目的基因T载体克隆实验步骤
分子克隆实验流程

分子克隆实验流程一、引物的稀释1、引物干粉冻存于-20℃,用前12000rpm离心1min;2、按引物管上的nmol数稀释,nmol=4.92,加49.2µL ddH2O至100µM;3、稀释至10µM(5µL引物F+5µL引物R+40µL ddH2O)二、目的基因的扩增实验前准备:生物安全柜紫外照射30min,模板DNA、水、引物、buffer,dNTP提前10min拿出解冻,用75%酒精擦拭移液器及台面。
扩增体系:Reagent 25µL 50µL10xbuffer (含Mg2+) 2.5µL 5µLdNTP (10mM) 0.5µL 1µLrT aq酶0.25μL0.5μLprimer (10μM) 1.25μL 2.5μLTemplate DNA 2μL4μLddH2O 18.5µL 37µL反应程序:(延伸时间按目的片段大小进行调整)95℃预变性3min(95℃变性30 s,60℃退火30s,72℃延伸45s)x3572℃后延伸7min4℃保持电泳:120V,加2µLloading buffer,上样5µL,1000bp marker 5µL小胶:2%,0.6g琼脂糖,30ml 1xTAE中胶:2%,1g琼脂糖,50ml 1xTAE大胶:2%,2g琼脂糖,100ml 1xTAE三、目的产物切胶回收(试剂盒)四、连接实验前准备:SolutionI在冰上融化连接体系:Reagent 10µL胶回收DNA(50ng/μL)4µLPDM-18T载体1µLSolution I 5µL反应条件:16℃,4h(PCR仪,热盖105℃)/ 4℃过夜五、转化实验前准备:开启42℃水浴锅实验步骤:样品+阴性对照(无质粒)+阳性对照(感受态带的质粒)1、把感受态细胞TOP10从-80℃冰箱拿出并放置于冰上解冻;2、每管分装30 - 50μL感受态细胞(冰浴);3、向感受态细胞中加入5μL连接产物,冰浴30min。
目的基因T载体克隆实验步骤

目的基因T载体克隆实验步骤目的基因T载体克隆实验步骤PCR产物的T载体克隆一. 重组质粒的构建:重组的DNA分子是在DNA连接酶的作用下,有Mg切的载体分子与外源DNA分子进行连接。
DNA连接酶有两种:T4噬菌体DNA连接酶和大肠杆菌DNA连接酶。
两种DNA连接酶都有将两个带有相同粘性末端的DNA分子连在一起的功能,而且T4噬菌体DNA连接酶还有一种大肠杆菌DNA连接酶没有的特性,即能使两个平末端的双链DNA分子连接起来。
但这种连接的效率比粘性末端的连接率低,一般可通过提高T4噬菌体DNA连接酶浓度或增加DNA浓度来提高平末端的连接效率。
T4噬菌体DNA 连接酶催化DNA 连接反应分为3 步:首先,T4 DNA 连接酶与辅因子ATP形成酶-ATP复合物;然后,酶-ATP复合物再结合到具有5’磷酸基和3’羟基切口的DNA上,使DNA腺苷化;最后产生一个新的磷酸二酯键,把切口封起来。
连接反应通常将两个不同大小的片断相连。
很多DNA聚合酶在进行PCR扩增时会在PCR产物双链DNA每条链的3’端加上一个突出的碱基A。
pUCm-T载体是一种已经线性化的载体,载体每条链的3’端带有一个突出的T。
这样,pUCm-T载体的两端就可以和PCR产物的两端进行正确的AT配对,在连接酶的催化下,就可以把PCR产物连接到pUCm-T载体中,形成含有目的片断的重组载体。
连接反应的温度在37℃时有利于连接酶的活性。
但是在这个温度下粘末端的氢键结合是不稳定的。
因此采取折中的温度,即12-16℃,连接12-16h(过夜),这样既可最大限度地发挥连接酶的活性,又兼顾到短暂配对结构的稳定。
2 、ATP存在的连接缓冲系统中,将分别经酶二. 感受态制备原理细菌在0 C CaCl2低渗溶液中胀成球形,丢失部分膜蛋白,成为容易吸收外源DNA的状态。
三. β-半乳糖甘酶显色反应选择法(蓝白筛选)原理 LacZ基因是大肠杆菌乳糖操纵子中的一个基因,可以编码β—半乳糖核苷酶。
基因克隆实验手册

插入片段 线性化载体
推荐使用的试剂盒
产品信息
DNA Ligation Kit <Mighty Mix> P4
Blunting Kination Ligation (BKL) Kit
P6
TaKaRa DNA Ligation Kit LONG P4
Mighty TA-cloning Kit
P6
Mighty TA-cloning Reagent Set for PrimeSTAR®
↓ ③42℃反应45秒后,在冰中放置1~2分钟
↓ ④加入已预先37℃保温的SOC培养基,使终体积为1 ml。
↓ ⑤37℃振荡培养1小时(160~225 rpm)
↓ ⑥取适量涂布于LB选择培养基、37℃过夜静置培养
5. PCR扩增确认插入片段DNA 6. 培养、纯化质粒
PCR扩增确认插入片段DNA请参考第5页
TCGA 5’ Hin d Ⅲ
线性化载体 ※
C TAG A G C T
※ 利用限制性内切酶酶将环状载体DNA切断后,切胶 回收、或必要时进行去磷酸化反应
转化
·感受态细胞
形成菌落
进行菌落PCR 确认插入片段
·EmeraldAmp® MAX PCR Master Mix等
·电泳相关制品
— 1—
基因克隆实验手册
※ PCR扩增产物是平滑末端时进行dA加尾反应。
【目的DNA片段】 带有限制性内切酶的
酶切末端
In-Fusion克隆
5×In-Fusion® HD In-Fusion酶使 Enzyme Premix 末端15个相同
碱基无缝融合
TA克隆
3’ A
A 3’
T载体
实验四_T_A_克隆及转化

PCR产物的连接及转化
一、实验目的
• • • • 弄清T载体的结构和T-A克隆的原理; 弄清蓝白斑筛选的原理; 学会PCR产物连接方法; 学会连接产物转化大肠杆菌感受态方法。
二、实验原理
• 1、T-A克隆原理
通过PCR的方法扩增目的基因片断,为了后续实验的需要,
往往需要将获得的目的片断通过TA克隆的方法,重组到T-载体 中。外源DNA与载体分子的连接称之为DNA重组,这样重新组 合的DNA叫做重组体或重组子。重组的DNA分子是在T4 DNA连 接酶作用下,有Mg2+、ATP存在的连接缓冲系统中,将载体分 子与外源DNA分子进行连接。
• • • • • • (低温、加温)水浴锅 离心机 PCR扩增仪 恒温培养箱 摇床 超净工作台等
主要试剂
• • • • pMD 18-T Vector试剂盒 PCR扩增回收纯化的片段 200 mg/ml的IPTG 过滤除菌 20 mg/ml的X-gal 溶于二甲基甲 酰胺,避光保存。(低温、加 温)水浴锅 • LB培养基( 含IPTG,X-gal, 50mg/ml 氨苄青霉素)
四、实验步骤
• 1、
产生不同末端的DNA聚合酶
• α互补 原理
许多载体(包括pMD18-T)都具有一段大肠杆菌β半乳糖苷酶的启动子和编码 其N端氨基酸(α肽链)的片断基因。宿主具有编码β半乳糖苷酶的C端基因。当 二者产物组合在一起,就具有活性酶的产生,此现象称为α互补。由α互补形成 的具有功能活性的β半乳糖苷酶,可以用X-gal显色测定出来,它能将无色的化合 物X-gal切割成半乳糖和蓝色底物。当外源DNA片断插入到多克隆位点后,就会
破坏α肽链的阅读框,从而不能合成与受体菌内的β半乳糖苷酶相互补的活性α肽
TA克隆简述

生物秀论坛『中国生物科学论坛』PCR产物的T载体克隆(一)重组T质粒的构建一.原理外源DNA与载体分子的连接就是DNA重组,这样重新组合的DNA叫做重组体或重组子。
重组的DNA分子是在DNA连接酶的作用下,有Mg2+、ATP存在的连接缓冲系统中,将分别经酶切的载体分子与外源DNA分子进行连接。
DNA连接酶有两种:T4噬菌体DNA连接酶和大肠杆菌DNA连接酶。
两种DNA连接酶都有将两个带有相同粘性末端的DNA分子连在一起的功能,而且T4噬菌体DNA连接酶还有一种大肠杆菌DNA连接酶没有的特性,即能使两个平末端的双链DNA分子连接起来。
但这种连接的效率比粘性末端的连接率低,一般可通过提高T4噬菌体DNA连接酶浓度或增加DNA浓度来提高平末端的连接效率。
T4噬菌体DNA 连接酶催化DNA 连接反应分为3 步:首先,T4 DNA 连接酶与辅因子ATP形成酶-ATP复合物;然后,酶-ATP复合物再结合到具有5’磷酸基和3’羟基切口的DNA上,使DNA腺苷化;最后产生一个新的磷酸二酯键,把切口封起来。
连接反应通常将两个不同大小的片断相连。
因为DNA片断有两个端点,所以切割时出现两种可能,一种是单酶切,另一种是双酶切,这两种酶切方法在基因工程操作中都经常用到。
对于单酶切来说,载体与供体的末端都相同,连接可以在任何末端之间进行,这样就导致了大量的自连接产物。
为了减少自环的高本底,可对载体进行5’除磷酸处理,原理是连接酶只能连接DNA片断的3’OH末端与5’端,所以除磷后载体不会自环。
一旦有外源片断插入时,由外源片断提供5’端就能与载体进行连接。
通过这种方法可大大减少由载体的自环造成的高本底。
对于双酶切来说,无论载体与供体同一片段上都有不同的末端,这样就避免了载体与供体的自环,能使有效连接产物大大增加。
双酶切的另一个特点是能将供体分子定向连接到载体上。
连接反应的温度在37℃时有利于连接酶的活性。
但是在这个温度下粘末端的氢键结合是不稳定的。
基因与载体连接方法

基因与载体连接方法及其原理基因与载体连接方法是分子生物学实验中常用的技术,它可以将目的基因或序列插入到合适的载体中,从而实现基因的克隆、表达或功能分析。
基因与载体连接方法主要依赖于DNA连接酶和限制性核酸内切酶的作用,以及DNA片段末端的互补配对。
根据DNA片段末端的性质不同,可以有以下几种基本的连接方法:一、粘性末端连接法粘性末端连接法是最常用的基因与载体连接方法,它利用限制性核酸内切酶在特定的序列上切割DNA,产生带有单链突出端的双链DNA片段,这些突出端称为粘性末端。
粘性末端可以与相同或相似的粘性末端互补配对,形成稳定的双链结构。
然后,DNA连接酶可以在这些配对的粘性末端上催化磷酸二酯键的形成,从而实现DNA片段的连接。
粘性末端连接法的优点是具有较高的特异性和效率,因为只有相同或相似的粘性末端才能配对,而且配对后的结构较为稳定,不易被水解。
粘性末端连接法的缺点是需要选择合适的限制性核酸内切酶来切割目的基因和载体,而且不同的限制性核酸内切酶可能有不同的反应条件和缓冲液,需要进行优化和调节。
粘性末端连接法的具体步骤如下:1. 选择合适的限制性核酸内切酶来切割目的基因和载体。
一般来说,选择能在目的基因两端和载体多克隆位点上切割出相同或相似粘性末端的限制性核酸内切酶。
如果没有这样的限制性核酸内切酶,可以通过引物设计来在目的基因两端添加所需的限制性核酸内切酶位点,然后通过PCR扩增来获取带有粘性末端的目的基因。
2. 配制酶切反应体系,并在适当的温度和时间下进行反应。
一般来说,每个限制性核酸内切酶都有其特定的反应条件和缓冲液,需要按照说明书或厂家推荐进行操作。
如果使用两种或以上的限制性核酸内切酶进行双酶切或多酶切,需要选择能够同时满足所有限制性核酸内切酶反应条件和缓冲液要求的组合,或者进行分步反应,并在每次反应后纯化DNA。
3. 通过琼脂糖凝胶电泳来分离和回收目的基因和载体片段。
一般来说,使用1%~2% 的琼脂糖凝胶电泳可以有效地分离不同大小的DNA片段,并通过紫外光照射来观察DNA条带。
T-载体连接方案

T-载体连接全过程第一步:PCR扩增目的基因及纯化1. PCR反应体系:10×扩增缓冲液 5.0ul10mMdNTP 1.0ul引物1 1.0ul引物2 1.0 ul模板DNA 1.0ulTaq DNA聚合 1.0ul加双蒸水至50ul(相同的引物体系可配置PCR MIX体系)2. PCR反应条件:PCR反应条件为温度、时间和循环次数。
95℃ 5min95℃ 45s40~60℃ 50s 循环 35次72℃ 55s72℃ 10min3. PCR期间配置琼脂糖凝胶:PCR结束以后跑胶验证,并照相,标记保存。
4. PCR产物回收(1)单一PCR产物用氯仿法●把PCR产物转移至1.5ml的离心管,加500ul无菌水,500ul氯仿异戊醇(24:1),混匀后10000rpm离心5-10分钟●转移上清(约400ul)至灭菌的新1.5 mL的Eppendorf管,加入2倍体积(780 µL体积即可)的无水乙醇与40ul3MNaAc。
置冰上或-80℃冰箱30分钟以上。
●10000 rpm离心10 min。
弃上清,沉淀用适量的70%的乙醇洗一次,于纸上扣干。
小心不要将沉淀洗掉。
●置于恒温器上干燥(55℃)至无色透明或无乙醇气味,大至要5~10min。
●加入40 µL 无菌水溶解沉淀备用。
(2)多条带PCR产物用切胶回收试剂盒。
●PCR产物进行琼脂糖电泳(大容量上样系统)●切胶并回收目的条带至1.5ml的离心管,称重。
●参照试剂盒说明书进行操作(附录3)。
●用50ul无菌水洗脱离心柱2次备用。
第二步:T-载体连接和转化准备工作:制备感受态细胞,方法见附录一,LB培养基,Amp母液,氯化钙的配制,方法见附录二;1.在微量离心管(PCR管)中配制以下体系:注:T-载体试剂盒开封前要向pMD18-T Vector 管中加入25 ul 无菌水,同时再管上面做标记。
pMD18-T Vector 1.0 ul外源DNA 1.5 ulSolutionI 2.5 ul2.16℃或4℃低温连接1h-2h(最好过夜);3. 连接产物全量加入50 ul 的DH5α感受态细胞中,轻轻摇匀后冰上放置30min;DH5α感受态细胞加5 ul 0.1M的无菌氯化钙做阴性对照;4. 热击:42℃水浴中热击90秒(注:精确计算时间,过长将对细胞造成伤害),热击后立即置于冰上冷2-5分钟(禁止摇动)。
原核表达实验流程

pGEM-T载体
目的基因获取 PCR扩增
重组载体1 转化
DH5a感受态制备 DH5a感受态细胞
第二步
转化细胞1 抽取质粒进行电泳 酶切验证检验阳性
(阳性)转化细胞1 酶切,获取目的基因
Pet28a/DH5a载体 重组
重组载体2
重组载体2 转化
DH5a感受态细胞
转化细胞2 抽取质粒进行电泳 酶切验证检验阳性
(6) 4℃用JA转头(或与之相当的转头) 以2500rpm/min离心20min,以回收 细胞。倒去培养液,用25mL冰冷的10%甘油重悬沉淀。(甘油有保存 细胞的作用)
(7) 4℃用JA转头(或与之相当的转头) 以2500rpm/min离心20min,以回收 细胞。倒去培养液,用1mL冰冷的10%甘油重悬沉淀。(倾倒培养液时 要小心,10%甘油中的细菌沉淀附着力降低。)
3 .方法
1 )0.8%琼脂凝胶板的配制:取 0.6g 琼脂糖加80ml TAE(1 × ),
20ml水,微波炉加热融化3 分
(8) 将细胞按40μL等份装入微量离心管,直接使用或-80℃保存
(9) 感受态细胞的检测:取感受态细胞分别涂布含有氨苄青霉素和卡那 霉素抗性的LB平板,
37℃培养12-16小时,观察感受态细菌是否有污染;
(10) 取 1μ g 超螺旋质粒转化制备的感受态,检测转化效率(一般情况下,没 有必要精确计算,可用根据经验估计)。
电转化法:外加于细胞膜上的电场造成细胞膜的不稳定,形成电穿 孔,不仅有利于离子和水进入细菌细胞,也有利于 DNA 等大分子进 入。同时 DNA 在电场中形成的极性对于它运输进细胞也是非常重要 的。 实验材料
质粒DNA,pGEM-T载体或PET表达载体,T4DNA连接酶,连接酶缓冲液 10×
实验三 目的基因的连接、转化

水浴摇床
收集菌体
4. 取1ml 菌液冰浴5min,6000rpm离心5 min;
5. 弃上清,除净剩余液体,轻轻弹匀菌体沉淀,加入 100μl 冰冷的100mmol/L CaCl2 致敏液悬浮细胞,冰 浴5分钟;
6. 6000rpm离心5min后,弃上清,轻轻弹匀菌体沉淀,
2. 如何估计DNA的含量?(DNA marker 参比法)
电泳样本:Marker 50ng/5μl、T载体1μl、回收片段 电泳后通过电泳样品的亮度,估算连接片段和载体的比例
二、PCR产物与T载体的连接
T载体一般从公司购买,载体DNA的含量和浓度已知。 因此,连接反应成败的关键是估计目的DNA的量。
M
三、感受态细胞的制备
感受态细胞的制备原理
培养至对数生长期的大肠杆菌,在低渗 CaCl2 存在的情况下,细菌变得膨胀而易于外源基因 的导入。 钙离子溶液处理的细胞对外源DNA的摄取能力 增强。
注意事项:
1)感受态细胞的膜已经很脆,应该超低温保存。
如果反复冻融会使细胞活力受影响,而且细胞 膜的变化可能复原,由感受态细胞状态回转到普通 细胞状态,失去接受外源DNA的能力。
公司及产品:
1、Invitrogen公司的Lipofectamine; 2、Promega的一系列阳离子转移剂:TransFast™,
Transfection Reagent, Tfx™ Reagents和 Transfectam® Reagent.
磷酸钙转染技术
原理:
磷酸钙共沉淀法 将氯化钙、DNA和磷酸盐缓冲液混合, 形成磷酸钙沉淀 ,附着于细胞膜并经过细胞内吞作用进入细 胞质。
2、生物制药、基因治疗
构建目的基因的真核表达载体实战操作

构建目的基因的真核表达载体实战操作最后更新:2010-5-16 阅读次数:451 【字体:小中大】目的:构建目的基因的真核表达载体一.PCR扩增得到目的片段1.引物的设计与合成:这里有很多血与泪的教训。
我前后一共送过九个载体测序,其中居然有五个是引物出错。
最后一次挑的两个克隆都是上游引物出错(此上游引物40个碱基),后来和这家引物公司交涉,他们拿走了我的板子,一次送了五个克隆测序,三个是上游引物出错,两个是正确的克隆,60%的出错率。
现在想想,有几点教训:(1)尽量避免长引物的合成。
越长意味着出错的几率越大,哪家公司都这样(2)选择一家质量可靠的公司,千万别在这省钱,引物再贵也花不了多少钱,可一轮构建下来,一测序发现引物错了,不知白扔了多少钱不说,那种心情实在是难以收拾(3)一旦发现引物有问题,及时换公司,我就让他们拿回去又是纯化又是重新合成过,结果还是错,时间咱耽误不起2.Pfu酶扩增:(1)扩增效率低的问题:往往taq酶扩的很好,一换成pfu就是扩不出来。
解决办法:a延长延伸时间(我1.2kb用3分15秒才扩出来),并适当增加循环数;b多试几家的pfu,总有一家能扩出来;c多设计几对引物试试,总有一对能扩出来,因为pfu有外切活性,可否适当的延长引物的3’互补端;d构建中间载体:一旦用某一对引物能扩出来,把此目的片段连接t-easy中间载体,测序正确后,即可以此中间载体做pcr扩增的模板。
实践证明以此中间载体为模板的pfu扩增效率远远高于以cDNA为模板。
(2)pfu保真性的问题:即使是pfu,也会出错。
现在各家公司都有号称高保真的pfu酶出售,价格不一。
虽然都是pfu,但质量不不见得一样,在都能扩增出来的情况下,选择信誉好的公司的pfu。
我用的是一家小公司的pfu,很便宜,100u才80块钱,但我1.2kb的片段却频频出错。
所以万不可在酶上省那百十块钱,最后花了大钱不说,还浪费了时间精力。
1.基因克隆的步骤

基因克隆的步骤一、RNA的提取1. 提取RNA前将洗净干燥的瓷研钵放入-70℃预冷10 min;2. 从液氮罐中取出样品组织放入预冷的研钵中,加液氮淹没后立即研磨,边研磨边加液氮,整个过程都不要使液氮挥干;3. 趁冷,将样品粉末50-80 mg加入1.5 mL离心管后再加入1 mL Trizol,样品体积应不超过所使用Trizol体积的10%,然后按照Trizol试剂盒说明操作;4. 室温静止5-15 min。
对于某些蛋白、多糖或脂含量很高的细胞或组织Trizol裂解后可能会有不溶物或油脂状漂浮物,需12000g、4℃离心10 min,然后吸取澄清的Trizol裂解产物至一新的离心管中;5. 每mL Trizol中加0.2 mL氯仿。
小心盖上离心管,剧烈地涡旋振荡15 sec,室温(24℃)静置3-5 min;(必需步骤)6. 12000 g、4℃离心15 min,此时混合物分成三部分:底层为苯酚-氯仿层,中间层,上层水相层,RNA完全存在水相中;7. 把小于80%的水相层(每mL Trizol约可吸0.5-0.55 mL)转移至一新的离心管中,并弃去下面的有机相(小心避免吸到中间层)。
往水相中加入异丙醇来沉淀RNA,每使用1 mL Trizol,便加500 uL的异丙醇,颠倒混匀,室温下孵育样品10 min;8. 4℃、12000 g离心样本10 min弃去上清,每mL Trizol加入1 mL 75%乙醇,涡旋混匀,室温沉淀10 min后,7500 g、4℃离心5 min,弃上清。
再用离心机甩一下(5000rpm,离心1 s);9. 小心吸走乙醇,短暂地在室温下置2-5 min,让RNA团风干,不要用离心干燥装置或真空干燥装置,因为过度干燥会导致很难用水重新溶解RNA,然后在55-60℃温浴10 min,然后-70℃保存。
[RNA](ng/uL)=A260×40×稀释倍数[DNA](ng/uL)=A260×50×稀释倍数纯DNA:OD260/OD280≈1.8(>1.9,表明有RNA污染;<1.6,表明有蛋白质、酚等污染)纯RNA:1.7<OD260/OD280<2.0(<1.7时表明有蛋白质或酚污染;>2.0时表明可能有异硫氰酸残存)二、M-MLV RT反转录1. oligo dT 1 uL + dNTP(2.5 mM)4 uL + 1-5 ug total RNA + 水= 12 uLHeat mixture to 65℃ for 5 min and quick chill on ice;2. 上述12 uL液体+ 5×Buffer 4 uL + 0.1 M DTT 2 uL + RNase OUT 1 uLMix contents of the tube,incubate at 37℃for 2 min3. 上述19 uL液体加M-MLV RT 1 uLIncubate tube at 25℃for 10 min;Incubate 50 min at 37℃;70℃for 15 min。
实验十 pUC57 T载体的制备

实验十 pUC57 T载体的制备一.原理PCR产物的克隆是一种有效的克隆目的基因的方法,但是在实际应用中,由于方法学上的一些技术问题可能限制其应用。
由于PCR产物3`末端突出一个A,因此克隆PCR产物的载体3`末端必须突出一个T,这样载体与PCR产物之间形成粘性长端。
现在许多公司开发出了此类专用于PCR产物克隆的T载体。
本实验中将平端内切酶EcoRv酶解的EcoRv,用Taq酶将其3`末端加上T,然后用T4连接酶将能自身环化的质粒连接。
用凝胶电泳将自身环化的质粒与3`末端带有T.不能自身环化的线性质粒分开,回收线性质粒片段即为pUC57 T载体。
二.方法1.制备pUC57质粒DNA。
(见实验一)2.用EcoR V酶解pUC57 DNA。
(见实验九)3.在酶解的pUC57 DNA末端加上T。
取一支0.5ml微离心管.加入:EcoR V酶解的pUC57 DNA 10μg10 X PCR缓冲液 15μldTTP 10mmol/L 20μlTaq DNA聚合酶(5U/μl) 1μlddH2O to 150μl10 000r/min离心5s混匀,72℃水浴2h。
4.DNA片段的纯化(1)在上述0.5ml微离心管中加入等体积(15μl)酚/氯仿混匀,放置数分钟,5 000r/min 离心5min.使液体分层。
(2)吸收酚/氯仿抽捉提之水相至另一1.5ml离心管中,加入1/10体积的乙酸钠,加2倍体积冷无水乙醇,混匀。
-20℃冰箱放置2h,15 000r/min离心15min。
(3)倾去乙醇,加入70%冷乙醇淋洗。
(4)倾去乙醇,滤纸吸干,真空抽吸2~3min。
(5)加人30μl ddH2O,溶解DNA。
5.载体DNA自身连接在DNA片段纯化管中,加入5μl 10x连接酶缓冲液和2 U T4 DNA连接酶,并用重蒸水补至总体积50μl。
16℃连接16小时。
6.琼脂糖凝胶电泳将DNA连接液在l%(Wt/V01)琼脂糖凝胶上电泳分离pUC57 T载体和原载体7.pUC57 T载体的纯化用琼脂糖凝胶DNA回收试剂盒纯化线性载体,井用电泳对此载体进行定量。
T载体连接获得重组DNA实验报告范本

T载体连接获得重组DNA实验报告范本
实验名称:T载体连接获得重组DNA实验
实验目的:通过T载体连接法获得重组DNA,掌握基本的分子生物学实验技能,提高实验操作能力。
实验原理:T载体连接是一种将目的DNA片段连接到T载体上的方法,其中T载体是一种双链DNA分子,具有两个限制性内切酶切割位点,可以在这些位点上连接目的DNA片段。
连接过程需要使用DNA 连接酶,在一定条件下,连接酶能够将目的DNA片段与T载体连接起来,形成重组DNA。
实验步骤:
1.制备T载体:将T载体质粒DNA与适当的限制性内切酶一起
切割,将切割后的DNA片段通过琼脂糖凝胶电泳分离,取出T
载体DNA片段。
2.制备目的DNA片段:将目的DNA片段通过PCR扩增得到。
3.进行连接反应:将T载体DNA片段与目的DNA片段在连接酶
作用下进行连接反应,反应条件为37℃,2小时。
4.离心筛选:将连接反应混合液用离心机进行离心分离,将重组
DNA沉淀下来。
5.洗涤重组DNA:用70%的乙醇洗涤沉淀下来的重组DNA,将其
溶于适当的缓冲液中。
6.验证重组DNA:通过琼脂糖凝胶电泳验证连接反应是否成功,
观察重组DNA是否出现预期的带状图谱。
实验结果:
经过连接反应和离心筛选,我们成功地获得了重组DNA。
通过琼脂糖凝胶电泳验证,可以看到重组DNA出现了预期的带状图谱,证明连接反应成功。
实验结论:
通过T载体连接法获得重组DNA是一种常用的分子生物学实验技术,通过本次实验,我们成功地掌握了这种方法,并获得了重组DNA。
这种方法可以应用于基因克隆、基因工程等领域,具有广泛的应用前景。
实验四 T-A连接(克隆)

三、T-A连接(克隆)的材料
四、T-A连接(克隆)的过程
1. 克隆DNA PCR反应体系(Reaction Assembly): 片段的准备: Component 50µL
Nuclease-free Water 23 µL (to 50 µL )
25 µL 1 µL 1 µL
PCR反应
2X PCR Mix 模板DNA (pC-mChr) Primer Mix
嘌呤核苷酸(A),与T载体之间的A-T互补性可提高PCR产物克隆的效率。
二、实验原理
二、实验原理
自制T载体:XcmI: 5’-CCANNNNNˇNNNNTGG-3’
二、实验原理
一些载体(如PUC系列质粒)带有β -半乳糖苷酶基 因(lacZ)的调控序列和N端α 片段146个氨基酸的编 码信息。该编码区中含有多克隆位点,可用于构建重 组子。这种载体适用于仅编码lacZ C端ω 片段的突变 宿主细胞。因此,虽然宿主和质粒编码的片段都没有 lacZ活性,但它们同时存在时,α 片段与ω 片段可通过 α -互补形成具有酶活性的β -半乳糖苷酶。 由α -互补而产生的LacZ+细菌在诱导剂IPTG(异丙 基硫代半乳糖苷)的作用下,在生色底物X-Gal存在 时产生蓝色菌落。
实验四
T-A连接(克隆)
赵建军 张龚炜 2018.10.20
一、实验目的
学习和掌握DNA连接的原理和方法 理解T-A连接的优缺点 理解蓝白斑筛选的原理和方法
二、实验原理
DNA连接反应(transformation)利用DNA连接酶把载体DNA和 要克隆的目的DNA片段连接在一起,成为一个完整的重组分子。 载体(vector) ,指在基因工程重组DNA技术中将DNA片段(目的 基因)转移至受体细胞的一种能自我复制的DNA分子。 T-载体:聚合酶链反应(PCR)产物的克隆载体,两侧3′端各多出一 个脱氧胸腺嘧啶核苷酸(T)。由于PCR产物两侧3′端通常含单个脱氧腺
实验4.T-A连接,感受态细胞制备,转化及筛选

4) 取1ml 菌液,冰浴5min,6000rpm离心5 min(收集菌 体) ;
5)弃上清,加入100μl冰冷的100mmol/L CaCl2溶液, 充分悬浮细胞,冰浴5分钟;
6)6000rpm离心5min; 7)弃上清,加入100μl冰冷的100mmol/L CaCl2溶液,
充分悬浮细胞; 8)将制备好的感受态细胞置于冰上待用。
午休
4) 6,000rpm离心10秒钟(收集菌体沉淀);
5)吸弃800l上清,用剩余的200lLB培养液重悬菌体;
6)将菌液均匀涂于含有氨基苄青霉素的LB琼脂培养板 上 , 室 温 放 置 5 - 1 0 min 待 菌 液 完 全 吸 收 后 , 倒 置 于 37℃孵箱中培养14-16小时。
注意:转化后的平皿倒置放于孵箱 (防止水蒸气形成的水滴影响细菌单
实验内容
1. PCR产物与T载体的连接 2. 感受态细胞的制备,连接产物的转化及筛选
实验1、PCR产物与T载体的连接
1. 连接反应
T vector
1 μl
10×ligation buffer
1 μl
PCR回收产物
7 μl
T4 DNA Ligase
1 μl
轻弹管底混合,离心机甩一下,置25oC水浴1h。 连接产物可直接用于转化,也可置-20oC保存备用。
如果反复冻融会使细胞活力受影响,而且细胞膜 的变化可能复原,由感受态细胞状态回转到普通细 胞状态,失去接受外源DNA的能力。
2)无菌操作。 在火焰附近操作,火焰附近是无菌区。 各种器具均需消毒。
无菌操作的注意事项
(超净台内操作)
• 枪头、EP管、试管、平皿等,各种试剂都是经过高压灭 菌后在超净台中专用的,但加样器需要自带。
基因克隆的一般流程
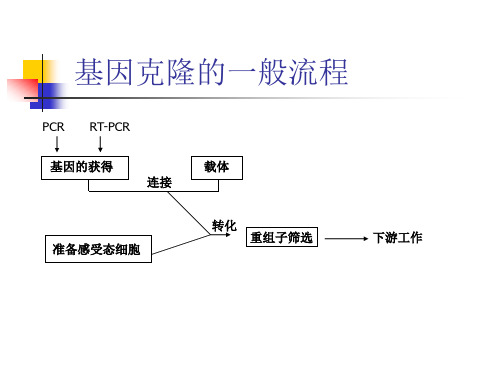
平端连接图示
匹配粘端连接图示
不匹配粘端连接图示
连接反应的建立
连接反应的温度:
粘性末端,按A-T,G-C含量计算,±5℃。 如 Pst I(CTGCA↓G,12℃ ),EcoRI(G↓AATTC,8℃)。 平末端,如EcoRⅤ(GAT↓ATC),可在室温进行,不超过30℃, 酶的用量要比粘性末端加大10-100倍 。
DNA量:
摩尔数之比 载体:目的 DNA = 1 : ( 1~3 ) 载体摩尔数 目标DNA摩尔数 目标基因片段碱基数×载体重量 目标基因片段重量×载体碱基数
载体和目标基因的连接反应
如未定量可以按回收效率75%计算DNA浓度,按载体与 目的基因摩尔数比1:3进行连接。
连接反应在灭菌的 0.5ml离心管中进行。 15 l 体积反
重组子筛选
下游工作
载体的选择
基因工程载体一般要求:
能在宿主细胞中复制繁殖,有较高的自主复制能力。 易进入宿主细胞,进入效率越高越好。 合适的多克隆位点(MCS)可接受外源片段,且不影响其进入宿主细 胞和在细胞中的复制。
易从宿主细胞中分离纯化出来, 便于重组操作。
有筛选标志,当其进入宿主细胞、或携带着外源序列进入宿主细胞都 能容易被辨认和分离出来。便于克隆操作。
NGF-sense: NGF-antisense:
6
5
pEGFP 荧光质粒表达载体
质粒DNA的酶切反应结果
质粒 M 酶切反应 M 酶切反应
pEGFP
pT-NGF
酶切产物凝胶回收操作步骤
(Gel Extraction Kit)
仔细切下含DNA的凝胶,置一称重的1.5ml离心管中。称重。 按300lS1溶液/100mg凝胶加入的比例加入S1溶液,置50℃水浴10分钟, 使凝胶完全溶化,每2分钟颠倒混匀一次。 将溶化后的凝胶溶液移入吸附柱,置收集管中,9000g×30秒,倒掉收集 管中的液体,再将吸附柱放入同一收集管中。 在吸附柱中加入500l W1溶液,9000g×15秒,倒掉收集管中的液体,再 将吸附柱放入同一收集管中。重复一次洗柱。 将吸附柱放入同一收集管中。9000g×1分钟。 将吸附柱放入一新1.5ml离心管中,在吸附膜中央加入30lddH2O,静置1 分钟,9000g×1分钟。备用。
基因克隆方法步骤

克隆步骤一、目的片段的回收纯化1.柱平衡步骤:向吸附柱CB2中(吸附柱放入收集管中)加入500ul 平衡液BL,12000rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
(请使用当天处理过的柱子)。
2.将单一的目的DNA条带从琼脂糖凝胶中切下(尽量切除多余部分)放入干净的离心管中,称取重量。
3.向胶块中加入加入溶液PC(0.1g加入100ul,注意没过胶块),50℃水浴放置10min左右,期间不断温和地上下翻转离心管,已确保胶块充分溶解。
4.将上一步所得溶液加入一个吸附柱(吸附柱放入收集管中),12000rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
5.向吸附柱CB2中加入600ul漂洗液(使用前请先检查是否已加入无水乙醇),12000rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
6.重复步骤5.7.将吸附柱CB2放入收集管中,12000rpm离心2min,尽量除去漂洗液,将吸附柱置于室温放置数分钟,彻底晾干。
8.将吸附柱CB2放入一个干净的1.5ml离心管中,向吸附膜中间位置悬空滴加40ul的无菌水。
室温放置2min,12000rpm离心2min,收集DNA溶液。
(可以将离心后的溶液重新加到柱子中再回收一次) 9.取2ulDNA加入loading buffer点样,电泳检测是否回收出来样品。
二、目的片段与载体的连接1. 将回收的目的片段与T 载体连接,反应体系如下:目的片段 4 .5µLpMD19-T simple Vector 0.5µLSolutionⅠ 5 µLTotal 10 µL按上述体系依次加入各组分后,旋涡混匀,稍加离心后置于16℃连接过夜。
2.转化1)从-80 ℃冰箱中取50 µL 感受态细胞在冰上解冻。
2)在超净工作台上,加入10 µL 连接产物,轻轻混匀,冰上放置30 min 。
TA克隆

PCR产物克隆大致分为两类,即平头连接和粘头连接。
平头连接是将制备好的平头载体和补平或削平的PCR产物直接进行连接。
载体可用EcoR V 或Sma I切成平头;PCR产物纯化后,可以在22℃用DNA聚合酶I作用30min(利用该酶所具有的3’→5’外切酶活性和5’→3’的聚合酶活性)。
如果要求不高,PCR产物也可不加处理。
如果使用Stratagene公司的pfu DNA聚合酶或New England Biolabs公司的Vent DNA聚合酶,这两种酶有5’→3’校对能力,扩增出来的PCR产物已经是平头,可以不作平端处理。
平端连接的一个显而易见的缺陷是连接效率低下,即使使用很高单位的连接酶,或在反应体系中加入PEG 8000,也只能很有限地提高效率。
粘头连接也可以大致分为两类,一类粘头连接是用某种方法,在载体和PCR产物上产生长的可互补的粘性末端。
最普遍的方法是在引物的5’端加入一段某种限制酶的识别序列。
如果两个引物选用不同的限制性内切酶识别序列,就可以做到定向连接。
另一类利用部分PCR产物3’端带有一个凸出的dAMP的特性,构建3’端带有凸出的dTMP的载体。
一般采用的方法是先把载体用某种限制性内切酶消化成平头,在70℃或72℃下在只加入一种dNTP、即dTTP的反应体系中用Taq DNA聚合酶处理半小时(也有人报道处理1~2小时能提高克隆效率,这样加T反应会更彻底)。
也可以用末端转移酶来完成加T反应。
载体自连、PCR产物串连可以忽略。
如果使用ddTTP,效果会更好。
这种方法一般称为加T/A法克隆,比平头连接效率高50~100倍。
一、PCR产物的TA克隆1. TA克隆构建原理:TA克隆系统由Invitrogen公司(San Diego,CA)发展而来的商业性试剂盒,它用于PCR产物的克隆和测序。
其原理是利用Taq酶能够在PCR产物的3’末端加上一个非模板依赖的A,而T载体是一种带有3’T突出端的载体,在连接酶作用下,可以快速地、一步到位地把PCR产物直接插入到质粒载体的多克隆位点(MCS)中。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
PCR产物的T载体克隆实验原理一.重组质粒的构建:重组的DNA分子是在DNA连接酶的作用下,有Mg2 、ATP存在的连接缓冲系统中,将分别经酶切的载体分子与外源DNA分子进行连接。
DNA连接酶有两种:T4噬菌体DNA连接酶和大肠杆菌DNA连接酶。
两种DNA连接酶都有将两个带有相同粘性末端的DNA分子连在一起的功能,而且T4噬菌体DNA连接酶还有一种大肠杆菌DNA 连接酶没有的特性,即能使两个平末端的双链DNA分子连接起来。
但这种连接的效率比粘性末端的连接率低,一般可通过提高T4噬菌体DNA连接酶浓度或增加DNA浓度来提高平末端的连接效率。
T4噬菌体DNA 连接酶催化DNA 连接反应分为3 步:首先,T4 DNA 连接酶与辅因子ATP形成酶-ATP 复合物;然后,酶-ATP复合物再结合到具有5’磷酸基和3’羟基切口的DNA上,使DNA腺苷化;最后产生一个新的磷酸二酯键,把切口封起来。
连接反应通常将两个不同大小的片断相连。
很多DNA聚合酶在进行PCR扩增时会在PCR产物双链DNA每条链的3’端加上一个突出的碱基A。
pUCm-T载体是一种已经线性化的载体,载体每条链的3’端带有一个突出的T。
这样,pUCm-T载体的两端就可以和PCR产物的两端进行正确的AT配对,在连接酶的催化下,就可以把PCR产物连接到pUCm-T载体中,形成含有目的片断的重组载体。
连接反应的温度在37℃时有利于连接酶的活性。
但是在这个温度下粘末端的氢键结合是不稳定的。
因此采取折中的温度,即12-16℃,连接12-16h(过夜),这样既可最大限度地发挥连接酶的活性,又兼顾到短暂配对结构的稳定。
二.感受态制备原理细菌在0 C CaCl2低渗溶液中胀成球形,丢失部分膜蛋白,成为容易吸收外源DNA的状态。
三.β-半乳糖甘酶显色反应选择法(蓝白筛选)原理LacZ基因是大肠杆菌乳糖操纵子中的一个基因,可以编码β—半乳糖核苷酶。
β—半乳糖核苷酶是由4个亚基组成的四聚体,可催化乳糖的水解.用X-Gal为底物进行染色时,呈蓝色。
现在一些特定的质粒(比如pUC/pBS等),常带有β—半乳糖核苷酶的调控序列和β—半乳糖核苷酶N端146个氨基酸(α肽段)的编码序列,在这个编码序列里还插入一个多克隆位点(MCS),它并不影响lacZ的表达。
另外,常用的大肠杆菌带有β—半乳糖核苷酶C端部分序列(β肽段),的编码序列。
在各自独立的情况下,这些质粒与大肠杆菌各自编码的β—半乳糖核苷酶片段都没有酶的活性。
只有当携带α肽编码信息的克隆载体成功进入宿主细胞,在培养基诱导物IPTG的诱导下,载体质粒能够合成β—半乳糖核苷酶N端(α肽段),这样就与宿主细胞合成的β—半乳糖核苷酶C端部分序列(β肽段)互补,形成完整的β—半乳糖核苷酶活性蛋白。
而当外源基因插入到此种载体质粒lacZ的多克隆位点后,会造成lacZ基因不能表达,从而不能合成β—半乳糖核苷酶;而对于空载体,lacZ基因正常表达,通过α互补合成β—半乳糖核苷酶,分解培养基里的色素底物X-gal,最终形成蓝色的化合物,出现蓝色菌斑。
X-Gal,中文名为5-溴-4-氯-3-吲哚-β-D-半乳糖苷,分子式为C14H15BrClNO6,分子量为408.63,它是β–半乳糖苷酶的底物,水解后呈蓝色。
IPTG,中文名,异丙基-β-D-硫代吡喃半乳糖苷,分子式: C9H18O5S ,分子量238.30 ,保存温度2-8°C冷藏保存,配制好后保存于-20°C,室温可放置一个月。
主要作用:异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用实验准备清洗,5个100ml锥形瓶(外加1个小的),6副培养皿,3个小试剂瓶,接种环,涂布棒准备100ml超纯水溶液:LB300ml(液体50ml+50ml,固体100ml),0.1mol/L CaCl2 10ml,100mg/mlAmp 1 ml,20mg/mlX-gal 1 ml,200mg/ml IPTG 1 ml。
大肠杆菌菌液1ml感受态细胞连接产物4管 4 x 5 = 20ul 做4份目的基因T载体T4连接酶10x冲液4 x 4uL 4x1uL 4x0.5uL 4x1uL灭菌:5个100ml锥形瓶(外加1个小的),6副培养皿,3个小试剂瓶,接种环,涂布棒,10ml超纯水,LB培养基,1.5mlEP管,大小枪头,过滤用具2副,,0.1mol/L CaCl2试剂20mg/ml X-gal X-gal为5-溴-4-氯-3-吲哚-β-D半乳糖苷。
用二基甲酰胺(DMF)溶解X-gal配制成的20mg/ml(取0.02gX-gal,用DMF定容为1ml)的贮存液。
保存于一玻璃管或聚丙烯管中,装有X-gal溶液的试管须用铝箔封裹以防因受光照而被破坏,并应贮存于-20℃。
X-gal溶液无须过滤除菌。
(DMF二甲基甲酰胺; Dimethylformamide; N,N-Dimethylformamide; DMF; CAS:68-12-2 理化性质:无色、淡的胺味的液体。
分子式C3-H7-N-O。
分子量73.10。
相对密度0.9445(25℃)。
熔点-61℃。
沸点152.8℃。
)200mg/ml IPTG在0.8ml蒸馏水中溶解0.2g IPTG后,用蒸馏水定容至1ml,用0.22μm滤器过滤除菌,贮存于-20℃。
LB培养基:配制每升培养基,应该在950 ml去离子水中加入:胰化蛋白胨10g酵母提取物5gNaCl 10g摇动容器直至溶质溶解.用5mol/LNaOH调pH至7.3.用去离子水定容至1L.在15psi高压下蒸汽灭菌20min.(100mlLB培养基加入1.5g琼脂粉为固体培养基)0.1mol/LCaCl2:取0.11098g氯化钙固体定容至10mlAmp(100mg/ml):溶解0.1g氨苄青霉素钠盐于足量的水中,最后定容至1ml,用0.22μm滤膜过滤除菌.实验步骤(一).目的基因片段与载体连接器材旋涡混合器,微量移液取样器,移液器吸头,1.5ml 微量离心管,双面离心管架,台式离心机,干式恒温气浴。
试剂T 载体,T4 DNA 连接酶,连接酶缓冲液,无菌dd Water操作步骤PCR产物与T载体直接连接:(1)事先将干式恒温仪(或冰盒里的水)温度设定在14~16︒C。
(2)取4个灭菌的200ul微量离心管,加入:(需要调整)4μl 目的基因1μl T载体0.5μl T4 DNA连接酶(TAKARA, 350U/ul)后加1μl 连接酶缓冲液10 x buffer3.5 μl dd Water总量10μl 体系(3)述混合液轻轻震荡后再短暂离心,然后置于14︒C干式恒温仪(或14︒C水中)中保温过夜(12-16h)。
(4)连接后的产物可以立即用来转化感受态细胞或置4︒C冰箱备用。
(二).大肠杆菌感受态细胞的制备细菌转化器材旋涡混合器,微量移液取样器,移液器吸头,50ml 微量离心管,1.5ml 微量离心管,台式冷冻离心机,制冰机,恒温摇床,分光光度计,超净工作台,恒温培养箱,摇菌试管,三角烧瓶,接种环,恒温摇床,培养皿(已铺好固体LB-Amp),酒精灯,玻璃涂棒,恒温培养箱,滤膜和过滤器试剂E. coli菌种,LB培养基,0.1 mol/L CaCl2溶液,无菌dd Water ,LB培养基(不加抗菌素),LB培养基(加抗菌素),无菌dd water,IPTG,X-gal。
步骤(1)在超净工作台中,将1ml大肠杆菌菌液加入100ml LB液态培养基(不含抗菌素),37℃摇床培养过夜。
(2)取0.5ml上述菌液转接到含有50mL LB培养基的三角烧瓶中,37℃下250r/min摇床培养2~3h,测定OD590为0.35~0.4左右(<0.4~0.6,细胞数<108/mL,此为关键参数!)。
(注意:此步摇菌的时候,要有一管不加菌的LB培养液同时摇菌)(3)将1ml菌液加入到4支1.5mL预冷无菌的聚丙烯离心管中,于冰上放置10min,然后于4℃,5000rpm离心5min。
(4)将离心管倒置以倒尽上清液,加入1ml 冰冷的0.1 mol/L CaCl2溶液,立即在涡旋混合器上混匀,插入冰中放置30min。
(5)4℃,5000rpm离心5min,弃上清液后,用100μL 冰冷的0.1 mol/L CaCl2溶液垂悬,插入冰中放置2h,可以直接用作转化实验,或立即放入-4摄氏度冰柜中保藏。
(6)事先将恒温水浴的温度调到42℃。
(7)从-70℃超低温冰柜中取出一管(100μL)感受态菌,立即用手指加温融化后插入冰上,冰浴5~10min。
(8)加入5μL连接好的质粒混合液(DNA含量不超过100ng),轻轻震荡后放置冰上20min。
(9)轻轻摇匀后插入42℃水浴中90s进行热休克,然后迅速放回冰中,静置3min。
(10)在超净工作台中向上述各管中分别加入300μL LB培养基(不含抗菌素)轻轻混匀,然后固定到摇床的弹簧架上37℃震荡1h。
(11)在超净工作台中取上述转化混合液200μL,分别滴到含合适抗菌素(Amp 100ug/L)的固体LB平板培养皿中,再在平板上滴加40μL 20mg/ml X-gal,7μL 200mg/ml IPTG,用酒精灯烧过的玻璃涂布棒涂布均匀(注意:一个不含抗生素作为对照组,玻璃涂布棒上的酒精熄灭后稍等片刻,待其冷却后再涂,菌液涂皿操作时,应避免反复来回涂布,因为感受态细菌的细胞壁有了变化,过多的机械挤压涂布会使细胞破裂,影响转化率。
)。
(12)在涂好的培养皿上做上标记,先放置在37℃恒温培养箱中30min直到表面的液体都渗透到培养基里后,再倒置过来放入37℃恒温培养箱过夜。
(13)在被细菌污染的桌面上喷洒70%乙醇,擦干桌面。
(14)观察平板上长出的菌落克隆,以菌落之间能互相分开为好。
注意白色菌斑。
(三).转化克隆的筛选和鉴定器材旋涡混合器,小镊子,微量移液取样器,移液器吸头,1.5ml 微量离心管,双面离心管架,干式恒温气浴(或恒温水浴锅),制冰机,恒温摇床,超净工作台,酒精灯,无菌牙签,摇菌管。
试剂LB培养基(加抗菌素),PCR用试剂,引物,质粒提取用试剂,酶切需要的限制性内且酶及其缓冲液,65%甘油(65%甘油,0.1mol/L MgSO4,0.025mol/L Tris Cl pH8.0)。
操作步骤方法一:快速PCR筛选法(1)在转化的平板培养基上随机选取4个边缘清晰的白色菌落,并用记号笔在其所在的培养皿底部玻璃背面画圈做标记编号。
(2)在0.2ml PCR 微量离心管中配制25μl反应体系。
dd water 16μl10×PCR buffer(不含MgCl2) 2.5μl25mM MgCl2 1.5μl2.5mmol/L dNTP 2μl(每种dNTP终浓度0.2mM)10μmol/L Primer1 1μl(12.5—25pmoles)10μmol/L primer2 1μl(12.5—25pmoles)模板质粒用小tip头轻轻粘一下选中的白色菌落,再伸入PCR混合液中洗一洗Taq酶0.5μl(1.5u)总体积25μl(2)根据厂商的操作手册设置PCR仪的循环程序(本实验室已经设置为WZ):①94℃5min②94℃1min③60℃1min④ 72℃1min50s⑤goto②29 times⑥72℃10min(2)PCR结束后,取10μl产物进行琼脂糖凝胶电泳(与原始插入片断同时比对)。