服从其使用期限。
任何药品的有效期和有效期的计算由权力机构经稳定性研究的试验结果决定。
除凡例和各论中另有说明,各论中的说明为强制要求;除了特定的引用信息,如果各论引用总论中内容时,该总论要求为法定要求。
各论中描述的活性物质,赋形剂,药物制剂和其它项目都是人用和兽用的(除非明确限制不可使用)。
药品项目必须符合各论的要求,否则不符合药典质量。
但并不要求产品放行前,生产商要做各论中的每项试验以满足药典要求。
生产商可通过原始数据,例如生产过程验证,和中间体控制,确保药品是否符合药典要求。
公布的环境参数,权力机构可适当采信,但不排除故意满足药典要求的可能。
检测和试验方法应基于药典标准的官方方法。
经权利机构允许可采用其它替代的分析方法以达到控制目的,并证明该方法是否能达到各论各标准。
若出现争论或异议,应以药典方法为准。
药典各论中的某些物质有多个等级可满足各种需要,除各论中另有说明,要求适用于各等级。
在一些各论中,特别是赋形剂,一系列相关的功能特性都有介绍,其中给出了一些特性的检测方法。
质量体系:在适宜的质量体系架构下,产生有疑问的项目时,应以各论中的质量标准为法定标准。
通则:各论中介绍的药物和制剂也应符合通则中的相关要求。
交叉引用的通则在各论中不特别指出。
除非限定了适用条件,如规定适用于药典各论中的物质,通则的内容适用于各论定义范围内的所有药物和制剂。
欧洲药典EP列表之三

EP(欧洲药典委员会)是欧洲的一个关键的组织,参与协调与合作的标准化、监管和质量控制的药物、输血、器官移植、制药和医药保健.安全药物以及它们安全使用的质量标准。
我们的标准品已经被认可为全球性的科学基准。
欧洲药典在欧盟成员国内具有法律约束力。
同样,EDQM开发用于输血、器官移植和消费者的身体健康问题领域的标准品和指导发展。
我们的使命是保障人类获取良好的药物和医疗保健的基本权利和促进、保护人类和动物的健康。
我们重视的是,首先,公共健康、科学技能、正直、客观和对欧洲委员会基本原则的尊重。
我们致力于与地区、国家以及国际机构、政府、机构和行业协会之间的合作以获取更大的收益。
我们还致力于通过不断改进,获取最高质量的产品和服务,为我们的客户、合作伙伴和员工争取更大的利益。
广州优瓦仪器有限公司是从事提供实验室分析领域内产品的专业公司;目前公司已是集研发、生产与销售为一体的综合性企业;在行业内具有良好的声誉;主要产品包括色谱产品、化学试剂、标准品、实验室用品、分析仪器配件及耗材等,总部位于香港。
我们代理的品牌有:USP、EP、BP、TLC、TRC、Molcan、LGC、Wellington、Chiron、Witega、NRC、ERM、Irmm、MBH、Fluorochem、TCI、Chemservice、Accstandard、 GmbH 、CIL 、C/D/N ISOTopes 、Inorganic Ventures、ULTRA Scientific 、NSI 、KeyOrganics、LC-Laboratoies 、Wibby 、APSC 、SPEX CertiPrep 、NIST、ACROS、Fluka、Matrix、Sigma、TCI、Strem、BP、Cerilliant 、Chromadex、Frontier、Echelon 、Serva 、Medical Isotope 、ChemBridge 、AMRESCO、SGE Analytical Science 、Brand BRAND 、VITLAB 、ISO 、Hamilton 、ISOLAB我们专注进口标准品。
欧洲药典附录译文

第二部分、附录附录1 溶液的澄清度 (2)附录2 溶液颜色检查 (3)附录3 旋光度 (7)附录4 铵盐检查法 (9)附录5 氯化物检查法 (11)附录6 硫酸盐灰分 (13)附录7 铁 (14)附录8 重金属 (16)附录9 干燥失重 (21)附录10 硫酸盐检查法 (23)附录11 红外吸收分光光度法 (25)附录12 pH测定 (29)附录13 滴定 (34)附录14 氯化物鉴别反应 (37)附录15 指示剂颜色与溶液pH 的关系 (38)附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取1.0g硫酸肼溶于水,加水稀释至100.0ml,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以25.0ml水溶解2.5g乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加25.0ml的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
表1-1附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
欧盟GMP附录

欧洲共同体:European Communities (EC)。
欧洲联盟:European Union (EU),简称欧盟。
人用药品注册技术标准国际协调会:ICH欧盟GMP附录1无菌药品的生产注:冻干瓶轧盖的条款自2010年3月1日开始实施。
原则为降低微生物、微粒和热原污染的风险,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法和规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
注:本指南没有对微粒、浮游菌和表面微生物等测试方法详细进行阐述,可参阅欧洲标准或国际标准(CEN/ISO)及药典资料。
总则1.无菌药品的生产必须在洁净区内进行,人员和(或)设备以及物料必须通过缓冲进入洁净区。
洁净区应当保持适当的洁净度,洁净区的送风须经具有一定过滤效率过滤器的过滤。
2.原料配制、产品加工和灌装等不同操作必须在洁净去内彼此分开的单独区域内进行。
生产工艺可分为两类:一类是最终灭菌工艺;第二类是部分或全部工序为无菌操作的工艺。
3.应按所需环境的特点确定无菌产品的洁净级别。
每一步生产操作都应达到适当的动态洁净度,以尽可能降低产品(或原料)被微粒或微生物污染。
洁净区的设计必须符合相应的“静态”标准,以达到“动态”的洁净要求。
“静态”是指安装已经完成并已运行,但没有操作人员在场的状态。
“动态”是指生产设施按预定的工艺模式运行并有规定数量的操作人员进行现场操作的状态。
应确定每一洁净室或每组洁净间的“动态”及“静态”标准。
无菌药品生产所需的洁净区一般可分为4个级别:A级:高风险操作区,如:灌装区,放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配/连接操作的区域。
通常用单向流操作台/罩来维护该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3% –1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3% –二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
–铝碳酸镁:限量:0.5%;–烷基酰胺:限量:0.5%;–烯烃酰胺:限量:0.5%;–硅铝酸钠:限量:0.5%;–二氧化硅: 限量:0.5%;–苯甲酸钠: 限量:0.5%;–脂肪酸酯及其盐类: 限量:0.5%;–磷酸三钠: 限量:0.5%;–液态石蜡: 限量:0.5%;–氧化锌: 限量:0.5%;–滑石: 限量:0.5%;–氧化镁: 限量:0.5%;–硬脂酸钠或硬脂酸锌或两者的混合物:限量:0.5%;–二氧化钛: 限量:4%;材料提供者须能证明样品组成的数量和质量都符合要求。
特性外观:粉末,珠子,颗粒或在转换后,不同厚度的片材或容器。
溶解性:几乎不溶于水,溶于热的芳香烃,几乎不溶于乙醇,乙烷和甲醇。
它们的软化温度在65 °C和165 °C之间,它们燃烧时都会有蓝色火焰。
鉴定:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
A.红外吸收分光光度法(2.2.24)准备:在0.25g样品中加入10ml的甲苯R并在回流冷凝器下加热约15分钟;放置几滴从氯化钠滑落获得的溶剂,然后在80°C的烘箱上将溶剂烘干。
最大吸收值:2920 cm− 1, 2850 cm− 1, 1475 cm− 1,1465 cm− 1, 1380 cm− 1, 1170 cm− 1, 735 cm− 1 和 720 cm− 1.所得出的光谱和材料选出的标准样品的光谱是一样的。
如果材料以表格的形式进行检查,鉴定应该直接决定裁剪成合适大小的片状。
B.它符合相应的现场添加剂的补充测试。
C.在一个铂钳锅内,用1g硫酸氢钾R混合20mg,然后加热直到完全熔化。
允许对其进行冷却并加入20ml的稀硫酸。
轻轻地进行加热。
过滤掉产生的溶剂。
在滤液中添加1ml的磷酸和1ml的强过氧化氢溶液R。
如果该物质是不透明的钛白色,那么将会变成橙黄色。
试验:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
S1试液.使用S1试液需要4小时的准备。
称取25g材料,置于带磨砂口的硼硅玻璃烧瓶中,加入500ml的注射用水R,然后加热回流约5个小时。
冷却并转移到烧杯中。
保留部分提取液用于测试S1试液的外观,然后用烧结玻璃过滤器过滤剩余溶液。
S2试液.精密称取2.0g材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入80ml的甲苯R然后加热搅拌回流约90分钟。
冷却至60°C然后加入120ml的甲醇R并持续搅拌。
用烧结玻璃过滤器将溶液进行过滤。
取25ml由40ml的甲苯R和60ml的甲醇R混合的溶液来冲洗烧瓶和过滤器,合并洗液和滤液,并用相同的混合溶剂稀释至250ml。
同法制备空白溶液。
S3试液.称取100g的材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入250ml的0.1M盐酸,加热搅拌回流1个小时。
冷却并收集溶液。
S1试液的外观。
清晰无色。
酸度或碱度。
量取100ml的S1试液,加入0.15ml的BRP指示剂R。
加入约1.5ml的0.01M氢氧化钠溶液,直到溶液颜色蓝色。
量取100ml的S1试液,加入0.2ml的甲基橙溶液R,加入约1ml的0.01M 盐酸,直到溶液变为黄色再变为橙色。
吸光率:S1试液在波长范围220nm-340nm之间的吸光率最大值不超过0.2。
还原性物质。
量取20ml的S2试液,加入1ml的稀盐酸R和20ml的0.002M高锰酸钾。
加热回流3分钟然后直接冷却。
加入1g碘化钾R然后直接用0.01M硫代硫酸钠进行滴定测量法测量,用0.25ml的淀粉溶液R作为指示液。
同法空白滴定。
滴定体积的区别小于3.0ml。
可溶于己烷的物质。
称取10g材料,置于250ml带磨砂口的的圆锥形硼硅酸盐玻璃烧瓶中。
加入100ml的己烷然后加热搅拌回流4个小时。
在冰水中冷却然后用烧结玻璃过滤器迅速过滤(过滤时间必须少于5分钟;如有必要可将溶液加压来加速过滤)并让溶液保持在0°C左右。
量取20ml的滤液,放在恒重过的硼硅酸盐玻璃皿上,在水浴中蒸发。
将滤渣放在100-105°C的烘箱上进行干燥一小时。
所得滤渣不得大于标准样品的10%,而且不超过5%。
可提取出的铝:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用铝标准溶液(200ppm Al)R加0.1M的盐酸稀释成标准溶液。
波长:铝发射的波长为396.15nm,光谱采用396.25nm。
确定所用的盐酸中没有铝。
可提取出的钛:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用钛标准溶液(100ppm Ti)R加0.1M的盐酸稀释成标准溶液。
波长:钛发射的波长为336.12nm,光谱采用336.12nm。
确定所用的盐酸中没有钛。
可提取出的锌:限量:1ppm。
原子吸收光谱测定法。
测试溶液:S3试液。
标准溶液。
用锌标准溶液(10ppm Zn)R加0.1M的盐酸稀释成标准溶液。
来源:锌空心阴极电子管。
波长:213.9nm雾化装置:空气乙炔火焰确定所用的盐酸中没有锌。
可提取出的重金属:限量:2.5ppm将50ml的溶液S3在水浴中蒸发至5ml然后加水R稀释成20.0ml。
取12ml溶液按照测试A进行。
用2.5ml的铅标准溶液(10ppm Pb)准备标准溶液。
硫酸灰分:限量:1.0%,称取5.0g试验。
此限制不适用于被二氧化钛处理过的材料。
补充测试只有在材料定期检查时,这些测试可全部或部分进行实施。
酚类抗氧化剂。
液相色谱法。
溶剂混合:氰化甲烷R,四氢呋喃R(50:50 V/V)S21试液。
精密量取50ml的S2试液,在45°C真空蒸干后,使残渣溶于5.0ml的混合溶剂中。
用S2试液的空白溶液同法制备空白样品。
S22试液。
精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的二氯甲烷中。
用S2试液的空白溶液同法制备空白样品。
S23试液.精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的相同量的氰化甲烷R和10g/l的四氢呋喃R里的叔丁基化过氧氢溶液的混合剂。
封上烧瓶静置1小时。
准备一个空白溶液。
仅在有必要对规定的所检查的物质组成进行酚类抗氧化剂分析时准备以下标准溶液。
标准溶液(a)。
溶解25.0mg的二叔丁基对甲酚CRS(增塑剂07)和60.0mg的增塑剂08CRS于10.0ml 的混合溶剂中。
用混合溶剂将2.0ml的该溶液稀释成50.0ml。
标准溶液(b)。
溶解60.0mg的增塑剂09CRS和60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释成50.0ml。
标准溶液(c)。
溶解60.0mg的增塑剂11CRS和60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(d)。
溶解25.0mg的增塑剂07CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(e)。
溶解60.0mg的增塑剂08CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(f)。
溶解60.0mg的增塑剂13CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(g)。
溶解60.0mg的增塑剂09CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(h)。
溶解60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶剂稀释至50.0ml。
标准溶液(i)。
溶解60.0mg的增塑剂11CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶剂(j)。
溶解60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(k)。
溶解20.0mg的增塑剂18CRS于10.0ml的同等量的氰化甲烷R和10g/l的四氢呋喃R 里的叔丁基化过氧氢溶液的混合剂。
允许放在密封容器内静置一个小时。
用混合溶剂将2.0ml 的此溶液稀释至50.0ml。
A.如果该物质检查出含有增塑剂07和/或增塑剂08,请实施以下测试:-尺寸:l = 0.25 m, O = 4.6mm;-固定相:十八烷基甲硅烷基硅胶的气相色谱R (5 μm).移动相:水R,氰化甲烷R流量:2 mL/min检测方法:分光光度计,280nm注入:20μL的 S21试液,相应的空白溶液,标准溶液(a),和要么标准溶液(d)要么标准溶液(e)要么标准溶液(d)和(e)。