常用药代动力学符注释
药代动力学参数汇总

稳态最低观测浓度
CL
清除率
CLss
静脉给药稳态清除率
CL/F
表观清除率
CLss/F
口服给药稳态清除率
CLR
肾清除率
DF
波动指数
fe
累积排泄分数
λz
消除速率常数
MIC
最低抑菌浓度
PD
药效学
PKS
药代动力学分析集
PKCS
药代动力学浓度分析集
PKPS
药代动力学参数分析集
Rac (AUC)、Rac (Cmax)
液相色谱-串联质谱
NCA
Non-compartmental analysis
非房室模型分析
t1/2
Elimination half life
半衰期,指血浆中药物浓度下降一半所需的时间。意义:①反映机体消除药物的能力与消除药物的快慢程度;②与药物转运和转化关系为,一次用药后经过5个t1/2后体内药物经消除仅剩下给药量的3.1%。同理,每隔一个t1/2用药一次,则给药5次后体内药物剩余量可达稳态浓度水平的96.9%;③按t1/2的长短常将药物分为5类:超短效为t1/2≤1 h,短效为1-4h,中效为4-8h,长效为8-24h,超长效为>24h;④肝、肾功能不良者,药物消除速率常数Ke值变小,药物的t1/2将相应延长,此时应依据患者肝、肾功能调整用药剂量或给药间隔。
Vd
Apparent volume of distribution
表观分布容积,指假设体内药物均匀分布时,由血药浓度推算得到的药物分布的体液容积,单位常用L·kg-1,也有用L。
λz
Elimination rate constant
末端消除速率常数,药时曲线末端进行对数转换后进行线性回归计算所得的斜率,如计算时包括的浓度点少于3个,或Rsq<0.800,λz被认为不可靠。进而t1/2,AUC0-inf,CL,CL/F,Vz,Vz/F,CLss,CLss/F,Vss,MRT也被认为不可靠。
药代动力学参数汇总
中文术语
AUC
浓度-时间曲线下面积
AUC0-t
从0时到最终可定量时间点的血药浓度-时间曲线下面积
AUC0-inf
从0到无穷大时间的血药浓度-时间曲线下面积
AUC0-tau
给药间隔内的血浆浓度-时间曲线下面积
Ae
累积排泄量
BLQ
低于定量下限
Cmax
峰浓度
Css,avg
稳态平均浓度
Css,max
稳态峰浓度
CL
Apparent clearance
表观清除率
CL
plasmaclearance
血浆清除率,是肝、肾和其他器官的药物清除率的总和,即单位时间内多少容积血浆的药物被清除干净,单位为:L/h。
Cmax
Maximum concentration
血药峰浓度
LC-MS/MS
Liquid chromatograpy tandem mass spectrometry
液相色谱-串联质谱
NCA
Non-compartmental analysis
非房室模型分析
t1/2
Elimination half life
半衰期,指血浆中药物浓度下降一半所需的时间。意义:①反映机体消除药物的能力与消除药物的快慢程度;②与药物转运和转化关系为,一次用药后经过5个t1/2后体内药物经消除仅剩下给药量的3.1%。同理,每隔一个t1/2用药一次,则给药5次后体内药物剩余量可达稳态浓度水平的96.9%;③按t1/2的长短常将药物分为5类:超短效为t1/2≤1 h,短效为1-4h,中效为4-8h,长效为8-24h,超长效为>24h;④肝、肾功能不良者,药物消除速率常数Ke值变小,药物的t1/2将相应延长,此时应依据患者肝、肾功能调整用药剂量或给药间隔。
常用药代动力学符号注释

常用药代动力学符号注释临床药物动力学符号注释A 体内药量(mg或μmol)Aa 在吸收部位有待于吸收的药量(mg或μmol)Ae 尿中累积原形药物排泄量(mg或μmol)Ael(m) 已消除的代谢物的量(mg或μmol)Aess 在稳态下,一个给药间隔期间尿中累积原形药物排泄量(mg或μmol)Ae∞单次给药后,至无穷大时间内尿中累积原形药物排泄量(mg 或μmol)A(m) 体内代谢物的量(mg或μmol)Amin 达到预期药物效应的所需最小药量(mg或μmol)AN,max;AN,min固定给药剂量和给药间隔,第N次给药后体内的最大和最小药量(mg或μmol)AN,t 在第N次给药后的t时间点的体内药量(mg或μmol)ARE 单次给药后,有待从尿中排泄的药量(mg或μmol)ASS 在恒速静脉滴注时,稳态时的体内药量(mg或μmol)ASS,AU 稳态时,一个给药间隔期间的平均体内药量(mg或μmol)ASS,max;Ass,min固定剂量,固定给药间隔,稳态时一个给药期间的最大和最小体内药量(mg或μmol)Ass,t 在固定剂量和给药间隔,稳态时一个给药剂量后t时间点的体内药量(mg或μmol)AUC 血浆药物浓度—时间曲线下面积,它代表一次用药后的吸收总量,反映药物的吸收程度。
(mg·h/L或μmol/L·h)AUCb 全血药物浓度—时间曲线下面积(mg·h/L或μmol/L·h)AUC(m) 血浆代谢物浓度—时间曲线下面积(mg·h/L或μmol/L·h)AUCSS 稳态时,一个给药间期的血浆药物浓度—时间曲线下面积(mg·h/L或μmol/L·h)AUMC 一阶矩—时间曲线下面积(mg·h2/L 或μmol/L·h2)C 在t时的血药浓度(mg/L或μmol/L)Ca 在吸收部位液体中的药物浓度(mg/L或μmol/L)CA 动脉血液中的药物浓度(mg/L或μmol/L)Cb 血液中的药物浓度(mg/L或μmol/h)Cbd 血浆中的结合药物浓度(mg/L或μmol/L)CD 离开透析器时透析液药物浓度(mg/L或μmol/L)Cinf 恒速滴注浓度(mg/L或μmol/L)CL 代谢抑制剂浓度(mg/L或μmol/L)CL 血浆药物总清除率(L/h)CLb 全血药物总清除率(L/h)CLbD 血液药物透析清除率(L/h)CLb,H 血液药物肝清除率(L/h)CLcr 肌酐肾清除率(L/h)CLD 血浆药物透析清除率(L/h)CLf 药物通过生成代谢物的清除率(L/h)CLH 血浆药物肝清除率(L/h)CL int 器官对药物的固有清除率(L/h)CL(m) 代谢物的总清除率(L/h)CLpD 血浆药物腹膜透析清除率(L/h)CLR 药物肾消除率(L/h)Clu 未结合药物消除率(L/h)C(m) 血浆代谢物浓度(mg/L或μmol/L)Cmax 一次给药后的最大血药浓度(mg/L或μmol/L)C(m)ss 持续恒速静脉滴注稳定时代谢物浓度(mg/L或μmol/L)Cmin 一次给药后的最小血药浓度(mg/L或μmol/L)C n在第n次给药间隔期间任何t时的血药浓度CN,max; CN,min 当固定给药剂量和给药间隔时,第N次给药后的最大和最小血浆药物浓度(mg/L或μmol/L)Cpc 腹腔中药物浓度(mg/L或μmol/L)Css 在零级过程滴注(即恒速静脉滴注)后达稳态时的血药浓度(mg/L或μmol/L)C0静脉注射后瞬时的血药浓度(C1)max 反复用药后第1次用药中最大血药浓度(C1)min 反复用药后第1次用药中最小血药浓度C∞反复用药达稳态时每一给药间隔期间内任一时间t时的血药浓度(C∞)max 稳态时每一给药间隔期间的最大血药浓度(C∞)min 稳态时每一给药间隔期间的最小血药浓度Css,av 固定给药剂给药间隔,当达到稳态时,任一给药间期的平均血浆药物浓度(mg/L或μmol/L)Css,max; Css,min 固定给药剂量及给药间隔,当到达稳态时,最大和最小血浆药物浓度(mg/L或μmol/L)CT 血浆外体液平均药物浓度(mg/L或μmol/L)CTW 总体液中未结合药物浓度(mg/L或μmol/L)Cupper;Clower血浆药物浓度的上、下限(mg/L或μmol/L)Cu 血液中未结合药物浓度(mg/L或μmol/L)Cu1血浆中未结合抑制剂浓度(mg/L或μmol/L)Cv 静脉血液中药物浓度(mg/L或μmol/L)D2 负荷剂量(mg或μmol/L)DM 固定剂量方案的维持剂量(mg或μmol/L)DM,max 最大维持剂量,以确保血浆药物浓度在稳态时于一个给药间隔内介于药物血浆浓度的上、下限之间(mg或μmol/L)E 抽提比(无单位)EC50 产生1/2最大效应的浓度(mg/L或μmol/L)EH 肝抽提比(无单位)Emax 最大效应(使用效应测量单位)fp 血浆中未结合药物的分数fs 唾液中未结合药物的分数F 药物生物利用度(无单位)Fbd 血浆中结合药物与总药物浓度比(无单位)fO 透析治疗期间,透析清除率占总清除率的分数(无单位)fe 原形药物在尿中的排泄分数(无单位)FEV1 一秒钟的强制呼吸量(L)FH 进入肝脏的药物逃过首过消除的分数(无单位)fm 进入体循环的药物转变为代谢物的分数(无单位)Fm 以代谢物形式进入体循环的药物占药物剂量的比例分数(无单位)FR 在肾小管重吸收的药物分数(无单位)fu 血浆未结合药物与总药物浓度的比例分数(无单位)fu 当结合力改变时,血浆未结合药物与总药物浓度的比例分数(无单位)fub 血浆未结合药物与血液中总药物浓度的比例分数(无单位)fup 血浆蛋白中未结合位点与总结合位点的比例分数(无单位)fuR 细胞内液中未结合药物与总药物浓度的比例分数(无单位)fuT 血浆外组织中未结合药物与总药物浓度的比例分数(无单位)γ浓度—效应关系曲线的形状因子(无单位)GFR 肾小球滤过率(mL/min或L/h)K 在一定模型的表观一级消除速率常数(h-1)ka 吸收速率常数(h-1)KA 药物与蛋白的结合常数(L/mol)kD 透析治疗中,患者的消除速率常数(h-1)Ke 尿排泄速率常数,也指表观一级吸收速率常数(h-1)K0在一室模型表现一级肾脏排泄速率常数,也指零级输入或滴注速率常数(h-1)K10从中央室消除的表观一级速率常数kf 代谢物形成速率常数(h-1)K1抑制平衡常数(mg/L或μmol)Km 在一室模型代谢转化的表观一级速率常数(即代谢物的消除速率常数),也有代表Michaelis常数(h-1)Ki 在肾衰竭时的处置速率常数(h-1)Kp 药物在血液或血浆与组织之间的平衡分布比例系数(无单位)RT 药物离开组织的速率分数(h-1)kt 转换速率分数(h-1)m 效应强度-对数浓度曲线中段的斜率(使用效应测量单位)MRT 一个分子在体内的平均驻留时间(h)Mu 到t时尿中排泄的代谢物量n 多次用药所给剂量的次数P 通透系数(cm/min或cm/h)Q 总体清除率(L/min或L/h)Q0肾清除率Qc 肌酐肾清除率QD 血液透析系统中的透析液流量(L/min或L/h)Qf 血液滤过系统中滤过液的流速(L/min或L/h)QH 肝血流量(门静脉加肝动脉)(L/min或L/h)ρ血细胞内药物与血浆未结合药物的浓度比(无单位)Rac 累积指数(无单位)Rd 某特殊病人未结合清除率与普通病人未结合清除率的比例(无单位)RF 某特殊病人的肾功能与普通病人肾功能的比例分数(无单位)Ro 恒速静脉滴注速率(mg/h)Rt 转换率(mg/h)S 纯度因子(无单位)SA 表面积(m2)τ给药间隔(h)τ1肾衰竭时的给药间隔Tmax 维持血浆浓度于Cupper和Clower之间的最大给药间隔时间(h)Tmax 血管外给药时,达到最大血药浓度时的时间(h)t d作用持续时间(h)t p一次给药后最大血药浓度出现的时间t'p反复用药达稳态时,在每一给药间隔期间最大血药浓度出现的时间Tinf 恒速滴注时间(h)Tm 药物转运(分泌)进入肾小管的最大速率(mg/h)t1/2,t0.5半衰期(h)t1/2α药物分布半衰期(h)t1/2β药物消除半衰期(h)T1/2Ka 药物吸收半衰期(h)Tt 转换时间(h)V 建立在血浆浓度基础上的表观分布容积,即在一室模型药物的表观分布容积(L)VB 血液容量(L)Vc 中央室的分布表观容积(L)Vd 在多室模型药物的表观分布容积,系指体内药量和后分布相血药浓度之间的一个比例常数(L)V1 起始稀释房室容积(L)Vm 用动力学Michaelis-Menten所描述过程的理论上的最大速率(mg/h或μmol/L)Vp 血浆容量(L)VPC 腹腔中透析液容量(L)VR 细胞内液中的水容量(L)VT 血浆外药物分布的生理容量(L)Vss 建立在血浆药物浓度基础上的稳态表观分布容积(L)VTW 血浆外药物分布的水容量(L)Vu 建立在血浆未结合药物浓度基础上的表观分布容积X 在t时体内的药量,稳态时体内的“平均”药量(mg或μmol)X A在t时吸收到体循环中的药量(mg或μmol)X a吸收部位的药量(mg或μmol)X c在t时中央室的药量(mg或μm ol)X n反复用药在第n个给药间隔期间任何时间t时的体内药量(mg 或μmol)X t在周边室的药量(mg或μmol)X u尿中排泄的原形药物累积药量(mg或μmol)X0负荷剂量或初剂量(mg或μmol)α在二室模型描述药物在体内分布的表观一级混合速率常数β在二室模型描述药物消除的表观一级混合速率常数。
药代动力学kp-概述说明以及解释

药代动力学kp-概述说明以及解释1.引言1.1 概述概述部分的内容:药代动力学(Pharmacokinetics,简称PK)是研究药物在机体内的吸收、分布、代谢和排泄过程的科学。
药代动力学研究对于评价药物的有效性和安全性至关重要,它可以帮助人们理解药物在体内的行为规律,为临床应用提供科学依据。
药代动力学研究的主要内容包括药物的吸收过程、分布过程、代谢过程和排泄过程。
药物的吸收过程研究药物从给药部位进入血液循环的过程,包括口服、注射、经皮等途径。
分布过程研究药物在体内的分布情况,包括药物在血液中的浓度分布以及药物与组织器官之间的互作。
代谢过程研究身体如何将药物分解和转化成代谢产物,通常由肝脏的酶系统参与。
排泄过程研究通过尿液、粪便、呼吸以及乳汁等途径,将药物及其代谢产物从机体内排出来。
药代动力学参数对于评价药物在体内的行为很重要,常用的参数有药物的生物利用度、血药浓度峰值、半衰期等。
这些参数可以帮助我们判断药物的疗效、剂量以及用药频率,从而更好地指导临床用药。
本文将就药代动力学的基本概念、研究方法以及应用领域进行详细阐述,旨在帮助读者更全面地了解药代动力学的重要性和价值,进而在临床实践中更科学地应用药代动力学知识。
1.2 文章结构本文将按照以下结构进行论述和分析:1. 引言:在文章引言部分,我们首先会概述药代动力学(Pharmacokinetics,简称PK)的基本概念和研究对象,介绍其在药物研发和合理用药中的重要性和应用价值。
同时,我们会明确写作的目的和意义,以及本文的主要内容安排。
2. 正文:正文部分是文章的核心部分,包括以下几个方面的内容:2.1 药代动力学的基础知识:在这一部分,我们将介绍药代动力学的基本原理和基础概念,如吸收、分布、代谢和排泄等过程。
同时,我们会阐述这些过程在药物治疗中的意义,以及药代动力学参数的测定方法和评价标准。
2.2 药代动力学的应用:在这一部分,我们将详细介绍药代动力学在临床药物治疗中的应用。
药代动力学参数汇总
稳态最低观测浓度
CL
清除率
CLss
静脉给药稳态清除率
CL/F
表观清除率
CLss/F
口服给药稳态清除率
CLR
肾清除率
DF
波动指数
fe
累积排泄分数
λz
消除速率常数
MIC
最低抑菌浓度
PD
药效学
PKS
药代动力学分析集
PKCS
药代动力学浓度分析集
PKPS
药代动力学参数分析集
Rac (AUC)、Rac(Cmax)蓄Biblioteka 比t1/2消除半衰期
Tmax
达峰时间
Tss,max
稳态达峰时间
Vz
分布容积
Vss
静脉输注稳态分布容积
Vz/F
表观分布容积
缩略语
英文全称
中文全称
%AUCex
Percentage ofthe area undertheconcentration-timecurvethathasbeenderived afterextrapolation
Rac(AUC)
AUC0-tau药物蓄积比,用AUC0—tau末剂/AUC0-24首剂计算
Rac(Cmax)
Cmax药物蓄积比,用Cssmax末剂/Cmax首剂计算
DF
波动指数,计算公式为100*(Css,max-Css,min)/Cavg
PD
Pharmacodynamics
药物效应动力学,就是研究药物对机体得作用及其规律,阐明药物防治疾病得机制,如,药物与器官得靶分子或靶细胞得相互作用等。
Tmax
Time toreach maximumconcentration
药代动力学参数

一、吸收溶出度:药物分子在消化道中溶解的程度生物利用度:药物吸收的程度绝对生物利用度最大血药浓度(Cmax)达峰时间(Tmax)二、分布由于体内环境的非均一性(血液、组织),导致药物浓度变化的速度不同。
隔室(compartment):同一隔室药物浓度的变化速度相同,均相。
一室模型:药物进入血液迅速分布全身,并不断被清除。
二室模型: 药物进入体内后,首先快速分布于组织中,然后进入较慢的消除过程。
表观分布体积(Vd)(aparent volume of distribution):表征药物在体内被组织摄取的能力。
表观容积大的药物体内存留时间较长。
药物浓度-时间曲线下面积(AUC);系统药物暴露(Systemic Exposure)血脑屏障;蛋白结合率;分布半衰期(t 1/2(α)三、消除消除(elimination):原药在体内消失的过程。
包括肾(尿)或胆汁(粪)或呼吸排泄及代谢转化的总和。
消除速率常数(elimination constants):反映药物在体内消失的快慢。
不完全反映药物的作用时间(代谢物也有活性)。
半寿期或半衰期(t1/2):药物浓度或药量降低50%所需的时间。
消除半衰期t1/2(β))Terminal Half-life ,Elimination Half-life。
清除率(clearance,廓清率)或肾清除率(renal clearance):反映药物或代谢物经肾被排出体外的速度。
一方面是药物对机体的作用,产生药效、毒性或副作用,表现为药物的药理作用或毒理作用,决定于特定的化学结构,具有较强的结构特异性。
另一方面是机体对药物的作用:吸收、分布,生物转化和排泄,表现为药物的药代动力学性质。
主要取决于药物的溶解性、脂水分配系数、电荷等药物分子整体的理化性质,结构特异性不强。
药物的吸收是药物由给药部位通过生物膜进入血液循环的过程。
吸收部位消化道(口服给药,口腔、胃、小肠、大肠)、呼吸道(鼻腔给药,肺)、肌肉()、粘膜(栓剂)。
常用药代动力学符号注释

中央室的分布表观容积(L)
Vd
在多室模型药物的表观分布容积,系指体内药量和后分布相血药浓度之间的一个比例常数(L)
V1
起始稀释房室容积(L)
Vm
用动力学Michaelis-Menten所描述过程的理论上的最大速率(mg/h或μmol/L)
Vp
血浆容量(L)
VPC
腹腔中透析液容量(L)
VR
细胞内液中的水容量(L)
Rt
转换率(mg/h)
S
纯度因子(无单位)
SA
表面积(m2)
τ
给药间隔(h)
τ1
肾衰竭时的给药间隔
Tmax
维持血浆浓度于Cupper和Clower之间的最大给药间隔时间(h)
Tmax
血管外给药时,达到最大血药浓度时的时间(h)
td
作用持续时间(h)
tp
一次给药后最大血药浓度出现的时间
t'p
反复用药达稳态时,在每一给药间隔期间最大血药浓度出现的时间
Emax
最大效应(使用效应测量单位)
fp
血浆中未结合药物的分数
fs
唾液中未结合药物的分数
F
药物生物利用度(无单位)
Fbd
血浆中结合药物与总药物浓度比(无单位)
fO
透析治疗期间,透析清除率占总清除率的分数(无单位)
fe
原形药物在尿中的排泄分数(无单位)
FEV1
一秒钟的强制呼吸量(L)
FH
进入肝脏的药物逃过首过消除的分数(无单位)
ASS
在恒速静脉滴注时,稳态时的体内药量(mg或μmol)
ASS,AU
稳态时,一个给药间隔期间的平均体内药量(mg或μmol)
药代主要参数
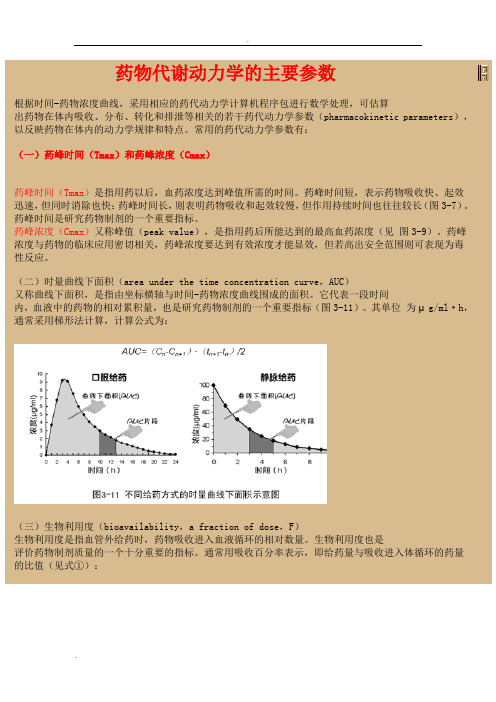
药物代谢动力学的主要参数根据时间-药物浓度曲线,采用相应的药代动力学计算机程序包进行数学处理,可估算出药物在体内吸收、分布、转化和排泄等相关的若干药代动力学参数(pharmacokinetic parameters),以反映药物在体内的动力学规律和特点。
常用的药代动力学参数有:(一)药峰时间(Tmax)和药峰浓度(Cmax)药峰时间(Tmax)是指用药以后,血药浓度达到峰值所需的时间。
药峰时间短,表示药物吸收快、起效迅速,但同时消除也快;药峰时间长,则表明药物吸收和起效较慢,但作用持续时间也往往较长(图3-7)。
药峰时间是研究药物制剂的一个重要指标。
药峰浓度(Cmax)又称峰值(peak value),是指用药后所能达到的最高血药浓度(见图3-9)。
药峰浓度与药物的临床应用密切相关,药峰浓度要达到有效浓度才能显效,但若高出安全范围则可表现为毒性反应。
(二)时量曲线下面积(area under the time concentration curve,AUC)又称曲线下面积,是指由坐标横轴与时间-药物浓度曲线围成的面积。
它代表一段时间内,血液中的药物的相对累积量,也是研究药物制剂的一个重要指标(图3-11)。
其单位为μg/ml·h,通常采用梯形法计算,计算公式为:(三)生物利用度(bioavailability,a fraction of dose,F)生物利用度是指血管外给药时,药物吸收进入血液循环的相对数量。
生物利用度也是评价药物制剂质量的一个十分重要的指标。
通常用吸收百分率表示,即给药量与吸收进入体循环的药量的比值(见式①):相对F 是评价厂家产品质量的重要标准之一。
如果制剂质量不合格,生物利用度低,临床疗效肯定差。
一般药典上都规定药厂生产的制剂,生物利用度的差距不应超过±10%。
生物利用度的意义在于:① 从制剂方面而言,剂量和剂型相同的药物,如果厂家的制剂工艺不同,甚至同一药厂生产的同一制剂的药物,仅因批号不同,都可以使药物的晶型、颗粒大小或其他物理特性,以及药物的生产质量控制等发生改变,从而影响药物的崩解和溶解度,使药物的生物利用度发生明显的改变,导致时间-药物浓度曲线的改变(见图3-12)。
药代动力学期末复习-本书数学符号注释.doc

—、本书数学符号注释(Glossary)u 药物体内分布的表观一级速度常数(Apparent first-order distribution rate constant fora drug that confers upon the body the characteristics of a multicompartent model)3 多房室模型描述药物消除的表观一级速度常数(Apparent first-order elimination rateconstant for a drug that confers upon the body the characteristics of amulticompartent model, obtained from the terminal slope of a semilogarithmicplot of drug concentration in the plasma versus time)T给药间隔时间(Dosing interval)A吸收表面积(The area of absorption)AUC 血药浓度■时间曲线下面积(The area under the drug concentration in the plasma versus time curve)C时间r 的血药浓度(Drug concentration in the plasma at time t)dC/dr 血药浓度的变化率(Rate of change of drug concentration in the plasma)J;Cd/ 血药浓度■时间曲线下总面积(Total area under the drug concentration in the plasma versus time curve)f^Cdr 血药浓度•时间曲线下时间从零到/的面积(Area under the drug concentration in the plasma versus time curve from time zero to T)C(l y稳态时“平均”血药浓度(Average drug concentration in the plasma at steady state upon multiple dosing)C max一次给药后'峰血药浓度(Maximum drug concentration in the plasmafollowing administration of a single dose)C n经n次给药,间隔期间任一时间/的血药浓度(drug concentration in the plasma at anytime t during the n-th dosing interval)Css零级过程输液后稳态时的血药浓度(drug concentration in the plasma at steady state following zero-order infusion)Co 静注后瞬间的药物浓度(drug concentration in the plasma immediately following intravenous injection)(Cj)max 多次给药首次的最高血药浓度(Maximum drug concentration in the plasmaafter the first of a series of repetitive doses)(Ci)m in 多次给药首次的最低血药浓度(Minimum drug concentration in the plasma after the first of a series of repetitive doses)J 多次给药达稳态时某一给药间隔的任一时刻的血药浓度(Drug concentration in the plasma at any time t during a dosing interval at steady state upon multiple dosing) JJC^dr稳态时某一给药间隔期内血药浓度■时间曲线下面积(Area under the drug concentration versus time curve during a complete dosing interval at steadystate upon multiple dosing)(C TO)max稳态时某一给药间隔期间最高血药浓度(Maximum concentration of drug inthe plasma during a dosing interval at steady state upon multiple dosing) (CJmin稳态时某—给药间隔期间最小血药浓度(Minimum concentration of drug in the plasma during a dosing interval at steady state upon multiple dosing) Cl 总清除率(Total clearance)Cl,-肾清除率(Renal clearance)C/G-肌酉干清除率(Creatinine clearance)F吸收率的通称,即剂量吸收的分数值(This is a rather general term used to define the extent of absorption, i.e., the fraction of the dose available)Fb体内牛物转化量占有效总量分数(Fraction of the drug biotransformed in the body to the drug actually absorbed)fss多次给药期间的血药浓度为稳态时血药浓度的分数值(Fraction of the steady-state plasma concentration after any given dose during multiple dosing)k房室模型中表观一级消除速率常数(Apparent first-order elimination rate constant of a drug that confers upon the body the characteristics of a one compartment model)k a表观一级吸收速率常数(Apparent first-order absorption rate constant)k e房室模型中表观一级肾排泄速率常数(Apparent first-order renal excretion rate constant of a drug that confers upon the body the characteristics of a one-compartment model)k h房室模型中代谢物形成的表观一级速率常数,即生物转化速度常数(Apparent first-order rate constant for a drug biotransformed that confers upon the body the characteristics of a one-compartment model)k hl房室模型中药物在肺内消除的表观一级速率常数(Apparent first-order lung excretion rate constant of a drug that confers upon the body the characteristics of a one-compartment model)kij房室间转运的表观一级速率常数式中=1, 2・・・,戶1, 2…,/(Apparent first-order intercompartmental transfer rate constants, where i= \, 2・・・,j=\, 2・・・, i工j)K,n米氏常数(Michaelis constant)k r肾衰病人处置速率常数(Apparent first-order elimination rate constant of a drug that confers upon the body the characteristics of a one compartment model in the patients under renal failure)ko零级输入或输注速率常数(Zero-order input or infusion rate constant)£i()从中央室消除的表观一级消除速率常数(Apparent first-order elimination rate constant from the central compartment)k\2' ^21 参阅褊•项内LBW 瘦体重(Low body weight)Tmax —次给药后达到最高血药浓度的时间,或称达峰时间(Time at which a maximum concentration of drug in the plasma occurs following a single dose)T;ax多次给药后达稳态时某一给药间隔内最高血药浓度,或称稳态达峰时间(Time at which a maximum concentration of drug in the plasma occurs during a dosing interval at steady state upon multiple dosing)T I/2或几.5 半衰期(Biologic half-life of a drug)V一房室模型的药物表观分布容积(Apparent volume of distribution of a drug that confers upon the body the characteristics of a one-compartment model)V c中央室的表观分布容积(Apparent volume of the central compartment)V tn米曼氏动力学过程中的最大速率的理论值(Theoretical maximum rate of process describable by Michaelis-Menten kinetics)X时间t的体内药量(Amount of drug in the body at time t)dX/dt体内药量的变化率(Rate of change of drug level in the body)X稳态时体内“平均”药量(“Average" amount of drug in the body at steady state) X A经时间t被吸收进入体循环内药M(Amount of drug absorbed into the systemic circulation at time t)X;时间00大时吸收进入体循环的药量(Amount of drug ultimately absorbed)X a吸收部位的药量(Amount of drug at absorption site)dX a/dt吸收速率(Absorption rate)X c时间 /时屮央室内药量(Amount of drug in the central compartment at time t)X E经吋间t通过各种途径消除的累积药量(Cumulative amount of drug eliminated by all routes to time t)d Xf/ck 药物从体内的消除速率(Rate of drug elimination from the body)X fl多次给药在第斤次给药间隔,在任一时刻f的体内药量(Amount of drug in the body at any time t during the n-th dosing interval of a series of repetitive doses)X ss静脉输注达稳态时的体内药量(Amount of drug in the plasma at steady state following zero-order infusion)X u排泄在尿中原形药物累积药量(Cumulative amount of unchanged drug excreted in the urine to time t)d XJAt原形药物肾排泄速率(Rate of renal excretion of unchanged drug)AX“ / Ar 一段时间内原形药物肾排泄的平均速率(Average rate of renal excretion of unchanged drug over a finite period of time)X:原形药物随尿排尽的累积总量(Cumulative amount of unchanged drug ultimately excreted in the urine)X()给药剂量(Administered dose)X()'负荷剂量(Loading dose)。
药代动力学参数及其意义

药代动力学参数及其意义药代动力学参数是描述药物在生物体内的传递、代谢和排泄过程的重要参数,他们是用来衡量药物作用效果的有效工具,可以帮助医生准确地把握病人药物治疗方案。
药代动力学参数主要有六种:入髓率(F),半衰期(T1/2),清除率(CL),平衡分布容积(Vd),多次剂量公式K,最大血药浓度(Cmax)。
入髓率(F):药物经过血脑屏障进入脑内的比例。
入髓率是衡量药物是否可以通过血脑屏障进入脑部的重要参数,它的值越大,表明药物经过血脑屏障进入脑内的可能性越大。
高入髓率的药物往往更容易有较好的疗效。
半衰期(T1/2):衡量药物真正从生物体中消失的时间长短,半衰期越短,药物在体内的滞留时间越短,就更容易实现“短效能”。
清除率(CL):衡量一天内药物从体内通过排泄所被清除的速度。
清除率与药物的性质直接关联,它影响药物在人体内的滞留时间,清除率越快,滞留时间越短,则药物血药量也越低。
平衡分布容积(Vd):是指一种药物在组织与细胞内的平衡态的浓度,它反映的是一种药物的可及性,如果一种药物的Vd值高,说明它的可及性较好,该药物更容易进入细胞内有效,能够发挥药效,达到治疗效果。
多次剂量公式K:是描述药物在某一时间和某一体重的平衡浓度和血药浓度之间的关系的参数。
它代表了一种药物在不断服药的情况下,血药浓度的稳定性,多次剂量公式K的值越小,表示每次新药物的剂量越大,总的血药浓度就越稳定。
最大血药浓度(Cmax):血药浓度的最大值,反映的是药物在服用后在肝脏的代谢水平,服药前后两次血药浓度最大值的差异,可以代表药物的滞留时间,Cmax是药物产生药理作用的标志,影响疗效。
以上这些药代动力学参数,共同反映了药物在人体内的传递、代谢和排泄的过程,是医生衡量药物作用效果的重要参考素。
使用这些参数,可以有效地把握病人的治疗方案,从而提高药物治疗效果,改善病人的治疗结果。
药代动力学(干货分享)

五、排泄 excretion
药物以原形或代谢产物形式通过排泄或分泌器官排出体外的过程,是体内药物消除的重要 组成部分。
排泄途径
尿液
胆汁
肠道
肺脏
汗腺
乳汁
36
(一)、肾排泄 1、肾小球滤过:以膜孔扩散方式 ① 绝大部分药物经肾脏排出体外 ②只有非与血浆蛋白结合的药物可被肾小球滤过
肾小球滤过率降低可使滤过药量减少。
跨膜转运可分为被动转运(passive transport)和载体转运(carriermediated transport)
n 被动转运 (passive transport)
➢特点:
顺膜两侧浓度差转运 高
低
不消耗能量
不需载体,无饱和性
各药间无竞争性抑制现象
5
被动转运包括: 滤过(filtration) 简单扩散(simple diffusion)
2、肾小管分泌:近曲肾小管 主动转运:酸性药物载体、碱性药物载体; 同类药物之间有竞争性,如丙磺舒,影响青霉素和头孢 菌素的作用强度及时间。
3、肾小管的重吸收: 主动重吸收:近曲肾小管;被动重吸收:远曲肾小管。 脂溶性药物在排泄过程中可被肾小管再吸收(与药物本 身的pKa、血和尿的pH有关)
37
n 尿液pH值对药物排泄的影响:弱酸性药物在碱性尿液中解离多,重吸收少,排泄快 (巴比妥类中毒时用碳酸氢钠解救) ,而在酸性尿液中解离少,重吸收多,排泄慢。 弱碱性药物(氨茶碱、哌替啶、阿托品)则相反。
11
➢ 在膜两侧处于不同pH状态时,弱酸性药物被动运转达平衡时,膜两侧浓度比 的计算方法
p pH H 胃 血侧 侧药 药物 物 1 1浓 浓 1 10 0p p度 度 H H 血 胃 --p pK Kaa
药代动力学参数总览

药代动力学参数总览简介药代动力学是研究药物在体内的吸收、分布、代谢和排泄过程的科学。
药代动力学参数是评估药物在体内行为的定量指标。
本文档将为您提供药代动力学参数的总览,帮助您了解药物的药代动力学特性。
主要的药代动力学参数1. 生物利用度(availability)生物利用度是指药物经口给药后在体内被吸收的程度,通常以口服给药后的AUC(曲线下面积)或F(生物利用度百分比)来表达。
2. 峰浓度(Peak n)峰浓度表示药物在给药后(通常为口服给药)达到的最高血浆浓度,以Cmax来衡量。
峰浓度直接影响药物的疗效和副作用。
3. 血浆半衰期(Plasma Half-life)血浆半衰期是指药物在血浆中浓度下降一半所需的时间,反映了药物在体内的清除速率。
血浆半衰期长短影响药物的给药频率和稳态浓度的达到时间。
4. 药物分布容积(Volume of n)药物分布容积描述药物在体内分布的范围,是药物分布到组织和器官的能力。
分布容积大表示药物更易进入组织,通常与药物的脂溶性相关。
5. 清除率(Clearance)清除率是指单位时间内从体内清除药物的量,反映了药物的消除速率。
清除率越大,药物从体内被排除的速度越快,通常与肝脏和肾脏的功能相关。
6. 生物转化率(n Rate)生物转化率是指药物在体内经过代谢转化的比例,通常以药物代谢后形成的代谢产物与未代谢药物的比值来表示。
结论药代动力学参数是评估药物在体内行为的重要指标,能够帮助我们了解药物的吸收、分布、代谢和排泄特性。
透过药代动力学参数的分析,可以优化药物的给药方案,提高药物疗效,减少副作用。
希望本文档的内容能够帮助您更好地理解药代动力学参数的意义和应用。
药代动力学参数的含义和计算

举例说明:
Time (h)
1 2 4 6 12 18 24
Conc (µg/L)
39.0 22.0 10.3 7.29 4.86 3.50 2.52
回归后各点β α 值 (µg/L) 值(µg/L) (实测 - β)
第一步:作C-T图
100
作图后发现,它
50
Conc (µg/L)
符合二室模型分
30
布。很明显,最
后三个点明显属
于消除相,对者3
10
个点进行回归分
5
析,并外推
3
0 4 8 12 16 20 24 1
100
50
30 B = 9.3 µg/L
10
得出直线回归方程,直 线和Y轴交点即B,直线 斜率为-β,T1/2β= ln2/β.所以获得全部消 除相参数。?
Conc (µg/L)
5
3
t ½β = 12.5h 10 4 8 12 16 20 24
• 计算:K or Ke=0.693/t1/2
表现分布容积 Vd
• 假设药物在体内各组织和体液中均匀分布 时,药物分布所需要的空间,常以L/kg表 示
• 表观分布容积仅是一理论容积 ,计算出欲 达某一血药浓度C所需使用的剂量X=CVd ,还可用于评估药物在体内的分布特点
• 计X0算/C:0而t=计0算时出的C0值及注射剂量X0,按V=
Conc (mg/L) Ln Conct = C0.e-Kt
2
00
2
4
1.0 LnCt = LnC0 - Kt
Ct
Ct
0.0
6 8 100 2 4 6
二室模型 D
V1
k12 k21
8个常用药代动力学参数
CL
plasmaclearance
血浆清除率,是肝、肾和其他器官的药物清除率的总和,即单位时间内多少容积血浆的药物n half life
半衰期,指血浆中药物浓度下降一半所需的时间。意义:①反映机体消除药物的能力与消除药物的快慢程度;②与药物转运和转化关系为,一次用药后经过5个t1/2后体内药物经消除仅剩下给药量的3.1%。同理,每隔一个t1/2用药一次,则给药5次后体内药物剩余量可达稳态浓度水平的96.9%;③按t1/2的长短常将药物分为5类:超短效为t1/2≤1 h,短效为1-4h,中效为4-8h,长效为8-24h,超长效为>24h;④肝、肾功能不良者,药物消除速率常数Ke值变小,药物的t1/2将相应延长,此时应依据患者肝、肾功能调整用药剂量或给药间隔。
缩略语
英文全称
中文全称
AUC
Area under the concentration-time curve
血药浓度-时间曲线下面积,给药后随着时间的改变血药浓度随之不断变化,以时间为横坐标,药物浓度(或对数浓度,logarithm concentration, logc)为纵坐标绘制的图形,称为药物浓度-时间曲线,也称时量曲线,由时量曲线与坐标横轴围成的面积称为曲线下面积|(area under the curve, AUC)与药物吸收的总量成正比。
F
bioavailability
生物利用度,指药物经血管外给药后能被吸收进入体循环的百分数。因为时量曲线下面积(AUC)与药物吸收的总量成正比,因此用血管外给药的AUC与静脉给药的AUC的比值的百分数表示绝对生物利用度。而相对生物利用度以相同给药途径比较测试药物与标准药物比值的百分数,可用于评价不同厂家同一种制剂或同一厂家的不同批号药品间的吸收率。
药代主要参数
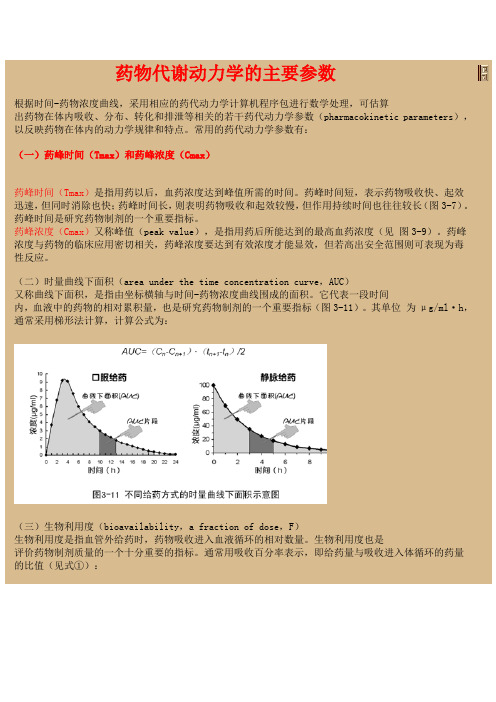
药物代谢动力学的主要参数根据时间-药物浓度曲线,采用相应的药代动力学计算机程序包进行数学处理,可估算出药物在体内吸收、分布、转化和排泄等相关的若干药代动力学参数(pharmacokinetic parameters),以反映药物在体内的动力学规律和特点。
常用的药代动力学参数有:(一)药峰时间(Tmax)和药峰浓度(Cmax)药峰时间(Tmax)是指用药以后,血药浓度达到峰值所需的时间。
药峰时间短,表示药物吸收快、起效迅速,但同时消除也快;药峰时间长,则表明药物吸收和起效较慢,但作用持续时间也往往较长(图3-7)。
药峰时间是研究药物制剂的一个重要指标。
药峰浓度(Cmax)又称峰值(peak value),是指用药后所能达到的最高血药浓度(见图3-9)。
药峰浓度与药物的临床应用密切相关,药峰浓度要达到有效浓度才能显效,但若高出安全范围则可表现为毒性反应。
(二)时量曲线下面积(area under the time concentration curve,AUC)又称曲线下面积,是指由坐标横轴与时间-药物浓度曲线围成的面积。
它代表一段时间内,血液中的药物的相对累积量,也是研究药物制剂的一个重要指标(图3-11)。
其单位为μg/ml·h,通常采用梯形法计算,计算公式为:(三)生物利用度(bioavailability,a fraction of dose,F)生物利用度是指血管外给药时,药物吸收进入血液循环的相对数量。
生物利用度也是评价药物制剂质量的一个十分重要的指标。
通常用吸收百分率表示,即给药量与吸收进入体循环的药量的比值(见式①):相对F 是评价厂家产品质量的重要标准之一。
如果制剂质量不合格,生物利用度低,临床疗效肯定差。
一般药典上都规定药厂生产的制剂,生物利用度的差距不应超过±10%。
生物利用度的意义在于:① 从制剂方面而言,剂量和剂型相同的药物,如果厂家的制剂工艺不同,甚至同一药厂生产的同一制剂的药物,仅因批号不同,都可以使药物的晶型、颗粒大小或其他物理特性,以及药物的生产质量控制等发生改变,从而影响药物的崩解和溶解度,使药物的生物利用度发生明显的改变,导致时间-药物浓度曲线的改变(见图3-12)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
血浆药物透析清除率(L/h)
CLf
药物通过生成代谢物的清除率(L/h)
CLH
血浆药物肝清除率(L/h)
CL int
器官对药物的固有清除率(L/h)
CL(m)
代谢物的总清除率(L/h)
CLpD
血浆药物腹膜透析清除率(L/h)
CLR
药物肾消除率(L/h)
Clu
未结合药物消除率(L/h)
C(m)
血浆代谢物浓度(mg/L或μmol/L)
血液滤过系统中滤过液的流速(L/min或L/h)
QH
肝血流量(门静脉加肝动脉)(L/min或L/h)
ρ
血细胞内药物与血浆未结合药物的浓度比(无单位)
Rac
累积指数(无单位)
Rd
某特殊病人未结合清除率与普通病人未结合清除率的比例(无单位)
RF
某特殊病人的肾功能与普通病人肾功能的比例分数(无单位)
Ro
恒速静脉滴注速率(mg/h)
Rt
转换率(mg/h)
S
纯度因子(无单位)
SA
表面积(m2)
τ
给药间隔(h)
τ1
肾衰竭时的给药间隔
Tmax
维持血浆浓度于Cupper和Clower之间的最大给药间隔时间(h)
Tmax
血管外给药时,达到最大血药浓度时的时间(h)
td
作用持续时间(h)
tp
一次给药后最大血药浓度出现的时间
t'p
反复用药达稳态时,在每一给药间隔期间最大血药浓度出现的时间
AUMC
一阶矩—时间曲线下面积(mg·h2/L或μmol/L·h2)
C
在t时的血药浓度(mg/L或μmol/L)
Ca
在吸收部位液体中的药物浓度(mg/L或μmol/L)
CA
动脉血液中的药物浓度(mg/L或μmol/L)
Cb
血液中的药物浓度(mg/L或μmol/h)
Cbd
血浆中的结合药物浓度(mg/L或μmol/L)
Cmax
一次给药后的最大血药浓度(mg/L或μmol/L)
C(m)ss
持续恒速静脉滴注稳定时代谢物浓度(mg/L或μmol/L)
Cmin
一次给药后的最小血药浓度(mg/L或μmol/L)
Cn
在第n次给药间隔期间任何t时的血药浓度
CN,max; CN,min
当固定给药剂量和给药间隔时,第N次给药后的最大和最小血浆药物浓度(mg/L或μmol/L)
ASS
在恒速静脉滴注时,稳态时的体内药量(mg或μmol)
ASS,AU
稳态时,一个给药间隔期间的平均体内药量(mg或μmol)
ASS,max; Ass,min
固定剂量,固定给药间隔,稳态时一个给药期间的最大和最小体内药量(mg或μmol)
Ass,t
在固定剂量和给药间隔,稳态时一个给药剂量后t时间点的体内药量(mg或μmol)
K1
抑制平衡常数(mg/L或μmol)
Km
在一室模型代谢转化的表观一级速率常数(即代谢物的消除速率常数),也有代表Michaelis常数(h-1)
Ki
在肾衰竭时的处置速率常数(h-1)
Kp
药物在血液或血浆与组织之间的平衡分布比例系数1)
kt
转换速率分数(h-1)
Emax
最大效应(使用效应测量单位)
fp
血浆中未结合药物的分数
fs
唾液中未结合药物的分数
F
药物生物利用度(无单位)
Fbd
血浆中结合药物与总药物浓度比(无单位)
fO
透析治疗期间,透析清除率占总清除率的分数(无单位)
fe
原形药物在尿中的排泄分数(无单位)
FEV1
一秒钟的强制呼吸量(L)
FH
进入肝脏的药物逃过首过消除的分数(无单位)
CD
离开透析器时透析液药物浓度(mg/L或μmol/L)
Cinf
恒速滴注浓度(mg/L或μmol/L)
CL
代谢抑制剂浓度(mg/L或μmol/L)
CL
血浆药物总清除率(L/h)
CLb
全血药物总清除率(L/h)
CLbD
血液药物透析清除率(L/h)
CLb,H
血液药物肝清除率(L/h)
CLcr
肌酐肾清除率(L/h)
临床药物动力学符号注释
A
体内药量(mg或μmol)
Aa
在吸收部位有待于吸收的药量(mg或μmol)
Ae
尿中累积原形药物排泄量(mg或μmol)
Ael(m)
已消除的代谢物的量(mg或μmol)
Aess
在稳态下,一个给药间隔期间尿中累积原形药物排泄量(mg或μmol)
Ae∞
单次给药后,至无穷大时间内尿中累积原形药物排泄量(mg或μmol)
Xc
在t时中央室的药量(mg或μmol)
Xn
反复用药在第n个给药间隔期间任何时间t时的体内药量(mg或μmol)
Xt
在周边室的药量(mg或μmol)
Xu
尿中排泄的原形药物累积药量(mg或μmol)
X0
负荷剂量或初剂量(mg或μmol)
α
在二室模型描述药物在体内分布的表观一级混合速率常数
β
在二室模型描述药物消除的表观一级混合速率常数
VT
血浆外药物分布的生理容量(L)
Vss
建立在血浆药物浓度基础上的稳态表观分布容积(L)
VTW
血浆外药物分布的水容量(L)
Vu
建立在血浆未结合药物浓度基础上的表观分布容积
X
在t时体内的药量,稳态时体内的“平均”药量(mg或μmol)
XA
在t时吸收到体循环中的药量(mg或μmol)
Xa
吸收部位的药量(mg或μmol)
ka
吸收速率常数(h-1)
KA
药物与蛋白的结合常数(L/mol)
kD
透析治疗中,患者的消除速率常数(h-1)
Ke
尿排泄速率常数,也指表观一级吸收速率常数(h-1)
K0
在一室模型表现一级肾脏排泄速率常数,也指零级输入或滴注速率常数(h-1)
K10
从中央室消除的表观一级速率常数
kf
代谢物形成速率常数(h-1)
Cpc
腹腔中药物浓度(mg/L或μmol/L)
Css
在零级过程滴注(即恒速静脉滴注)后达稳态时的血药浓度(mg/L或μmol/L)
C0
静脉注射后瞬时的血药浓度
(C1)max
反复用药后第1次用药中最大血药浓度
(C1)min
反复用药后第1次用药中最小血药浓度
C∞
反复用药达稳态时每一给药间隔期间内任一时间t时的血药浓度
Vc
中央室的分布表观容积(L)
Vd
在多室模型药物的表观分布容积,系指体内药量和后分布相血药浓度之间的一个比例常数(L)
V1
起始稀释房室容积(L)
Vm
用动力学Michaelis-Menten所描述过程的理论上的最大速率(mg/h或μmol/L)
Vp
血浆容量(L)
VPC
腹腔中透析液容量(L)
VR
细胞内液中的水容量(L)
(C∞)max
稳态时每一给药间隔期间的最大血药浓度
(C∞)min
稳态时每一给药间隔期间的最小血药浓度
Css,av
固定给药剂给药间隔,当达到稳态时,任一给药间期的平均血浆药物浓度(mg/L或μmol/L)
Css,max; Css,min
固定给药剂量及给药间隔,当到达稳态时,最大和最小血浆药物浓度(mg/L或μmol/L)
A(m)
体内代谢物的量(mg或μmol)
Amin
达到预期药物效应的所需最小药量(mg或μmol)
AN,max; AN,min
固定给药剂量和给药间隔,第N次给药后体内的最大和最小药量(mg或μmol)
AN,t
在第N次给药后的t时间点的体内药量(mg或μmol)
ARE
单次给药后,有待从尿中排泄的药量(mg或μmol)
Tinf
恒速滴注时间(h)
Tm
药物转运(分泌)进入肾小管的最大速率(mg/h)
t1/2,t0.5
半衰期(h)
t1/2α
药物分布半衰期(h)
t1/2β
药物消除半衰期(h)
T1/2Ka
药物吸收半衰期(h)
Tt
转换时间(h)
V
建立在血浆浓度基础上的表观分布容积,即在一室模型药物的表观分布容积(L)
VB
血液容量(L)
CT
血浆外体液平均药物浓度(mg/L或μmol/L)
CTW
总体液中未结合药物浓度(mg/L或μmol/L)
Cupper; Clower
血浆药物浓度的上、下限(mg/L或μmol/L)
Cu
血液中未结合药物浓度(mg/L或μmol/L)
Cu1
血浆中未结合抑制剂浓度(mg/L或μmol/L)
Cv
静脉血液中药物浓度(mg/L或μmol/L)
D2
负荷剂量(mg或μmol/L)
DM
固定剂量方案的维持剂量(mg或μmol/L)
DM,max
最大维持剂量,以确保血浆药物浓度在稳态时于一个给药间隔内介于药物血浆浓度的上、下限之间(mg或μmol/L)
E
抽提比(无单位)
EC50
产生1/2最大效应的浓度(mg/L或μmol/L)
EH
肝抽提比(无单位)
fup
血浆蛋白中未结合位点与总结合位点的比例分数(无单位)
fuR
细胞内液中未结合药物与总药物浓度的比例分数(无单位)
fuT
血浆外组织中未结合药物与总药物浓度的比例分数(无单位)