药物应用的药动学基础
药物动力学

药物动力学药物动力学是研究药物在体内的吸收、分布、代谢和排泄等过程的学科,它对药物在人体内的行为进行定量研究,为合理用药提供科学依据。
药物的吸收过程药物在体内的吸收途径药物可以通过口服、注射、吸入、外用等方式进入体内,其中口服是最常见的给药途径。
影响药物吸收的因素药物的理化性质、给药途径、药物的制剂形式、肠道内容物的影响等都会影响药物的吸收速度和程度。
药物的分布过程药物在体内的分布药物在体内主要通过血液输送到各组织器官,分布到组织器官的速度和程度取决于药物的脂溶性、离子性等因素。
影响药物分布的因素药物与血浆蛋白结合的程度、血流动力学、血-脑屏障等因素都会影响药物在体内的分布。
药物的代谢过程药物的代谢目的药物代谢是为了加快药物的排泄,使药物更容易从体内排除。
代谢通路药物代谢主要通过肝脏中的细胞内酶系统完成,包括细胞色素P450等。
药物的排泄过程药物的排泄途径药物主要通过肾脏排泄,也可通过胆汁排泄、乳汁排泄等途径。
影响药物排泄的因素肾功能状态、药物的分子大小、极性等性质都会影响药物在体内的排泄速度。
药物动力学参数表述药物动力学的参数有哪些常用的药物动力学参数包括药动学半衰期、药物清除率、生物利用度、药物浓度-时间曲线等。
参数的意义这些参数能够定量描述药物在体内的行为,为临床用药监测和药物研发提供参考。
药物动力学在临床上的应用用药策略制定根据药物的动力学特点,制定合理的用药策略,包括给药途径、用药剂量等。
临床用药监测通过监测药物浓度,根据药物动力学参数进行用药调整,确保疗效和安全性。
结语药物动力学是研究药物在体内行为的重要学科,对于提高药物治疗效果,减少不良反应具有重要意义。
科学掌握药物动力学知识,有助于提高临床用药的合理性,实现个体化用药的目标。
临床药学中的药物药动学与药效学研究

临床药学中的药物药动学与药效学研究药物药动学与药效学是临床药学领域非常重要的两个研究方向。
药物药动学研究药物在人体内的吸收、分布、代谢和排泄过程,而药效学则研究药物在机体内的作用机制以及对疾病的治疗效果。
这两个学科研究的深入,能够为新药开发和合理用药提供科学依据。
本文将从理论基础、研究方法以及应用价值三个方面探讨临床药学中药物药动学与药效学的重要性。
一、理论基础药物药动学是研究药物在机体内的吸收、分布、代谢和排泄的科学,是药物治疗过程中的基础。
药物在体内的药动学过程决定了药效学参数的形成,进而影响药物的临床疗效。
药物药动学对于理解药物在人体内的药效学表现和解释药物治疗的效果至关重要。
药物吸收是药物进入血液的过程,主要发生在消化道。
药物分布是指药物在体内各组织和器官之间的分布情况。
代谢是指药物在体内经过化学代谢转化的过程,通过代谢药物的机制能够使药物更易于排泄。
药物排泄是指药物从体内排出的过程,主要通过肾脏、肝脏和肺脏完成。
这些药物药动学过程相互作用,共同影响药物在体内的行为。
药效学是研究药物在机体内具体作用机制以及对疾病的治疗效果的科学。
药物通过与特定受体结合或影响机体内的特定生理过程,发挥治疗作用。
药效学研究能够帮助我们了解药物在机体内具体的作用方式和对于疾病的治疗效果,为临床医生选择合适的药物提供科学依据。
二、研究方法药物药动学与药效学的研究需要深入了解药物在体内的行为以及药物对于机体的作用机制。
因此,研究方法也非常多样和复杂。
在药物药动学方面,常见的研究方法包括药物吸收动力学、药物分布动力学、药物代谢动力学和药物排泄动力学等。
通过采集药物在不同时间点的血药浓度数据,可以绘制药物的药时曲线,从而了解药物在体内的行为。
此外,也可以通过药物的药代动力学参数来评估药物在体内的代谢情况。
药效学的研究方法多样,常用的包括体外药物活性测定、细胞实验和动物实验等。
体外药物活性测定通过测定药物对于特定靶标的活性来评估药物的作用机制。
药物治疗中的药物动力学与药效学

药物治疗中的药物动力学与药效学药物动力学和药效学是药物治疗领域的两个重要概念。
药物动力学研究药物在体内的吸收、分布、代谢和排泄过程,而药效学则关注药物在体内的作用机制及其产生的效应。
深入了解药物动力学和药效学对于合理用药、优化治疗方案以及降低药物不良反应具有重要意义。
一、药物动力学药物动力学研究药物在体内的动态过程,包括吸收、分布、代谢和排泄四个环节。
1. 药物吸收药物吸收是指药物经过给药途径进入体内的过程。
吸收途径主要包括口服、皮肤贴敷、注射等。
吸收速度与给药途径相关,不同途径吸收速度和程度不同。
药物吸收还受到一系列因素的影响,如药物的物理化学性质、给药部位的血流情况、药物的溶解度和肠胃pH值等。
2. 药物分布药物分布是指药物在体内各个组织器官之间的传递过程。
药物在体内分布呈现复杂的动力学特点,包括组织亲和性、血浆蛋白结合率和血脑屏障、胎盘屏障等。
这些因素会影响药物在体内的浓度分布,从而影响药物的疗效和不良反应。
3. 药物代谢药物代谢是指药物在体内被生物转化成代谢产物的过程。
药物主要经过肝脏进行代谢,也可在肾脏、肺、肠道等器官中进行代谢。
药物代谢的主要作用是使药物更容易排泄,同时还能转化为有活性的代谢产物。
药物代谢主要由酶催化发生,包括细胞色素P450酶和非细胞色素P450等。
4. 药物排泄药物排泄是指药物及其代谢产物通过肾脏、肠道、肝脏、呼吸道等途径从体内排出的过程。
主要由肾脏和肝脏承担,其中肾脏是主要排泄途径。
药物排泄速度和方式会影响药物在体内的半衰期和药物浓度。
二、药物效应学药物效应学研究药物在体内的作用机制及其产生的效应。
1. 作用机制药物的作用机制是指药物与生物体内特定靶点发生相互作用,从而引起生理或病理效应的过程。
药物可以通过与受体结合、激活酶或抑制酶等方式发挥作用。
药物作用的具体机制对于指导合理用药和开发新药具有重要意义。
2. 产生的效应药物通过作用机制产生一系列效应,包括治疗效应和不良反应。
生物药剂学与药动学—药物应用的药动学基础
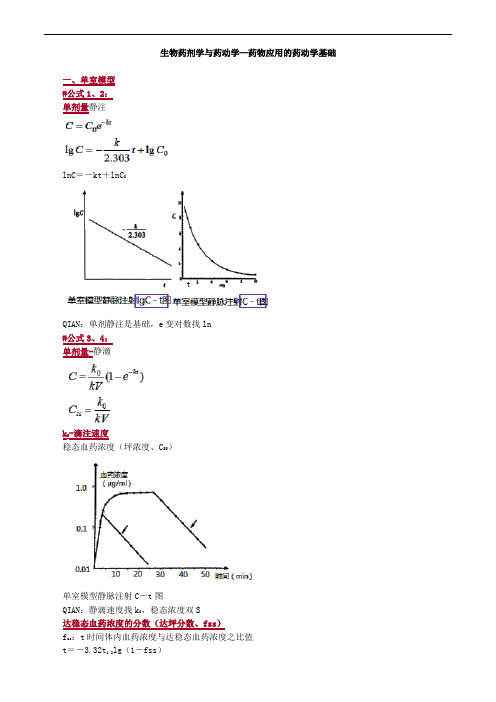
生物药剂学与药动学—药物应用的药动学基础一、单室模型#公式1、2:单剂量静注lnC=-kt+lnC0QIAN:单剂静注是基础,e变对数找ln#公式3、4:单剂量-静滴k0-滴注速度稳态血药浓度(坪浓度、C SS)单室模型静脉注射C-t图QIAN:静滴速度找k0,稳态浓度双S达稳态血药浓度的分数(达坪分数、fss)f ss:t时间体内血药浓度与达稳态血药浓度之比值t=-3.32t1/2lg(1-fss)达到稳态某一分数fss所需要的时间与药物的t1/2成正比,t1/2越短,达到稳态的时间越快;而与滴注速度k0的快慢完全无关补充:n=-3.32lg(1-fss),n为半衰期的个数n=1 →50%n=3.32 →90%n=6.64 →99%n=10 →99.9%#公式5:单剂量-血管外F:吸收系数吸收量占给药剂量的分数单次口服给药后的血药浓度时间曲线达峰时间:t max峰浓度:C maxC-t曲线下面积:AUC二、双室模型混杂参数——A、B:经验常数α:分布速度常数、快处置速度常数β:消除速度常数、慢处置速度常数α、β:描述两个指数项即分布项和消除项的特征QIAN:双室模型AB杂中央消除下标10三、多剂量给药固定剂量、固定给药间隔是临床最常用的给药方案。
这种给药方法,血药浓度和体内药量是波动的。
第二次给药前体内的药物尚未被完全清除,所以后一次给药使体内的药量在前一次的基础上逐渐累积,血药浓度逐渐升高。
随着给药次数的不断增加,血药浓度几乎不再升高,而是恒定在一定的水平上随每次给药作周期性的变化。
血药浓度达到稳态其主要有三个特征性参数,即稳态最大血药浓度、稳态最小血药浓度和稳态血药浓度的波动范围(简称坪幅)。
#公式7-10:多剂量给药(重复给药)QIAN:多剂量需重复,间隔给药找τ值既不是与的算术平均值,也不是其几何平均值,仅代表与之间的某一血药浓度值。
系指血药浓度达到稳态后,在一个给药间隔时间τ内,药-时曲线下面积除以τ得的商。
药代动力学的数学基础(中文)

药物动力学的数学基础:介绍因为药物动力学和生物药剂学有很强的数学基础,在代数、微积分、指数、对数和单位分析这些数学原理上有一个坚实的基础对学生学习这两门学科至关重要。
本章中自检方法提供对一个人基本数学技能中弱点的自我评估。
在自我检测中问题处理困难表明回顾数学本质是必要地。
在这里总结数学原理以仅达到回顾的目的。
为了一个更加完整的基本原理的讨论,应该参考一本合适的数学教科书。
数学自检方法1.浓度的单位是什么?2.一药物溶液浓度为50mg/ml,在20.5ml溶液中需药物量为多少?在0.4升?若30mg药物需溶剂量多少?3.将上述溶液转换单位:从mg/ml到g/L和μg/μl,如果药物分子质量为325Da,换算为浓度单位为多少?4.如果将20mg药物加入装水容器使浓度为0.55mg/L,容器中水的体积是多少?5.下列方程:y=0.5x+2a.画出方程的曲线b.描述该方程各部分的相关性c.如果x=0.6,y=?d.如果y=4.1,x=?6.求下列方程的解a.logx=0.95b.e x=0.44c.lnx=1.227.连接两点直线斜率是多少?y=0.5x+28.根据下图,若x=2,x=12,C=?评估与计算器和计算机的使用药物动力学需要的大多数学计算和这本书中其他计算可能通过铅笔,方格纸和逻辑思维过程完成。
拥有对数和指数运算功能的科学计算器使计算不再繁琐。
特殊的计算机软件(见附录B)可用于临床药物动力学疾病状态计算。
每当计算影响药物剂量时,应该在给定的信息条件下评估结果是否正确。
比如,对于给定的问题,考虑结果数据是否有正确的范围和单位,如果正确的结果应该在100mg到200mg之间,像结果12.5mg和1250mg就是错误的。
一个问题的答案的单位应该仔细检查,比如,如果期望值是浓度单位,那mg/L或μg/ml是可以的,像L或mg/hr一定是错误的。
错误的单位可能由错误的替换或选择了错误的公式引起。
在药物动力学计算中,只要数据和单位都正确,结果也会正确。
药理学3药动学

药理学3药动学
生 物 转化 (biotransformation)
又称代谢或药物转化,是指 药物在体内发生的化学结构 改变。转化后的药物活性降 低或失去药理活性,极性增 加,易于排泄。
药理学3药动学
生物转化的部位及其催化酶
•生物转化的主要部位是肝脏。 •生物转化由肝微粒体细胞色
药理学3药动学
药物通过生物膜的转运
• 细胞膜主要由脂类(磷脂、胆固醇与糖脂) 和蛋白质组成。
• 大多数极性(离子化程度较强)药物难于 通过脂质双层,而脂溶性药物可以通透。 小分子药物可从膜孔透过膜。
• 药物通过生物膜的能力主要决定于药物的 脂溶性、解离度及分子量,其转运机制可 分为被动转运和载体转运两大类。
---是指体内药物或其代谢物排出体外 的过程,它与生物转化统称为药物消 除(elimination)。
药理学3药动学
载体转运 ---是指细胞膜上的载体与药物结合,并
载运它到膜另一侧的过程。
• 主动转运:又称“上山”或逆流转运。 特点是:①逆浓度梯度或逆电化学梯度 透过细胞膜 ②细胞膜的载体对药物有特 异的选择性 ③消耗细胞能量 ④以同一载 体转运的两种化合物可出现竞争性抑制 ⑤转运速度有最高限度。
代谢酶的活性,增加自身或其他药物的代谢速率。 苯巴比妥是典型的酶诱导剂。
(2) 酶的抑制:某些化学物质能抑制肝微粒体药物
代谢酶的活性,减慢其他药物的代谢速率。红霉 素是酶抑制剂。
3. 生理因素与营养状态 生物转化有明显的种属差 异、种族差异和个体差异
4. 病理因素
药理学3药动学
排 泄(excretion)
药理学3药动学
pKa值的概念
• pKa值——是弱酸性或弱碱性药物在50% 解离时溶液的pH值。注意:pKa值不是药 物自身的pH值。药物离子化程度受pKa值 及所在溶液的pH值决定。
药物动力学的研究及其应用

药物动力学的研究及其应用药物动力学是研究在体内药物作用的变化规律和药物消除过程的动力学过程。
药物动力学的研究不仅有助于解析药物在体内的动力学变化规律,也能为探讨药物的安全性和疗效提供科学依据。
以下将详细介绍药物动力学的相关内容和应用。
I. 药物在体内的代谢和消除过程药物在体内的代谢和消除过程是药物动力学研究的关键点。
这些过程涉及到药物的生物转化、药物作用靶点、药物的分布等方面。
1. 药物的生物转化药物的生物转化通常包括药物代谢和药物分解两个方面。
药物代谢是指药物和生体内化学反应所形成的代谢产物,通常经过肝脏的代谢。
药物分解则是指药物被水解或酸解,产生分解产物。
2. 药物作用的靶点药物在体内的作用靶点通常与体内分子的特异性结合有关。
药物的作用靶点范围广泛,包括细胞表面受体、酶、离子通道、细胞膜以及细胞核等。
3. 药物的分布药物在体内的分布通常受到以下多种因素的影响:药物的溶解度、分子量、药物的蛋白结合率和各个器官的血流速度。
II. 药物动力学的应用药物动力学不仅是药物的研究和开发的重要手段,还可以为药物疗效评价、安全评价以及药物的现代化管理提供科学的依据。
1. 临床药物开发药物动力学研究是临床药物开发中的重要一环。
通过分析药物在体内的代谢过程,可以了解药物在体内的转化和消除规律。
同时通过分析药物在体内的药效学特性,可以为药物疗效评价和药物治疗方案的制定提供科学依据,从而提高药物的疗效和安全性。
2. 药物疗效及安全性评价药物动力学研究可以评估药物的疗效和安全性。
药物的生物利用度、药物的代谢代谢以及药物在体内的分布情况等因素,都会影响药物的疗效和安全性。
通过药物动力学研究,可以评估药物的疗效并寻找副作用的原因,为药物的治疗方案和安全评价提供依据。
3. 药物现代化管理药物动力学与药物现代化管理密切相关。
药物在临床应用中的安全性和有效性往往是药物上市管理审批的重要标准。
药物动力学研究可以为药物安全性评价和治疗效果评价提供科学依据,有助于提高药物现代化管理水平,促进医学领域的持续发展。
药理学基础理论

静脉滴注
分次给药
0
t
CSS的特点
①Css的高低取决于每次给药的剂量。
②静脉恒速滴注时量曲线平滑上升达Css;分次给药可 使Css随吸收、分布、和消除的过程有明显的上下波 动。曲线波动的大小与给药间隔时间有关。
③不论恒滴还是分次给药,在达到Css后,如中途改变
给药速度或给药量,则需再经5个t1/2才能达到新的
静脉滴注的DL Ass= CssVd = RA/K = RA/(0.693/t1/2)
= 1.44 t1/2 RA 将第一个t1/2内静脉滴注量的1.44倍在静脉滴注开始 时就推注入静脉即可达到并维持Css
分次恒速给药的DL
分次恒速给药达到Css 时,体内是维持剂量 (maint enance dose , Dm)与体内上一次剂量残 留药物的总和
Ass = Dm+ Asse-kt , ∵ DL = Ass= Dm/(1-e-kt) 当给药间隔时间τ = t1/2 时, DL=Dm/(1-e-0.693)=Dm/0.5=2Dm
即每隔一个t1/2给药一次,首剂加倍剂量可以使 血药浓度迅速达到稳态
C Css
0
i.v.gtt. 分次恒速给药
t
药物消除动力学
恒比消除的药物在恒速恒量给药过程中,经5个t1/2,给药速 度等于消除速度,血药浓度维持在一个基本稳定的水平称 为Css。
(恒量消除的药物因吸收速度大于消除速度,使体内药物 蓄积,血药浓度会无限制的增高)
连续多次给药的药物血浆浓度变化
按一级动力学消除的药物连续给药的时量曲线图
Cp Css
5×t1/2
统(hepatic microsomal mixed function oxidase system)
第一章 药物学基础总论

(二)药物的其他作用机制
1. 改变理化环境 2. 参与或干扰机体的代谢过程 3. 影响生物膜的通透性或离子通道 4. 影响酶的活性 5. 影响递质的释放或激素的分泌 6. 影响核酸的代谢 7. 影意”是指用药后观察用药反应
3. 用药后
(1)要密切观察用药后病人的病情变化,继续观察药物的疗效 (2)根据药物出现的不良反应,给出护理诊断,采取相应的护理措施 (3)对病人进行用药指导,强调必须严格执行医嘱,不可擅自调整用
药方案,科学合理用药、确保用药安全
停药就会出现戒断症状,表现为烦躁不安、流泪、出汗、疼痛、 恶心、呕吐、惊厥等,甚至危及生命,再次用药后症状消失
三、药物的作用机制
(一)药物-受体作用机制 1. 受体与配体
• 受体是位于细胞膜或细胞内一些具有 识别、结合特异性配体并产生特定效 应的大分子物质
2. 药物与受体结合
• 药物与受体结合引起生物效应,需具 备两个条件:即亲和力和内在活性
(四)防治作用和不良反应
1. 防治作用分为预防作用和治疗作用 (1)预防作用 (2)治疗作用:是指凡符合用药目的或能达到治疗疾病效果的作用
治疗作用
对因治疗 对症治疗
2. 不良反应
• 对防治疾病无益甚至有害的反应,称为不良反应 (1)副作用:是指药物在治疗量时与治疗作用同时出现,
与用药目的无关的作用 • 特点:副作用是药物本身固有的作用,一般危害不大,
(4)继发反应
• 由药物的治疗作用引起的不良后果 • 例如:“二重感染”
(5)后遗效应又称后遗作用
• 是指停药后血药浓度降至最低有效浓度(阈值)以下时残存的药理 效应
药师职称考试药理学知识点总结药物应用的药动学基础

药师职称考试药理学知识点总结药物应用的药动学基础一、单室模型血管内给药的药动学(一)单室模型血管内给药的药动学1.单室模型静脉注射单次给药的药动学单室模型药物静脉注射后很快在体内达到分布平衡,药物的体内过程基本上只有消除过程,血药浓度与时间的关系式为:C=C0·e-kt式(3-2-5)式中,C0为静脉注射后的血药初始浓度;C为t时刻的血药浓度,其对数方程为:lnC=-kt+lnC0式(3-2-6)对数方程为线性方程,因此由血药浓度数据的回归直线的斜率可以求得消除速率常数k,由其截距可以求得C0。
由此可以根据(式3-2-2)计算消除半衰期t1/2,根据式(3-2-3)计算表观分布容积V,根据式(3-2-4)计算总清除率Cl,药-时曲线下面积可按式(3-2-7)计算。
式(3-2-7)2.单室模型静脉滴注单次给药的药动学静脉滴注是指以缓慢、近乎恒定的速度向静脉血管内给药的一种方式。
单室模型药物静脉滴注后血药浓度与时间的关系式为:式(3-2-8)式中,C为t时刻的血药浓度;k0为滴注速度。
由式(3-2-8)可见,滴注初始C值迅速增大,随后增大速度逐渐减小,最后趋于一个恒定的浓度值,称为稳态血药浓度或坪浓度,用Css表示。
式(3-2-9)静脉滴注时,达到C ss以前的血药浓度C始终小于C ss,任何时间的C值都可用C ss的某一分数来表示,称为达稳态分数或达坪浓度,用f ss表示。
临床需关心静脉滴注持续多少时间才能趋近稳态浓度,该时间的计算公式为:式(3-2-10)可以看出,达到稳态某一分数f ss所需要的时间与药物的t1/2成正比,t1/2越短,达到稳态的时间越快;可与滴注速度K0的快慢完全无关。
(二)单室模型血管外给药的药动学药物除了直接通过血管进入血液循环外,还可以通过血管外给药,如口服、肌内注射、透皮给药和黏膜给药等。
血管外给药后,药物均需通过给药部位的生物膜吸收进入血液循环,然后在体内分布并进一步被消除。
生物药剂学和药动学及其给药方案设计

一、静脉注射
积累因子:R = 1/(1-e-kτ) 负荷剂量:XL = X维/(1-e-kτ) = RX维
可见负荷量和维持量呈正比,比例系数恰好等于R
若τ= t1/2,则 R = 2,即 XL = 2X维 这就是所谓的Thumb原则
例:已知某药注射剂量为0.5克,Vd为128升,半衰期 为 3.5 小 时 , 有 效 浓 度 为 1.6ug/ml , 试 确 定 给 药 间 隔 τ.解: 根据 Cssmin= C0e-kτ/(1-e-kτ),求τ
(t= 0)
Cssmin= C0e-kτ/(1-e-kτ) (t=τ)
波动范围: 坪幅 = Cssmax - Cssmin = C0 = X0/V
可见剂量可调节波动幅度,而与给药间隔无关
一、静脉注射
平均稳态血药浓度
Css
X0
VK
可见给药间隔可调节血药浓度的水平
达稳态血药浓度所需时间:
nτ= -1.443t1/2ln(1-fss)
C2 = K0/Vk(1-e-kt) = 20 40/50 0.693(1- e-0.6934/40)
= 1.546 (μg/ml) 所以:4h后体内血药浓度为:
C = C1 + C2= 0.373 + 1.546 = 1.919 (μg/ml)
例4.已知某药体内最佳治疗浓度为13ug/ml, k = 0.1/h,V = 10L。请设计静脉给药的方案。
Vd = FX0/ AUC Cl = Vd = K10Vc = FX0/AUC
AUCLM KNa
重复给药
多剂量函数
(1enk ) R
(1ek )
一、静脉注射
Ct = C0e-kt(1-e-nkτ)/(1-e-kτ) (0≤t≤τ)
抗生素药动学

阿洛西林、氨曲南、头孢唑啉、头孢甲肟、头孢 哌酮、头孢噻肟、头孢西丁、头孢曲松、头孢呋辛、 美洛西林、萘啶酸、呋喃妥因、苯唑西林、青霉素
13
编辑ppt
三、药物消除
❖ 半衰期(half life) 定义: 血浆中药物浓度下降一半所需要的时间 意义:
表明药物消除快慢
抗菌药物在体内的动态过程
药物
周边室组织
iv im,sc
分布
中央室 游结
po 肝
离合 型型
作用 部位
游离型
生物效应
排再 泄吸
收
类脂质屏障
3
编辑ppt
一、药物吸收
口服固体制剂体内吸收过程示意图
制 剂 崩解或 颗粒或 释放 药
药物+辅料
解凝聚
粗分散 体
物
溶于 体液
药物 透过 血药
溶液
生物膜
浓度
溶出相
吸收相
青霉素类 头孢菌素类 其他β-内酰胺类 大环内酯类(除外酯化物) 磷霉素
异烟肼
磺胺药+TMP 呋喃妥因
碘苷
阿糖腺苷
12
编辑ppt
抗菌药物在乳汁中的浓度
❖ 乳汁药物浓度>母体血清药物浓度25%~50%者
磺胺药、TMP、异烟肼、甲硝唑、红霉素、克林 霉素、氯霉素、四环素、阿米卡星、庆大霉素、 卡那霉素、链霉素、妥布霉素、氨苄西林、羧苄西林
青霉素 羧苄西林 头孢唑啉 头孢噻吩 氨曲南 SMZ 万古霉素
苯唑西林 头孢氨苄 庆大霉素 妥布霉素 阿米 卡星 氯霉素 头孢他啶 头孢噻肟 头孢呋辛 亚胺培南
17
编辑ppt
肝功能减退时抗菌药物的应用
第二章 药动学

目的要求:
掌握机体对药物处置的过程-------吸收、分布、 代谢和排泄的影响因素。 掌握肝肠循环、肝药酶诱导剂与抑制剂、首过 效应、半衰期、生物利用度、稳态血药浓度等 基本概念。 熟悉药时曲线、一级动力学、零级动力学概念 和意义。
药物代谢动力学(简称药动学、药物 的体内过程)
脏, 被代谢一部分,使进入血循的有效
药量减少,效应降低。
药
首过效应示意图
代谢 代谢
血 液 循 环
首过效应强的药 举例
•硝酸甘油(90%)、杜冷丁、异丙肾上腺
素、等——舌下给药
(2)舌下给药:舌静脉吸收, 快,但不规则
(3)直肠给药:直肠粘膜吸收,吸
收快,不规则,给药不方便;
2、注射给药:
•(1)皮下注射或肌肉注射(sc或im): • 用于:病情较严重者——PG、庆大; •(2)静脉注射:包括iv和ivgtt • 用于:急诊、休克病人;
3、乳腺排泄:
乳液——弱酸性,弱碱性药物易排出 哺乳期妇女用药注意。如吗啡
4、其他: 如汗液、泪液、唾液
第三节 血药浓度的动态变化 及主要药动学参数
一、药时曲线
达峰时间
药峰浓度
最小中毒浓度
安全范围
最小有效浓度
吸 收 分 布 过 程
代谢排泄过程
潜伏期
持续期
残留 期
1、潜伏期: 用药后到开始出现疗效的时间
(二)影响药物吸收的因素
1、药物的理化性质; 2、吸收环境;
3、首过效应;
1、药物的理化性质
(1)分子大小
(2)脂溶性;
(3)剂型(注射剂、片剂)
2、吸收环境:
第一章药物学基础概论

影响药效的因素
(一)机体方面的因素 1.年龄 2.性别 3.心理因素 4.遗传因素 5.病理因素
(二)药物 方面的因素 1.药物的化学结构 2.剂量 3.剂型 4.给药时间和次数 5.联合用药及药物的相互作用
二、药物的体内过程
1.吸收:药物从给药部位进入血液循环的过程。
二、药物的体内过程
2.不同给药途径的吸收特点
(1) 口服给药:
① 主要吸收部位:小肠
②
特点:最常用、最安全、最简便的给药途径。
③ 吸收途径:药物经消化道吸收后经门静脉进入肝脏,最后
进入体循环。
④首关消除(又称首关效应):药物在吸收过程中部分被
肝脏和胃肠道的某些酶灭活代谢,使进入体循环的药量减少。
注意:首关消除较多的药物不口服给药,如硝酸甘油。
二、药物的体内过程
(2)舌下含服: ①特点:吸收迅速,无首关消除
②例子:硝酸甘油舌下给药。 (3)直肠给药
①经直肠或肠粘膜吸收
二、药物的体内过程
(4)注射给药: ①静脉注射可迅速准确地进入体循环,没有吸收过程。 ②由于肌肉组织血流量较皮下组织丰富,故肌肉注射比皮下 注射吸收快。 ③当休克时,因周围循环不良,皮下和肌肉注射速度均大大减 慢,需静脉注射才能达到急救目的。
药物的作用机制
(4)受体的调节:受体数目和亲和力的变化称为受体的调节 向下调节: 见于:长期使用激动剂,使受体数目减少 (受体脱敏) 表现:耐受性
向上调节 见于:长期使用受体阻断药,使受体数目增 (受体增敏) 表现: 反跳现象
第三节
药物代谢动力学
药物代谢动力学
一、药物的跨膜转运:简单扩散是药物转运的最主要方式。
5.不良反应
(8)依赖性:精神依赖性 生理依赖性
药物应用的药动学基础

药物应用的药动学基础一、单室模型血管内给药的药动学(一)单室模型血管内给药的药动学1.单室模型静脉注射单次给药的药动学单室模型药物静脉注射后很快在体内达到分布平衡,药物的体内过程基本上只有消除过程,血药浓度与时间的关系式为:C=C0·e-kt式(3-2-5)式中,C0为静脉注射后的血药初始浓度;C为t时刻的血药浓度,其对数方程为:lnC=-kt+lnC0式(3-2-6)对数方程为线性方程,因此由血药浓度数据的回归直线的斜率可以求得消除速率常数k,由其截距可以求得C0。
由此可以根据(式3-2-2)计算消除半衰期t1/2,根据式(3-2-3)计算表观分布容积V,根据式(3-2-4)计算总清除率Cl,药-时曲线下面积可按式(3-2-7)计算。
式(3-2-7)2.单室模型静脉滴注单次给药的药动学静脉滴注是指以缓慢、近乎恒定的速度向静脉血管内给药的一种方式。
单室模型药物静脉滴注后血药浓度与时间的关系式为:式(3-2-8)式中,C为t时刻的血药浓度;k0为滴注速度。
由式(3-2-8)可见,滴注初始C值迅速增大,随后增大速度逐渐减小,最后趋于一个恒定的浓度值,称为稳态血药浓度或坪浓度,用Css表示。
式(3-2-9)静脉滴注时,达到C ss以前的血药浓度C始终小于C ss,任何时间的C值都可用C ss的某一分数来表示,称为达稳态分数或达坪浓度,用f ss表示。
临床需关心静脉滴注持续多少时间才能趋近稳态浓度,该时间的计算公式为:式(3-2-10)可以看出,达到稳态某一分数f ss所需要的时间与药物的t1/2成正比,t1/2越短,达到稳态的时间越快;可与滴注速度K0的快慢完全无关。
(二)单室模型血管外给药的药动学药物除了直接通过血管进入血液循环外,还可以通过血管外给药,如口服、肌内注射、透皮给药和黏膜给药等。
血管外给药后,药物均需通过给药部位的生物膜吸收进入血液循环,然后在体内分布并进一步被消除。
因此,与血管内给药的药动学模型不同的是,血药浓度的变化还受到药物从给药部位进入体循环的速率的影响,通常以吸收速率常数k a来描述这个速率过程。
药理学 药动学

时间延长,中毒时可采 用洗胃,导泻等方法,
促进排泄。
• 有些药物可在肠腔重吸收,重新回到肝,形
成肝肠循环。
4.乳汁排泄
❖ 乳汁偏酸,有利于 弱碱的排出。
❖碱性药物如吗啡、奎 宁等较易进入乳腺管 内,达到比血浆高数 倍的浓度。有乳汁排 泄的药物,应考虑对 乳儿的影响
5.其他 • 唾液、泪液和汗液、头发、皮肤。 • 呼吸道可排出挥发性药物和气体。
CL=Vd·K(消除速率常数 )
意义:CL小,首关消除少,F大; CL大, 首关消除多,F小。
(七)多次用药和给药方案
Css
• 大多数药物均需多次
Cp
给药,如每隔一个t1/2
2
等量给药一次,则经
过5~7个t1/2血药浓度 可达到一个稳态/坪值
1
(Css) 。药时曲线
开始呈峰值与谷值交
替组成的锯齿形上升 0
量有限 • 但血流丰富,吸收
也迅速
3.注射给药 静脉、肌肉、皮下和皮内注射等 避免被破坏,起效快 4.吸入给药,气雾剂、粉雾剂等 经肺吸收进入血液循环 或治疗鼻咽局部疾病 5.皮肤给药,膜剂,贴剂,膏剂等 可发挥局部或全身作用
二.药物分布
药物从血循环系统到达机体各个部位和组织的 过程。 影响因素包括 1.药物与血浆蛋白结合
1
2
3
4
5
6 t1/2
,然后逐渐趋于平稳
。
2、负荷量与维持量
Cp
首次剂量增加,后改维持剂量
维持剂量
t1/2
•负荷量加倍可迅速产生药效 •在一个t½ 内达到坪浓度 •然后改用维持量
• 给药方案个体化
❖每个病人的病情和体质各不相同,选择最佳 给药方案称为给药方案个体化。
药理基础知识

吡唑酮类 :安乃近 (抗体肽、炎敌头孢Ⅱ、全能孢) 其它抗炎有机酸:吲哚美辛(荆防败毒散)
2.抗绦虫药:吡喹酮,理想的新型广谱驱绦虫药、抗血吸虫 药和驱吸虫药。毒性极低,应用安全,主要用于动物血吸 虫病,也用于绦虫病和囊尾蚴病。
3.抗吸虫药:三氯苯达唑,对牛羊片形吸虫、前后盘吸虫有 良效,为理想的杀肝片吸虫药。
4.抗球虫药:妥曲珠利、地克珠利、磺胺氯丙嗪钠、磺胺喹 噁啉、磺胺喹噁啉钠
5.抗滴虫病:甲硝唑、地美硝唑
2.抗消化性溃疡药:消化性溃疡的发病与黏膜局部损伤和保护机制之 间的平衡失调有关。损伤因素增强或保护因素减弱,均可引起消化性 溃疡。如碳酸钙,抗酸作用强、快而之久。
3.泻药与止泻药:泻药是能增加肠内水分、促进蠕动、软化粪便或润 滑肠道促进排便的药物。如硫酸镁和硫酸钠。
4.止吐药:如甲氧氯普安。
本公司用于消化道系统的药有:新肠乐、劲腾肠毒清、四季痢康、消炎 增蛋多、劲腾痢百、劲腾痢停、劲腾痢拉克。
药物 作用 的两 重性
治疗作用
对因治疗(治本) 对症治疗(治标) 补充治疗(替代)
不良反应:
(对机体不利 不符用药目的)
副反应 毒性反应 后遗效应 停药反应 变态反应 特异质反应 继发反应
第二节 机体对药物的作用——药动学
(一)概念
药物代谢动力学,简称药动学,研究 药物体内过程及体内药物浓度随时间变化 的规律。
药理基础知识
第一章 总论
1.药物对机体的作用——药效学 2.机体对药物的作用——药动学 3.影响药物作用的因素
药物动力学研究内容及意义

药物动力学药物动力学(pharmacokinetics)亦称药动学,系应用动力学(kinetics)原理与数学模式,定量地描述与概括药物通过各种途径(如静脉注射,静脉滴注,口服给药等)进入体内的吸收Absorption、分布Distribution、代谢Metabolism和消除Elimination,即吸收、分布、代谢、消除(ADME)过程的“量-时”变化或“血药浓度-时”变化的动态规律的一门科学。
药物动力学研究各种体液、组织和排泄物中药物的代谢产物水平与时间关系的过程,并研究为提出解释这些数据的模型所需要的数学关系式。
药物动力学已成为生物药剂学、药理学、毒理学等学科的最主要和最密切的基础,推动着这些学科的蓬勃发展。
它还与基础学科如数学、化学动力学、分析化学也有着紧密的联系。
从它发展较快的近20 年来,其研究成果已经对指导新药设计、优选给药方案、改进药物剂型、提供高效、速效、长效、低毒、低副作用的药剂,发挥了重要作用。
临床意义药物动力学近年来的发展和应用,日益证明了它在药学领域中所占的特殊重要地位。
首先,药物动力学作为一门用数学分析手段来处理药物在体内的动态过程的科学,具有重大的理论价值,是“数学药学”的重要组成部分,它的基本分析方法已经渗放到生物药剂学,临床药剂学,药物治疗学,临床药理学,分子药理学,生物化学,分析化学,药剂学,药理学及毒理学等多种科学领域中,已成为这些学科的最主要和最密切的基础,推动着这些学科的蓬勃发展。
同时,药物动力学还有着析为广泛的实用意义,它的发展将对现有的药物的客观评价、新药的能动设计、改进药物剂型、提供高效、速效、长效、低毒副作用的药剂,特别是对于临床指导合理用药,通过药物动力学特征的研究,要挟临床治疗所需有效血药浓度选择最适剂量,给药周期,负荷剂量的计算,以及连续用药是否会在体内发生蓄积,设计最优给药方案等具有重大的实用价值。
总之,药物动力学已成为一种新的有用的工具,已被广泛地应用于药学领域中和各个学科,成为医药研究人员和广大医药工作者都需要了解和掌握的学科。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药物应用的药动学基础一、单室模型#公式1、2:单剂量静注lnC=-kt+lnC0QIAN:单剂静注是基础,e变对数找ln#公式3、4:单剂量-静滴k0-滴注速度稳态血药浓度(坪浓度、C SS)单室模型静脉注射C-t图QIAN:静滴速度找k0,稳态浓度双S达稳态血药浓度的分数(达坪分数、fss)f ss:t时间体内血药浓度与达稳态血药浓度之比值t=-3.32t1/2lg(1-fss)达到稳态某一分数fss所需要的时间与药物的t1/2成正比,t1/2越短,达到稳态的时间越快;而与滴注速度k0的快慢完全无关补充:n=-3.32lg(1-fss),n为半衰期的个数n=1 →50%n=3.32 →90%n=6.64 →99%n=10 →99.9%#公式5:单剂量-血管外F:吸收系数吸收量占给药剂量的分数单次口服给药后的血药浓度时间曲线达峰时间:t max峰浓度:C maxC-t曲线下面积:AUC二、双室模型混杂参数——A、B:经验常数α:分布速度常数、快处置速度常数β:消除速度常数、慢处置速度常数α、β:描述两个指数项即分布项和消除项的特征QIAN:双室模型AB杂中央消除下标10三、多剂量给药固定剂量、固定给药间隔是临床最常用的给药方案。
这种给药方法,血药浓度和体内药量是波动的。
第二次给药前体内的药物尚未被完全清除,所以后一次给药使体内的药量在前一次的基础上逐渐累积,血药浓度逐渐升高。
随着给药次数的不断增加,血药浓度几乎不再升高,而是恒定在一定的水平上随每次给药作周期性的变化。
血药浓度达到稳态其主要有三个特征性参数,即稳态最大血药浓度、稳态最小血药浓度和稳态血药浓度的波动范围(简称坪幅)。
#公式7-10:多剂量给药(重复给药)QIAN:多剂量需重复,间隔给药找τ值既不是与的算术平均值,也不是其几何平均值,仅代表与之间的某一血药浓度值。
系指血药浓度达到稳态后,在一个给药间隔时间τ内,药-时曲线下面积除以τ得的商。
药物治疗时,总是希望将平均稳态血药浓度调整到安全有效的治疗浓度范围内。
因此,可根据的计算公式,选定τ调整X0,或选定X0调整τ,由此进行给药方案设计。
由设计给药方案的局限性是不能说明血药水平波动的情况。
临床常常需要知道经过多少次给药、经过多少时间(或多少个半衰期)体内的血药浓度才能接近或达到稳态。
因此与前述的静脉滴注相似,要估算达到稳态的程度可引入达坪分数(f ss)的概念。
通过公式推导,通常可以认为给药7个半衰期后,体内的药物浓度约达到稳态的99%。
多剂量给药之蓄积、血药浓度波动①体内药量的蓄积蓄积系数R与k和τ有关τ越小,蓄积程度越大半衰期大,易蓄积②血药浓度波动程度波动度DF多剂量给药后,血药浓度总是在一定的范围内波动。
对于那些安全有效浓度范围很窄的药物,若血药浓度波动太大,则易引起中毒。
药物消除越快,给药频率越慢,波动度越大。
总结:隔室模型这些年我们一直在追的公式QIAN药动学经典公式总结:单剂静注是基础,e变对数找ln 静滴速度找k0,稳态浓度双S血管外需吸收,参数F是关键双室模型AB杂,中央消除下标10 多剂量需重复,间隔给药找τ值A.C maxB.t1/2C.AUCD.MRTE.1.平均稳态血药浓度是『正确答案』E2.平均滞留时间是『正确答案』DA.B.C.C=-kt+C0D.lnC=-kt+lnC0E.1.双室模型静脉注射给药血药浓度-时间关系式的方程为『正确答案』A2.单室模型血管外给药血药浓度-时间关系式的方程为『正确答案』B1.单室静脉滴注给药过程中,稳态血药浓度的计算公式是『正确答案』B2.药物在体内的平均滞留时间的计算公式是『正确答案』A四、非线性药动学(酶、载体参与时出现饱和,速度与浓度不成正比)非线性药动学主要由酶或载体饱和所致,故可采用表示酶动力学过程的米氏方程拟合动力学过程。
C:血药浓度V m:药物体内消除的理论最大速率K m:米氏常数,反映酶或载体系统的催化或转运能力K m不是消除常数,而是酶动力学的一个混合速率常数,是指药物体内的消除速率为V m一半时的血药浓度。
A:关于线性药物动力学的说法,错误的是A.单室模型静脉注射给药,lgC对t作图,得到直线的斜率为负值B.单室模型静脉滴注给药,在滴注开始时可以静注一个负荷剂量,使血药浓度迅速达到或接近稳态浓度C.单室模型口服给药,在血药浓度达峰瞬间,吸收速度等于消除速度D.多剂量给药,血药浓度波动与药物半衰期、给药间隔时间有关E.多剂量给药,相同给药间隔下,半衰期短的药物容易蓄积『正确答案』E五、给药方案设计1.一般原则——安全、有效、经济2.基本要求:除应考虑文献资料提供的有效血药浓度范围外,还应充分考虑血药浓度影响因素,如因年龄、性别、疾病状态、遗传因素等不同导致的患者之间的差异,具体情况具体分析。
3.血药浓度监测,实现个体化给药治疗窗窄的药物,要求血药浓度波动范围控制在最低中毒浓度与最低有效浓度之间; 治疗剂量即表现出非线性药动学特征的药物,给药剂量微小变化可能导致血药浓度的较大差异,甚至产生严重的毒副作用;对于生理活性很强的药物,患者体内个体差异导致血药浓度水平的显著改变,则极易引起严重的不良后果。
4.影响因素①药效学因素:药物对机体的作用——药物的效应和毒性②药动学因素:人体对所用药物制剂的作用——吸收、分布、代谢、排泄③患者自身的生理因素及病理状态④患者的心理因素——对医师的信赖程度、用药的依从性等5.基本步骤①选择最佳给药途径和药物制剂②确定期望的血药浓度③确定必要的药动学参数④计算、确定初步的给药方案⑤试用方案并进行方案调整6.根据药动学参数设计给药时间临床上可根据药物的t1/2来确定适当的给药间隔时间(或每日的给药次数),预计连续用药时血浆药物浓度达到相对稳定的时间及停药后药物从体内消除的时间。
一般情况下,代谢快、排泄快的药物t1/2短,而代谢慢、排泄慢的药物t1/2较长。
多数药物的t1/2通过查阅文献资料可获得。
7.根据药动学参数设计给药剂量①静滴给药,为了在短时间内使血药浓度接近稳态浓度(1)常规静脉滴注以前先静脉注射一个负荷剂量X负荷,使血药浓度能够迅速达到或接近稳态血药浓度Css。
X负荷=C ss·V=k0/k(2)常规静脉滴注以前先快速滴注一个负荷剂量,先以速度k1快速滴注T时间,使血药浓度迅速达到或接近稳态浓度,然后再按常规速度k0滴注。
只要知道药物的消除速率常数k,根据预先设定的k0和T,就能确定快速滴注的速度k1。
k1=k0/(1-e-kT)②口服多剂量给药到达稳态血药浓度需要一定的时间,而稳态血药浓度往往就设置为治疗浓度。
如果按正常方案治疗,可能在治疗初期的很长一段时间内都没有疗效,特别是半衰期较长的药物,不利于临床疗效的迅速发挥。
为了尽快达到有效血药浓度,临床治疗中常常在首次给药(首剂)时加大剂量,使血药浓度在短时间内达到期望值。
此后,以适当的剂量维持血药浓度在稳态范围内。
这种首剂给予的较大剂量称为负荷剂量。
计算可知当τ=t1/2时,给予2倍维持剂量的负荷剂量后即可达到最小稳态浓度,之后再按给药周期给予维持剂量,即可维持血药浓度不低于最小稳态浓度。
A:关于单室静脉滴注给药的错误表述是A.k0是零级滴注速度B.稳态血药浓度C ss与滴注速度k0成正比C.稳态时体内药量或血药浓度恒定不变D.欲滴注达稳态浓度的99%,需滴注3.32个半衰期E.静滴前同时静注一个负荷剂量,可使血药浓度一开始就达稳态『正确答案』D静脉滴注给药方案设计实例体重为75kg的患者用利多卡因治疗心律失常,利多卡因的表观分布容积V=1.7L/kg,消除速率常数k=0.46h-1,希望治疗一开始便达到2μg/ml的治疗浓度,请确定静滴速率及静注的负荷剂量。
解:负荷剂量X0=C0V=2×1.7×75=255(mg)静滴速率k0=C ss kV=2×0.46×1.7×75=117.3(mg/h)六、个体化给药1.治疗指数小,治疗剂量表现出非线性药动学特征——血药浓度波动需在安全范围内2.测定血药浓度,计算参数,制定安全有效方案3.方法:wagner法、一点法、重复一点法4.肾功减退患者药物主要经肾排泄时,肾清除率Cl r与肌酐清除率Cl cr成正比,根据患者肾功,预测Cl、k,调整剂量或τ成年男性Cl cr=(140-年龄)×体重/(72×Scr)成年女性=男性×0.85七、治疗药物监测(TDM)治疗药物监测临床意义①指导临床合理用药、提高治疗水平②确定合并用药的原则③药物过量中毒的诊断④医疗差错或事故的鉴定依据⑤评价患者用药依从性A:治疗药物监测的目的是保证药物治疗的有效性和安全性,在血药浓度、效应关系已经确立的前提下,不需要进行血药浓度监测的有A.治疗指数小,毒性反应大的药物B.一般抗高血压药物C.在体内容易蓄积而发生毒性反应的药物D.合并用药易出现异常反应的药物E.个体差异很大的药物『正确答案』B。