理论计算
钢板常用理论重量计算公式

钢板常用理论重量计算公式钢板是一种常见的金属材料,在工业生产和建筑领域中被广泛应用。
在实际应用中,我们经常需要计算钢板的重量,以便进行材料采购、运输和设计等工作。
钢板的重量计算是一个基本而重要的问题,它涉及到钢板的尺寸、材质和厚度等因素。
在本文中,我们将介绍钢板常用的理论重量计算公式,希望能够帮助读者更好地理解和应用这一知识。
一、钢板的重量计算公式。
钢板的重量计算公式是根据钢板的尺寸、材质和厚度等因素进行推导和建立的。
在实际应用中,我们通常使用以下两种常用的理论重量计算公式:1. 钢板的重量(kg)=长度(m)×宽度(m)×厚度(mm)×钢板密度(g/cm³)×0.001。
2. 钢板的重量(kg)=长度(m)×宽度(m)×厚度(m)×钢板密度(g/cm ³)。
这两种公式都是根据钢板的体积和密度来计算钢板的重量的,其中钢板的密度是一个常数,通常为7.85g/cm³。
根据这两种公式,我们可以很方便地计算出钢板的重量,为后续的工作提供了重要的参考数据。
二、钢板的重量计算实例。
为了更好地说明钢板的重量计算方法,下面我们通过一个具体的实例来进行说明。
假设有一块钢板的尺寸为2m×1m×10mm,那么根据上述的第一种公式,我们可以计算出这块钢板的重量为:钢板的重量(kg)=2m×1m×10mm×7.85g/cm³×0.001=15.7kg。
通过这个实例,我们可以看到,通过上述的公式,我们可以很方便地计算出钢板的重量,为后续的工作提供了重要的参考数据。
三、钢板重量计算的注意事项。
在进行钢板重量计算时,需要注意以下几个问题:1. 钢板的尺寸应该按照实际测量的尺寸进行计算,避免出现误差。
2. 钢板的密度通常为7.85g/cm³,但在实际应用中也可能会有一定的误差,因此在进行计算时需要注意。
常用计算的基本理论和方法

积分是微积分的另一重要概念,用于计算曲 线与x轴所夹的面积,是解决实际问题的重 要方法。
线性代数基础
向量
向量是一组有序数,可以表示空间中的点或方向,是线性代数中 的基本概念。
矩阵
矩阵是由数字组成的矩形阵列,可以表示向量之间的关系和变换, 是线性代数中的重要工具。
线性方程组
线性方程组是描述多个未知数之间线性关系的方程组,通过矩阵和 向量运算可以求解线性方程组。
代数运算
代数运算包括加法、减法、乘法、 除法以及指数、对数等运算,是 数学中基本的运算方法。
代数式
代数式是由数字、字母通过有限 次的四则运算得到的数学表达式, 是代数中常用的表达方式。
微积分基础
极限
极限是微积分的基本概念,描述了函数在某 点的变化趋势,是研究连续函数的重要工具 。
导数
导数描述了函数在某点的切线斜率,是研究函数变 化速度和极值点的关键概念。
阐述本主题的目的,即掌握常用计算 的基本理论和方法,提高计算思维和 解决问题的能力。
意义
强调常用计算基本理论和方法的重要 性,包括在计算机科学、数学、工程 等领域的应用价值。
02 数学基础
代数基础
代数方程
代数方程是数学中的基本概念, 包括一元一次方程、一元二次方 程、多元一次方程组等,用于描 述数学关系和解决实际问题。
方差分析
总结词
方差分析是一种通过比较不同组数据的变异程度来分 析因素对结果的影响的方法。
详细描述
方差分析的基本思想是将数据的变异分解为两部分:一 部分是由实验操作或处理引起的变异,另一部分是由随 机误差引起的变异。通过比较不同组数据的变异程度, 可以判断不同因素对结果的影响是否显著。方差分析需 要满足一定的假设条件,如各组数据的方差齐性、正态 性等。在应用方差分析时,需要注意数据的分布特征和 处理方式,以及选择合适的统计方法和软件进行数据分 析。
理论重量的计算方法

理论重量的计算方法理论重量(theoretical weight)是指根据物质的密度和几何尺寸等参数计算出来的预测值。
它在工程领域中广泛应用于材料选择、设备设计等方面。
以下是计算理论重量的常用方法:1.线材和板材的理论重量计算:对于线材和板材,我们首先需要知道材料的密度ρ(单位为千克/立方米)和尺寸参数,例如长度L(单位为米)和横截面积A(单位为平方米)。
线材的理论重量W可通过公式:W=ρxAxL进行计算。
板材的理论重量W可通过公式:W=ρxAxt进行计算,其中t表示板材的厚度(单位为米)。
2.管材的理论重量计算:对于管材,其理论重量计算需要考虑管径d(单位为米)、壁厚t(单位为米)以及管长L(单位为米)。
管材的理论重量W可通过公式:W=[π/4x(d^2-(d-2t)^2)]xLxρ进行计算。
3.型材的理论重量计算:型材的理论重量计算需要考虑材料的密度ρ(单位为千克/立方米)、长度L(单位为米)、横截面积A(单位为平方米)以及材料的形状。
型材的理论重量W可通过公式:W=ρxAxL进行计算。
另外,对于复杂形状的物体,可能需要将其分解为简单的几何体计算各部分的理论重量,再取其之和得到整体的理论重量。
需要特别注意的是,此处提到的计算方法中所用的单位需要保持一致,常用的单位有千克(kg)、克(g)、立方米(m^3)、平方米(m^2)、米(m)等。
确保在计算过程中使用正确的单位可以避免出现计算错误。
总结起来,理论重量的计算方法主要基于材料的密度和几何尺寸等参数。
对于不同形状的材料,我们需要根据不同的公式进行计算。
在实际应用中,需要注意单位的一致性以及对于复杂形状的物体可能需要进行分解计算。
理论计算ppt课件

(三)、应用领域
Mateirals Studio 是 Acce lrys 公司专为材料科学领域开发的 新一代材料计算软件。它能方便地建立 3D 分子模型,深入分 析有机晶体、无机晶体、无定形材料以及聚合物,可以在催 化剂、聚合物、固体化学、结晶学、晶粉衍射以及材料特性 等材料科学研究领域进行性质预测、聚合物建模
三、运用
采用 Materials Studio4.0 软件对ZnO的能带及电子能态密度进行 了模拟计算 1 模拟计算步骤 1.1 建立晶体 需要计算ZnO晶体参数通过相关文献查阅得到,如表1所示。 表1 相关晶胞参数表
晶体 ZnO 晶体种类 a b c 3D Hexagonal 3.24927 3.24927 5.20544
理论计算
1
几个重要概念
2
MS(Materials Studio)软件介绍
3
MS软件的运用
一、几个重要概念
1、理论计算:应用现有的定律、定理及规律对问题进行分析推理,找 出符合其尊循的规律公式(推导出来的公式)进行计算,其整个过程 叫理论计算 2、理论化学:是运用纯理论计算而非实验方法研究化学反应的本质 问题,主要以理论物理为研究工具(如热力学、量子力学、统计力学 、量子电动力学、非平衡态热力学等),并且大多辅以计算机模拟。 3、计算化学(computational chemistry):是理论化学的一个分支 。计算化学的主要目标是利用有效的数学近似以及电脑程序计算分子 的性质(例如总能量,偶极矩,四极矩,振动频率,反应活性等)并 用以解释一些具体的化学问题。 计算化学这个名词有时也用来表示计算机科学与化学的交叉学科。
聚丙烯的结构图
9
应用Visualizer模块构造体系结模块对苯分子 的电荷、分子轨道和能量等方面进行计算。
国际理论计算公式

国际理论计算公式国际理论计算公式是指用于解决各种科学、工程和经济问题的数学公式,能够描述现实世界中发生的物理、化学和经济等各种现象。
这些公式是基于理论和经验数据推导得到的,可以通过计算机等数值方法进行求解。
国际理论计算公式在科技领域具有广泛的应用,对于研究和解决实际问题具有重要的指导意义。
一、物理学公式1.速度公式:V=d/t,其中V表示速度,d表示距离,t表示时间。
2.加速度公式:a=(v-u)/t,其中a表示加速度,v表示末速度,u表示初速度,t表示时间。
3.力公式:F=m*a,其中F表示力,m表示质量,a表示加速度。
4.万有引力公式:F=G*(m1*m2)/r^2,其中F表示引力,G表示万有引力常量,m1和m2表示两个物体的质量,r表示两个物体之间的距离。
二、化学公式1.质量守恒定律:反应前和反应后的总质量相等。
2.摩尔质量公式:M=m/n,其中M表示摩尔质量,m表示物质的质量,n表示物质的摩尔数。
3.摩尔比公式:CxHyOz+nO2→nCO2+mH2O,其中n和m分别表示生成的二氧化碳和水的摩尔数。
4.理想气体状态方程:PV=nRT,其中P表示气体的压力,V表示气体的体积,n表示气体的摩尔数,R表示气体常数,T表示气体的温度。
三、经济学公式1.边际效用公式:MU=ΔU/ΔQ,其中MU表示边际效用,ΔU表示效用的变化,ΔQ表示消费量的变化。
2.边际成本公式:MC=ΔC/ΔQ,其中MC表示边际成本,ΔC表示成本的变化,ΔQ表示生产量的变化。
3.各种成本公式:-总成本公式:TC=FC+VC,其中TC表示总成本,FC表示固定成本,VC表示变动成本。
-平均成本公式:AC=TC/Q,其中AC表示平均成本,Q表示产量。
-边际成本公式:MC=ΔTC/ΔQ,其中MC表示边际成本,ΔTC表示总成本的变化,ΔQ表示产量的变化。
4.弹性公式:-价格弹性公式:E=ΔQ/ΔP*P/Q,其中E表示价格弹性,ΔQ表示需求量的变化,ΔP表示价格的变化,P表示平均价格,Q表示平均需求量。
钢材理论重量计算公式

1.钢板重量计算公式公式: 7.85×长度(m)×宽度(m)×厚度(mm)例: 钢板6m(长)×1.51m(宽)×9.75mm(厚)计算: 7.85×6×1.51×9.75=693.43kg2.钢管重量计算公式公式: (外径-壁厚)×壁厚mm×0.02466×长度m 例: 钢管114mm(外径)×4mm(壁厚)×6m(长度)计算: (114-4)×4×0.02466×6=65.102kg3.圆钢重量计算公式公式: 直径mm×直径mm×0.00617×长度m例: 圆钢Φ20mm(直径)×6m(长度)计算: 20×20×0.00617×6=14.808kg4.方钢重量计算公式公式: 边宽(mm)×边宽(mm)×长度(m)×0.00785例: 方钢50mm(边宽)×6m(长度)计算: 50×50×6×0.00785=117.75(kg)5.扁钢重量计算公式公式: 边宽(mm)×厚度(mm)×长度(m)×0.00785例: 扁钢50mm(边宽)×5.0mm(厚)×6m(长度)计算: 50×5×6×0.00785=11.7.75(kg)6.六角钢重量计算公式公式: 对边直径×对边直径×长度(m)×0.00068例: 六角钢50mm(直径)×6m(长度)计算: 50×50×6×0.0068=102(kg)7.螺纹钢重量计算公式公式: 直径mm×直径mm×0.00617×长度m例: 螺纹钢Φ20mm(直径)×12m(长度)计算: 20×20×0.00617×12=29.616kg8.扁通重量计算公式公式: (边长+边宽)×2×厚×0.00785×长m例: 扁通100mm×50mm×5mm厚×6m(长)计算: (100+50)×2×5×0.00785×6=70.65kg9.方通重量计算公式公式: 边宽mm×4×厚×0.00785×长m例: 方通50mm×5mm厚×6m(长)计算: 50×4×5×0.00785×6=47.1kg10.等边角钢重量计算公式公式: 边宽mm×厚×0.015×长m(粗算)例: 角钢50mm×50mm×5厚×6m(长)计算: 50×5×0.015×6=22.5kg(表为22.62)11.不等边角钢重量计算公式公式: (边宽+边宽)×厚×0.0076×长m(粗算) 例: 角钢100mm×80mm×8厚×6m(长)计算: (100+80)×8×0.0076×6=65.67kg(表65.676)其他有色金属12.黄铜管重量计算公式公式: (外径-壁厚)×厚×0.0267×长m例: 黄铜管20mm×1.5mm厚×6m(长)计算: (20-1.5)×1.5×0.0267×6=4.446kg13.紫铜管重量计算公式公式: (外径-壁厚)×厚×0.02796×长m例: 紫铜管20mm×1.5mm厚×6m(长)计算: (20-1.5)×1.5×0.02796×6=4.655kg14.铝花板重量计算公式公式: 长m×宽m×厚mm×2.96例: 铝花板1m宽×3m长×2.5mm厚计算: 1×3×2.5×2.96=22.2kg黄铜板: 比重8.5紫铜板: 比重8.9锌板: 比重7.2铅板: 比重11.37计算方式: 比重×厚度=每平方的重量注:公式中长度单位为米, 面积单位为平方米, 其余单位均为毫米长方形的周长=(长+宽)×2正方形的周长=边长×4长方形的面积=长×宽正方形的面积=边长×边长三角形的面积=底×高÷2平行四边形的面积=底×高梯形的面积=(上底+下底)×高÷2直径=半径×2 半径=直径÷2圆的周长=圆周率×直径=圆周率×半径×2圆的面积=圆周率×半径×半径长方体的表面积= (长×宽+长×高+宽×高)×2长方体的体积=长×宽×高正方体的表面积=棱长×棱长×6正方体的体积=棱长×棱长×棱长圆柱的侧面积=底面圆的周长×高圆柱的表面积=上下底面面积+侧面积圆柱的体积=底面积×高圆锥的体积=底面积×高÷3长方体(正方体、圆柱体)的体积=底面积×高平面图形周长—C, 面积—S,正方形:C=4a ;S=a2长方形:a、b—边长C=2(a+b) ;S=ab三角形:a、b、c—三边长, H—a边上的高, s—周长的一半, A,B,C-内角其中s=(a+b+c)/2 S=ah/2=ab/2·sinC=[s(s-a)(s-b)(s-c)]1/2=a2sinBsinC/(2sinA)四边形:d,D-对角线长, α-对角线夹角S=dD/2·sinα平行四边形:a,b-边长, h-a边的高, α-两边夹角S=ah=absinα菱形:a-边长, α-夹角, D-长对角线长, d-短对角线长S=Dd/2=a2sinα梯形:a和b-上、下底长, h-高, m-中位线长S=(a+b)h/2=mh圆:r-半径, d-直径C=πd=2πrS=πr2扇形:r—扇形半径, a—圆心角度数C=2r+2πr×(a/360)S=πr2×(a/360)弓形:l-弧长, b-弦长, h-矢高, r-半径, α-圆心角的度数S=r2/2·(πα/180-sinα)=r2arccos[(r-h)/r] - (r-h)(2rh-h2)1/2=παr2/360 - b/2·[r2-(b/2)2]1/2=r(l-b)/2 + bh/2≈2bh/3圆环:R-外圆半径, r-内圆半径, D-外圆直径, d-内圆直径S=π(R2-r2)=π(D2-d2)/4椭圆:D-长轴, d-短轴S=πDd/4立方图形面积S和体积V正方体a-边长S=6a2V=a3长方体a-长, b-宽, c-高S=2(ab+ac+bc)V=abc棱柱:S-底面积, h-高V=Sh棱锥:S-底面积, h-高V=Sh/3棱台:S1和S2-上、下底面积, h-高V=h[S1+S2+(S1S1)1/2]/3拟柱体:S1-上底面积, S2-下底面积, S0-中截面积, h-高V=h(S1+S2+4S0)/6圆柱:r-底半径, h-高, C—底面周长, S底—底面积, S侧—侧面积, S表—表面积C=2πrS底=πr2S侧=ChS表=Ch+2S底V=S底h=πr2h空心圆柱:R-外圆半径, r-内圆半径, h-高V=πh(R2-r2)直圆锥:r-底半径, h-高V=πr2h/3圆台:r-上底半径, R-下底半径, h-高V=πh(R2+Rr+r2)/3球:r-半径, d-直径V=4/3πr3=πd2/6球缺:h-球缺高, r-球半径a-球缺底半径V=πh(3a2+h2)/6=πh2(3r-h)/3a2=h(2r-h)球台:r1和r2-球台上、下底半径, h-高V=πh[3(r12+r22)+h2]/6圆环体:R-环体半径, D-环体直径, r-环体截面半径, d-环体截面直径V=2π2Rr2=π2Dd2/4桶状体:D-桶腹直径, d-桶底直径, h-桶高V=πh(2D2+d2)/12(母线是圆弧形,圆心是桶的中心)V=πh(2D2+Dd+3d2/4)/15(母线是抛物线形)。
气体摩尔热容的计算

气体摩尔热容的计算气体的摩尔热容是指单位摩尔物质在恒压下温度变化单位度时所吸收或释放的热量。
气体摩尔热容的计算可以通过理论推导和实验测定两种方法来进行。
一、理论计算方法:1.理想气体的摩尔热容:对于理想气体,摩尔热容可通过以下公式计算:Cp=(f/2+1)R(理论计算的公式1)Cv=(f/2)R(理论计算的公式2)其中,Cp为恒压摩尔热容,Cv为恒容摩尔热容,f为气体分子自由度的个数,R为气体常数。
对于双原子分子气体(如氧气、氮气等),分子自由度f=5,带入公式1和公式2可得相关的摩尔热容值。
2.实际气体的摩尔热容:对于实际气体,可以通过计算多原子分子振动、转动和电子能级的贡献来计算摩尔热容。
这个过程需要使用量子力学理论。
具体的计算公式比较复杂,这里不展开讨论。
二、实验测定方法:实验测定摩尔热容的方法有很多,下面介绍两种常用的方法。
1.等压热容法:等压热容方法是指在恒定的压力下测量气体温度的变化,从而计算出摩尔热容。
实验过程如下:a.将一定质量的气体加入到容器中,保持恒定的压力。
b.将测量温度的热电偶或温度计放入容器中,记录初始温度。
c.在恒压条件下加热或冷却气体,测量气体温度的变化。
d.测得的温度变化量与所加的热量之间的比值即为摩尔热容。
2.等容热容法:等容热容法是指在恒定的体积下测量气体压强的变化,从而计算出摩尔热容。
a.将一定质量的气体加入到容器中,保持恒定的体积。
b.将测量压强的压力计放入容器中,记录初始压强。
c.在恒容条件下加热或冷却气体,测量气体压强的变化。
d.测得的压强变化量与所加的热量之间的比值即为摩尔热容。
以上是关于气体摩尔热容的计算方法的介绍,包括理论计算和实验测定的方法。
根据需要选择合适的方法进行计算,可以更好地了解和研究气体的热力学性质。
理论重量计算方法

钢材理论重量计算(单位:公斤)等边角钢:每米重量=0.00785×(2边宽-边厚)×边厚不等边角钢:每米重量=0.00785×(长边宽+短边宽-边厚)×边厚圆钢、螺纹钢、线材、钢丝:每米重量=0.00617×直径×直径扁钢、钢板、钢带:每米重量=0.00785×厚度×边宽管材:每米重量=0.02466×壁厚×(外径-壁厚)黄铜管:每米重量=0.02670×壁厚×(外径-壁厚)工字钢:每米重量=0.00785×腰厚〔高+f(腿宽-腰厚)〕(f值系数:一般型号带a的为3.34,带b的为2.65,带c的为2.26)槽钢:每米重量=0.00785×腰厚〔高+e(腿宽-腰厚)〕(e值系数:一般型号带a的为3.26,带b的为2.44,带c的为2.24)紫铜管:每米重量=0.02796×壁厚×(外径-壁厚)铝花纹板:每平方米重量=2.96×厚度有色金属比重:紫铜板8.9黄铜板8.5锌板7.2铅板11.37有色金属板材的计算公式为:每平方米重量=比重×厚度角钢:每米重量=0.00785*(边宽+边宽-边厚)*边厚圆钢:每米重量=0.00617*直径*直径(螺纹钢和圆钢相同)扁钢:每米重量=0.00785*厚度*边宽管材:每米重量=0.02466*壁厚*(外径-壁厚)板材:每米重量=7.85*厚度黄铜管:每米重量=0.02670*壁厚*(外径-壁厚)紫铜管:每米重量=0.02796*壁厚*(外径-壁厚)铝花纹板:每平方米重量=2.96*厚度有色金属比重:紫铜板8.9黄铜板8.5锌板7.2铅板11.37有色金属板材的计算公式为:每平方米重量=比重*厚度数据采集和对于《实用钢铁材料便查手册》H型钢理论重量表螺旋缝高频焊钢管(单位米重)理论计算表螺旋缝埋弧焊钢管(单位米重)理论计算表钢轨理论重量槽钢理论重量、规格表焊管理论单重焊接钢管包括一般焊接钢管、吹氧钢管、电线套管、镀锌钢管、变压器管、电焊异型管等热轧结构用无缝钢管理论重量。
计算理论基础知识

计算理论基础知识计算理论是计算机科学的核心领域之一,它研究的是计算过程的本质和限制。
在计算机科学的发展过程中,计算理论提供了重要的理论基础和方法,为计算机科学和技术的发展奠定了坚实的基础。
本文将简要介绍计算理论的基础知识。
一、自动机理论自动机是计算理论中的重要概念之一,它用于描述计算过程的抽象模型。
自动机可以分为有限自动机和非确定性有限自动机等多种类型。
有限自动机是一种最简单的计算模型,它由状态、输入字母表、转换函数和初始状态等组成。
通过状态的转换和输入的驱动,有限自动机可以执行特定的计算任务。
非确定性有限自动机则相对更加复杂,它在进行状态转换时可以有多个可能的选项。
二、形式语言与文法形式语言和文法是计算理论中研究自动机行为规律的重要工具。
形式语言是由符号组成的集合,用于表示计算过程中的输入、输出和中间结果等信息。
文法则定义了形式语言的句子生成规则。
常见的文法类型有上下文无关文法、上下文相关文法等。
形式语言和文法的研究使得我们能够通过规则来描述和分析计算过程,从而更好地理解计算机科学中的一些重要概念和问题。
三、图灵机和可计算性理论图灵机是计算理论中最重要的概念之一,它由一个无限长的纸带和一个读写头组成。
图灵机通过读写头在纸带上的移动和改写来模拟计算过程。
图灵机的提出使得我们能够更深入地研究计算过程的本质和限制。
可计算性理论是计算理论中的一个重要分支,它研究的是什么样的问题可以通过某种计算模型解决。
根据可计算性理论,存在一些问题是不可计算的,即无法用任何计算模型来解决。
四、复杂性理论复杂性理论是计算理论中的另一个重要分支,它研究的是计算问题的复杂度。
复杂性理论主要关注计算问题的难解性和可解性。
常见的复杂性类别有P类、NP类等。
P类问题是可以在多项式时间内解决的问题,而NP类问题是可以在多项式时间内验证解的问题。
复杂性理论的研究使得我们能够更好地理解计算问题的本质,从而设计更高效的算法和方法。
五、计算复杂性和可计算性的关系计算复杂性和可计算性是计算理论中两个重要的概念。
型钢理论重量计算公式

.一、H350*350*12*19理论计算公式(350*12+350*19*2)/1000 *7.85 =139.73kg/m(腹板长度*腹板厚+翼缘宽度*翼缘厚度*2)*7.85请注意单位最后是kg/m二、钢板的理论重量计算公式是怎样的?最后举个例子。
重量=厚度*宽度*长度*7.85 比如:20mm*2000mm*10000mm单重:0.02*2*10*7.85=3.14T 注:单位要换算为米(M) ...三、钢管重量(公斤)=0.02466×壁厚×(外径-壁厚)×长度四、角钢重量(公斤)=0.00785×(边宽+边宽-边厚)×边厚×长度五、圆钢重量(公斤)=0.00617×直径×直径×长度六、C型钢每米重量计算公式:举例C100*50*20*2每米重量=(100+50*2+20*2)*2*7.85/1000C型钢计算公式 (宽+高*2+小边*2)*厚度*7.85/1000例:外输泵房GZ1 H450*250*6*10计算公式:(450*6+250*10*2)/1000*7.85=60.445kg/m外输泵房GZ8 H(450-300)*250*6*10计算公式:<(450+300)/2*6+250*10*2>/1000*7.85=56.912 kg/m外输泵房角钢∠63*5计算公式:(63+63-5)*5*0.00785=4.75 kg/m外输泵房 XG1 Φ114*5计算公式:0.02466*5*(114-5)=13.4397 kg/m外输泵房LT1 Φ12 计算公式:0.00617*12*12=0.88848 kg/m外输泵房 QL1 C180*60*20*3计算公式:(180+60*2+20*2)*3*0.00785=8.007 kg/m..。
理论产量计算公式

理论产量计算公式
在制造业中,理论产量计算公式通常涉及到设备的生产能力、工时和
效率等因素。
例如,工厂的设备产能为每小时100个产品,工作时间为
10小时,实际生产效率为80%时,计算理论产量的公式为:
理论产量=设备产能×工作时间×实际生产效率
=100×10×0.8
=800个产品
在农业中,理论产量计算公式通常涉及到单位面积或单位农田的产量。
例如,作物的每亩产量为500公斤,农田面积为100亩,那么理论产量的
公式为:
理论产量=单位面积产量×农田面积
=500×100
理论产量=本金×(1+年化收益率)^投资时间
理论产量=市场规模×市场份额
需要注意的是,以上公式仅为理论产量的计算公式,不考虑实际生产、经营或市场中的各种复杂因素。
实际情况中,可能会出现各种不确定因素、资源限制、技术限制、市场需求变化等情况,导致实际产量与理论产量有
所差别。
因此,在实际应用中需要结合实际情况进行调整和修正。
理论重量计算公式
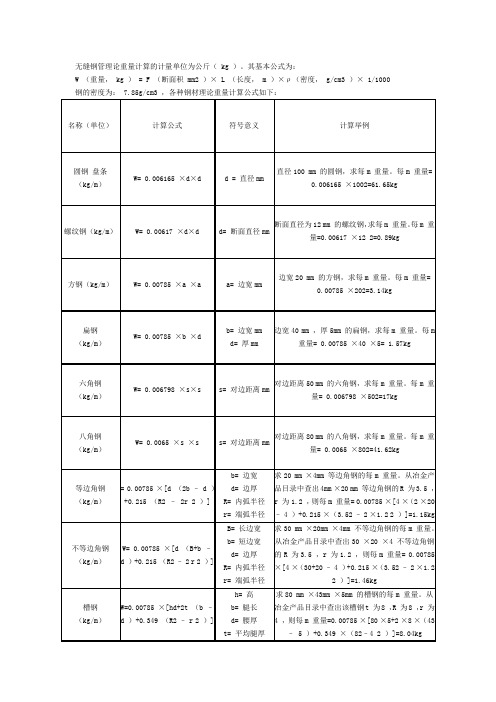
(kg/m)
W= 0.006798 ×s×s
s= 对边距离mm
对边距离798 ×502=17kg
八角钢
(kg/m)
W= 0.0065 ×s ×s
s= 对边距离mm
对边距离80 mm 的八角钢,求每m 重量。每m 重量= 0.0065 ×802=41.62kg
方钢(kg/m)
W= 0.00785 ×a ×a
a= 边宽mm
边宽20 mm 的方钢,求每m 重量。每m 重量= 0.00785 ×202=3.14kg
扁钢
(kg/m)
W= 0.00785 ×b ×d
b= 边宽mm
d= 厚mm
边宽40 mm ,厚5mm 的扁钢,求每m 重量。每m 重量= 0.00785 ×40 ×5= 1.57kg
槽钢
(kg/m)
W=0.00785 ×[hd+2t (b – d )+0.349 (R2 – r 2 )]
h= 高
b= 腿长
d= 腰厚
t= 平均腿厚
R= 内弧半径
r= 端弧半径
求80 mm ×43mm ×5mm 的槽钢的每m 重量。从冶金产品目录中查出该槽钢t 为8 ,R 为8 ,r 为4 ,则每m 重量=0.00785 ×[80 ×5+2 ×8 ×(43 – 5 )+0.349 ×(82–4 2 )]=8.04kg
W= 0.006165 ×d×d
d = 直径mm
直径100 mm 的圆钢,求每m 重量。每m 重量= 0.006165 ×1002=61.65kg
螺纹钢(kg/m)
W= 0.00617 ×d×d
d= 断面直径mm
断面直径为12 mm 的螺纹钢,求每m 重量。每m 重量=0.00617 ×12 2=0.89kg
标准工时理论计算公式

标准工时理论计算公式在现代工业生产中,为了更好地管理生产过程和提高生产效率,人们引入了标准工时理论。
标准工时理论是指根据生产过程中所需的时间和资源,计算出完成一项工作所需的标准工时,并以此作为衡量生产效率和成本的指标。
标准工时理论的计算公式是生产过程中必不可少的工具,它可以帮助企业合理安排生产计划,控制生产成本,提高生产效率。
标准工时理论的计算公式主要包括以下几个要素,标准工时、实际工时、标准工时产量、工作效率等。
下面我们将详细介绍标准工时理论的计算公式及其应用。
1. 标准工时的计算公式。
标准工时是指完成一项工作所需的标准时间,它是根据生产过程中所需的时间和资源来计算的。
标准工时的计算公式如下:标准工时 = (总工作时间 + 休息时间) / 标准产量。
其中,总工作时间是指完成一项工作所需的总时间,包括准备时间、加工时间、装配时间等;休息时间是指工人在工作过程中的休息时间,一般按照一定的比例进行计算;标准产量是指在标准工时内完成的产品数量。
2. 实际工时的计算公式。
实际工时是指完成一项工作所需的实际时间,它是根据生产过程中实际耗费的时间和资源来计算的。
实际工时的计算公式如下:实际工时 = 总工作时间 + 加班时间 + 损耗时间。
其中,总工作时间是指完成一项工作所需的总时间,包括准备时间、加工时间、装配时间等;加班时间是指工人在工作时间外额外加班的时间;损耗时间是指由于各种原因导致的生产过程中的时间损耗。
3. 标准工时产量的计算公式。
标准工时产量是指在标准工时内完成的产品数量,它是根据生产过程中所需的时间和资源来计算的。
标准工时产量的计算公式如下:标准工时产量 = 总工作时间 / 标准工时。
其中,总工作时间是指完成一项工作所需的总时间;标准工时是指完成一项工作所需的标准时间。
4. 工作效率的计算公式。
工作效率是指在单位时间内完成的工作量,它是根据生产过程中实际完成的工作量来计算的。
工作效率的计算公式如下:工作效率 = 实际工时产量 / 实际工时。
理论计算在工科的重要性

理论计算在工科的重要性理论计算是指研究计算方法、计算模型、计算过程和计算效率等方面的学科。
它涉及数学、计算机科学、信息论等多个领域。
在工科中,理论计算发挥着重要的作用。
首先,理论计算为工科提供了数学基础。
在工程问题中,往往需要进行数学建模和分析。
理论计算可以提供这些数学手段和方法,使得工程问题可以量化、模拟和预测。
比如,计算流体力学可以利用数学模型和计算方法来模拟流体在特定条件下的运动和行为,从而帮助工程师设计飞机、汽车、水电站等工程项目。
其次,理论计算为工科提供了算法和数据结构的基础。
在工程问题求解过程中,往往需要进行大量的计算和数据处理。
理论计算可以研究和设计高效的算法和数据结构,提高工程问题的求解效率。
比如,图像处理领域中的算法可以通过理论计算来优化图像处理过程,使得图像处理速度更快、效果更好。
另外,理论计算在信息科学和通信工程中也具有重要意义。
信息科学和通信工程要求高效地传输和处理信息,理论计算可以提供这些信息处理和传输的方法和技术。
比如,编码理论可以通过理论计算来设计和分析编码方案,提高信息传输的可靠性和效率。
此外,理论计算也对工科的创新起到了重要的推动作用。
理论计算可以帮助工程师发现问题的新方法和新思路,提供新的技术和设计方案。
通过理论计算的研究和开发,可以不断提高工程的质量和效率,推动工科的发展。
最后,理论计算还对工程教育有重要的影响。
在工科教育中,理论计算作为一门学科,可以培养学生的数学思维和计算思维能力。
理论计算的学习可以帮助学生提高抽象思维能力和问题解决能力,培养创新意识和创造力,为学生未来的工程实践打下坚实的基础。
综上所述,理论计算在工科中具有重要的作用。
它为工程问题提供了数学基础,研究和设计了高效的算法和数据结构,推动工程创新,对工程教育起到了重要的作用。
在工科领域中,理论计算的研究和应用将会持续发展,为工程问题求解提供更多的理论支持和技术支持。
汽车理论计算公式

汽车理论计算公式汽车的运行原理和性能由多个因素决定,通过理论计算可以对汽车的性能和效益进行预测和评估。
以下是一些常见的汽车理论计算公式。
1.马力和扭矩马力和扭矩是衡量发动机输出功率的指标。
常见的计算公式如下:马力(HP)= 扭矩(lb-ft)× 发动机转速(rpm)/ 52522.动力输出汽车的动力输出受到驱动系统的影响。
常见的计算公式如下:动力输出(kW)=马力(HP)×0.74573.转速和车速的关系转速和车速的关系取决于车辆的传动比和车轮半径。
常见的计算公式如下:车速(mps)= 2 × 3.1416 × 轮胎半径(m)× 转速(rpm)× 60 / 10004.油耗油耗是衡量汽车燃油效率的指标。
常见的计算公式如下:油耗(L/100km)= 油耗(升)/ 行驶距离(km)× 1005.推力推力是衡量汽车加速性能的指标。
常见的计算公式如下:推力(N)= 车辆质量(kg)× 加速度(m/s^2)6.停车距离停车距离取决于刹车系统和路面摩擦力。
常见的计算公式如下:停车距离(m)=0.5×刹车系统效率×车辆初速度(m/s)^2/路面摩擦力(m/s^2)7.行驶阻力行驶阻力包括空气阻力、滚动阻力和爬坡阻力。
常见的计算公式如下:行驶阻力(N)=空气阻力(N)+滚动阻力(N)+爬坡阻力(N)空气阻力(N)=0.5×空气密度×面积(m^2)×空气阻力系数×车速(m/s)^2滚动阻力(N)= 车辆质量(kg)× 9.8 × 滚动阻力系数爬坡阻力(N)= 车辆质量(kg)× 9.8 × sin(坡度)8.加速时间加速时间是衡量汽车加速性能的指标。
加速时间(s)=车速(m/s)/加速度(m/s^2)9.弯道转向力弯道转向力是衡量汽车在弯道行驶时的操控性能的指标。
理论频数的计算公式

理论频数的计算公式
期望频数计算公式有多种形式,常用的有以下两种:
1.理论频数的计算公式(公式一):
期望频数=(行总频数×列总频数)/总频数
在公式一中,行总频数是指其中一行中各分类的频次之和,列总频数是指其中一列中各分类的频次之和,总频数是指所有分类的频次之和。
这种计算公式假设行和列之间是相互独立的。
2.理论频数的计算公式(公式二):
期望频数=(行总频数×列总频数)/样本总数
在公式二中,样本总数是指所有观察样本的数量。
这种计算公式假设行和列之间是相互独立的,并且样本总数是确定的。
这两种公式可以根据具体问题的要求进行选择和应用。
在使用公式计算理论频数时,需要注意以下几点:
1.在计算理论频数之前,需要先收集实际的观测数据,得到各分类的实际频数。
2.公式中的行总频数和列总频数可以根据实际数据直接统计得到。
3.公式中的总频数或样本总数也可以根据实际数据直接统计得到。
4.公式计算得到的期望频数是在其中一种理论假设下的预期结果,与实际观测数据进行比较可以评估理论假设的合理性。
国际理论计算公式

计算公式1、对外贸易地理方向与世界贸易地位对外贸易地理方向表明一国出口货物和服务的去向地、进口货物和服务的来源地。
一国与其他国家和地区之间的经济贸易联系程度。
计算公式:对某国家和地区的出口或进口贸易额/对世界出口或进口贸易额×100% 对外贸易地位表明世界各洲、各国或地区在国际贸易中所占的比重。
计算公式为:或服务)整个世界贸易额(货物务)对世界出口(货物或服×100% 2、对外贸易依存度对外贸易依存度也叫对外贸易系数,是指一国货物与服务进口额在该国国民生产总值中所占的比重。
计算公式:Z=(X +M )/GDP ×100%Z 为对外贸易系数,X 为出口贸易总额,M 为进口贸易总额。
对外贸易系数为:(进口额+出口额)/国民生产总值(GNP )或国内生产总值(GDP )×100%出口贸易系数为:出口贸易额(货物出口或服务出口或两者相加)/GNP 或GDP ×100% Zx=X /GDP ×100%3、对外贸易乘数理论凯恩斯把反映投资增长和国民收入扩大之间的依存关系称为乘数或倍数理论。
新增加的投资引起对生产资本的需求增加,从而引起从事生产资料的人们的收入增加,结果有次增加了的国民收入总量会等于原增加投资量的若干倍。
乘数K 的计算公式是:K =边际消费倾向—11 国民所得的增加量(ΔY )=乘数(K )×投资的增加量(ΔI )计算对外贸易顺差对公民收入的影响倍数公式为:ΔY -[ΔI+(ΔX -ΔM )] ·K其中,ΔY 代表国民收入的增加额,ΔI 代表投资的增加额,ΔX 代表出口增加额,ΔM 代表进口增加额,K 代表乘数。
4、增值标准 澳大利亚和美国等国家都采用这项标准,它规定使用这进口成分占最终产品价值的百分比来确定其是否达到实质性变化的标准。
澳大利亚规定:①产品的最后加工工序是在该受惠国内进行②本国成分的百分比不得小于产品出厂成本的50%,其公式为: 产品出口价格本国原料和劳务价值×100>50%美国规定:本国成分的价值不得低于产品出厂价值的35%,其公式是 产品出口价格不包括一般行政费用)本国原料和劳务价值(×100>35%5、从量税从量税是以商品的重量、数量、容量、长度和面积等计量单位为标准计征的关税。
计算机科学中的计算理论

计算机科学中的计算理论计算理论是计算机科学中的一门重要学科,它研究计算过程和计算机的本质,涉及到算法、自动机、形式语言、复杂性理论等方面。
计算理论的发展和研究影响了计算机科学的发展和应用,为计算机科学的发展提供了理论基础和支撑。
一、基本概念1. 计算:计算是指一切机器可以完成的信息处理活动,包括数据的采集、传输、存储和加工等过程。
计算理论关注的是这些信息处理活动本质上的规则和方法。
2. 计算机:计算机是一种能够按照预定程序自动进行算术或逻辑运算、处理和存储大量数据的电子设备。
计算机在现代社会中扮演着重要的角色,广泛应用于各个领域。
3. 算法:算法是指计算问题求解的一系列步骤或规则。
算法可以是手工的,也可以是自动计算机程序的形式,是计算理论中的重要基础。
4. 自动机:自动机是指在给定输入和状态下的状态转移系统。
自动机在计算机科学中有广泛的应用,包括正则表达式、文法、语言的描述和识别等方面。
5. 形式语言:形式语言是为了精确地描述计算问题而设计的一种语言。
形式语言通常是通过符号和规则来描述的,是计算机科学中的核心内容之一。
二、研究方向1. 算法算法是计算理论中的基础,研究算法可以帮助我们更好地理解计算过程和计算机的本质。
算法的分析和设计是算法研究的核心内容,其目的是寻找效率高、正确性保证的算法。
常见的算法有排序算法、查找算法、最短路径算法等。
2. 自动机自动机是计算机科学中的重要工具,它可以用来描述计算问题、语言和文法。
自动机理论是计算机科学、通信工程、电路设计等领域的基础知识,是计算机自动化技术的核心之一。
3. 形式语言形式语言是形式化描述计算问题的一种语言,包括正则语言、上下文无关语言、上下文敏感语言等。
形式语言理论是计算机科学中的重要分支,它在编译器设计、自然语言处理、计算机安全等领域有广泛的应用。
4. 复杂性理论复杂性理论是计算理论中的一个重要分支,主要研究计算问题的复杂度和可解性。
复杂性理论的目标是确定计算问题的最优算法,进而推导出计算问题的难度。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(二)、模块简介
Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许 用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目 前,Materials Studio软件包括如下功能模块: 1、Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以 操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并 提供软件的基本环境和分析工具以及支持Materials Studio的其他产 品。是Materials Studio产品系列的核心模块。 2、Discover: Materials Studio的分子力学计算引擎。使用多种分子力学和动力学方 法,以仔细推导的力场作为基础,可准确地计算出最低能量构型、分子 体系的结构和动力学轨迹等。 3、COMPASS: 支持对凝聚态材料进行原子水平模拟的功能强大的力场。是第一个由凝 聚态性质以及孤立分子的各种从头算和经验数据等参数化并经验证的从 头算力场。可以在很大的温度、压力范围内精确地预测孤立体系或凝聚 态体系中各种分子的结构、构象、振动以及热物理性质。
3、计算化学(computational chemistry):是理论化学的一个分 支。计算化学的主要目标是利用有效的数学近似以及电脑程序计算分 子的性质(例如总能量,偶极矩,四极矩,振动频率,反应活性等) 并用以解释一些具体的化学问题。 计算化学这个名词有时也用来表示计算机科学与化学的交叉学科。
二、MS软件 (一)概况
(三)、应用领域
Mateirals Studio 是 Acce lrys 公司专为材料科学领域开发的 新一代材料计算软件。它能方便地建立 3D 分子模型,深入分 析有机晶体、无机晶体、无定形材料以及聚合物,可以在催 化剂、聚合物、固体化学、结晶学、晶粉衍射以及材料特性 等材料科学研究领域进行性质预测、聚合物建模
6、DMol3: 独特的密度泛函(DFT)量子力学程序,是唯一的可 以模拟气相、溶液、表面及固体等过程及性质的商业 化量子力学程序,应用于化学、材料、化工、固体物 理等许多领域。可用于研究均相催化、多相催化、分 子反应、分子结构等,也可预测溶解度、蒸气压、配 分函数、熔解热、混合热等性质。 7、CASTEP: 先进的量子力学程序,广泛应用于陶瓷、半导体、金 属等多种材料,可研究:晶体材料的性质(半导体、 陶瓷、金属、分子筛等)、表面和表面重构的性质、 表面化学、电子结构(能带及态密度)、晶体的光学 性质、点缺陷性质(如空位、间隙或取代掺杂)、扩 展缺陷(晶粒间界、位错)、体系的三维电荷密度及 波函数等。
三、运用
采用 Materials Studio4.0 软件对ZnO的能带及电子能态密度进行 了模拟计算 1 模拟计算步骤 1.1 建立晶体 需要计算ZnO晶体参数通过相关文献查阅得到,如表1所示。 表1 相关晶胞参数表
晶体 ZnO 晶体种类 a b c 3D Hexagonal 3.24927 3.24927 5.20544
2.1.1 能带结构 由图1可以看出:能带结构图和电子能态密度图是相对应的。能带结构图 中曲线越密集,则电子能态密度图中相应能量位置峰值越大,表明电子 越多。反之,曲线越稀疏,则峰值越小,电子越少。本图中,电子在4.75~ -3.95 eV及-6.00 ~-4.95 eV范围密度最大。
图1 ZnO能带结构及电子能态密度
Materials Studio是专门为材料科学领域研究者开发的一款 可运行在PC上的模拟软件。它可以帮助你解决当今化学、 材料工业中的一系列重要问题。支持Windows 98、 2000、NT、Unix以及Linux等多种操作平台的Materials Studio使化学及材料科学的研究者们能更方便地建立三维 结构模型,并对各种晶体、无定型以及高分子材料的性质 及相关过程进行深入的研究。 多种先进算法的综合应用使Materials Studio成为一个强有 力的模拟工具。无论构型优化、性质预测以及复杂的动力 学模拟和量子力学计算,我们都可以通过一些简单易学的 操作来得到切实可靠的数据
高等无机
理论计算
化学11-1 马雪佳
1
几个重要概念
2
MS(Materials Studio)软件介绍
3
MS软件的运用
一、几个重要概念
1、理论计算:应用现有的定律、定理及规律对问题进行分析推理,找 出符合其尊循的规律公式(推导出来的公式)进行计算,其整个过程 叫理论计算 2、理论化学:是运用纯理论计算而非实验方法研究化学反应的本质 问题,主要以理论物理为研究工具(如热力学、量子力学、统计力 学、量子电动力学、非平衡态热力学等),并且大多辅以计算机模 拟。
Materials Studio生成的结构、图表及视频片断 等数据可以及时地与其它PC软件共享,方便与 其他同事交流,并能使你的讲演和报告更加引 人入胜。 Materials Studio软件能使任何研究者达到与世 界一流研究部门相一致的材料模拟的能力。模 拟的内容包括了催化剂、聚合物、固体及表 面、晶体与衍射、化学反应等材料和化学研究 领域的主要课题。
能带结构图和态密度图可以解决哪些问题
禁带宽度 5.976eV
Sio2能带结构图
Sio2的电子态密度图
偏态密度图
般常见晶体材料的结构,可以从MS自带的晶体结构 库中调出来,如半导体硅的晶体结构,还可以通过C ASTEP模块计算该晶体的能带结构等性质
硅的晶体结构和能带结构图
9
聚合物如聚丙烯,这里聚丙烯是采用10个单体聚合连接 构成,聚合物两端以氢原子结束。该体系可以通过DMol3 模块进行计算
聚丙烯的结构图
9
应用Visualizer模块构造体系结模块对苯分子 的电荷、分子轨道和能量等方面进行计算。
苯分子的电荷、轨道和结构图
7
1.2 设置计算任务 由于建立的晶体需要进行结构优化,对于优化的方式选有 GGA/PBE;GGA/RPBE;GGA/PW91;LDA/CA-PZ以上四种, 优化完成后,进行能量以及电子密度的模拟计算,最后得到电子 能态密度和能带结构等数据和相关图表。这些数据可以表明晶体 电子结构的一个快速定性图象,这些结论可以直接和实验光谱结 果相关联。 2结果与讨论
4、Amorphous Cell: 允许对复杂的无定型系统建立有代表性的模型,并对主要性 质进行预测。通过观察系统结构和性质之间的关系,可以对 分子的一些重要性质有更深入的了解,从而设计出更好的新 化合物和新配方。可以研究的性质有:内聚能密度 (CED)、状态方程行为、链堆砌以及局部链运动等。 5、Equilibria: 可计算烃类化合物单组分体系或多组分混合物的相图,溶解 度作为温度、压力和浓度的函数也可同时得到,还可计算单 组分体系的virial系数。适用领域包括石油及天然气加工过程 (如凝析气在高压下的性质)、石油炼制(重烃相在高压下 的性质)、气体处理、聚烯烃反应器(产物控制)、橡胶 (作为温度和浓度的函数的不同溶剂的溶解度)。
模拟的方法
1、量子力学的密度泛函理论; 2、半经验的量化计算方法; 3、分子力学; 4、分子动力学; 5、介观模拟方法等.
(四)、Ms 软 件 的 优 点
更容易地创建研究分子模型或 材料结构 操作灵活方便,并且能够最大 限度地运用网络资源。
1
优点
2
计算能带大小及分析能带组成
3Hale Waihona Puke 可以与其它标准PC软件共享 这些数据