多中心临床试验的分中心小结表

多中心临床试验的分中心小结表
良事件发生情
药物临床试验基本流程(总结)
药物临床试验基本流程 1.临床试验启动阶段 1.1获得药物临床试验批件(有效期3年) 1.1制作研究者手册 理化性质、药理、药化、药效、毒理以及已有的临床研究资料,制作研究者手册。 研究者手册是临床试验开始前的资料汇编。研究者手册的内容一般包括:目录、序言、化学和物理性质、临床前研究、药理学研究、药代动力学研究、毒理学研究、已有临床资料、药品使用信息。 1.2筛选主要研究者 CFDA 网站中浏览临床药理基地名录,筛选医院(一般来说不是整个医院而是 某个科室),然后选择合适的主任级医生; 联系主任医生(看是否有意向,在给予任何资料前,签保密协 议,然后发放可行性问卷,收集可行性问卷,确认完整无误,回复感谢信, 感谢其参与问卷调查) 如果可行,可安排访视,具体考察该基地是否有能力执行临床试验; (电话预约研究者,给予方案和研究者手册,agenda 确认要讨论的具体问题) Agenda:研究人员的分配 方案的可行性 能否入够病人及如何保证其依从性 药品管理 档案管理 研究者职责 主要研究者资质的确认 相关研究人员的资质 硬件设施考察 伦理委员会的访视(承不承认中心伦理、伦理开会频率、送审清单、伦理委员会人员清单) 书写访视报告; 再次拜访,与其讨论方案、试验时间、试验费用等问题; 1.3试验文件准备 会同CRO 、申办者、主要研究者,共同制定临床试验方案、病例报告表、知情同意书样本、原始文件;CRO 制定临床试验中其他用表; 1.4其他研究者筛选 从临床基地名录中选择其他可能的研究者,可根据首研者的推荐以及公司曾经合作的情况进行综合判断; 准备资料:方案、研究者手册、知情同意书样本、病例报告表; 与其谈论方案,要求提供其医院所能提供的病例数、时间进度和经费预算; 选定合适研究者,征得其医院管理部门的同意; 1.5召开多中心临床方案认论会(研究者会议),讨论试验方案、CRF 等 注:是否需要召开研究者会议要看具体情况 1.6取得伦理委员会批件 注:IRB (Independent Review Board )/IEC (Independent Ethics Committee )——IRB 为美国的伦理委员会,IEC/EC 为欧盟的伦理委员会 按照GCP 的要求,所有临床试验必须得到伦理委员会的批准。在实际进行临床试验时,首先必须取得牵头单位伦理委员会的批准,其他参加单位是否要过伦理根据各家单位的具体要求而定。 伦理委员会将对有关批件、药检报告、研究者手册、知情同意书样本、试验方案(protocal )、病例报告表进行审批; 1.7试验药品准备 督促申办方进行试验用药品的送检; 生物统计师设计随机分组方案; 根据随机分组方案,设计药品标签; 设计应急信件; 药品包装:为每一个受试者准备好一盒药物,药盒上贴好标签,并装入相应的应 先拜访研究者还是医院相关部门视具体情况而定,没有统一标准 1.7及1.8可在研究者会议后开始准备,同时申请伦理委员会批件
临床试验总结报告的体例格式和内容要求内容
临床试验总结报告体例和容要求 1.题目封面 封面题页应包括如下容: ——试验题目 ——试验药物/研究产品的名称 ——试验用药品的适应症 ——如不能在题目中表明、则简要描述(以1~2句话)设计(平行、交叉、双盲、随机)比较(安慰剂、活性成分、剂量/反应),间隔,剂量和病例数。 ——申办者 ——原始规划与计划的确认(编码或数目,确认日期) ——试验预期进度与进程安排 ——研究开始数据(受试的第一位病人,或任何其它数据) ——后期研究终末数据(最终一个受试者的名称、或终末研究者完成的数据) ——试验研究完整的数据(最后的受试者) ——参加者或合作者或主办者责任医务官等人士的名称和职责。 ——药业公司名称/主办方签名(在公司/主办者中负责研究报告的人,公司/主办方联络者的,和传真,以备在回顾研究报告提出问题时,在此页码或应用的字母有所表明)。 ——表明该项研究是否在优良临床试验管理规(GCP)条件下进行的包括应达到的基本文件、研究设施、人员知识结构,统一培训等。 ——报告的资料(通过题目和资料,确定与该项研究相同的任何其它较早报告) 2.容概述 应提供—个临床试验的主要容提要(通常限于3页容),该提要应包括试验题目、研究人员、研究出版物名称、试验时间、试验目的、试验方法、试验研究样本、诊断及进入研究的主要标准、试验的产品、剂量、给药方式及批号、治疗持续时间、参考的治疗、剂量、给药方式及批号、评价标准(有效性、安全性)、统计方法、总结——结论(效能结论、安全性结论、结论)及报告日期。该提要应包括表明结果的数字资料,而不仅仅是文字和P值(见附件l,研究容概述)。 3.容目录 容表格应包括: ——页码或其它每一个部分的局部资料,包括归纳性表格、图和图表。 ——附录、列表和任何能提供的病例报告形式的汇总和定位。 4.名词、术语缩写和定义 应提供报告中使用的—系列缩写符、特殊的或不常用的术语,或测量单位定义,应拼写出缩写术语,并在文中首次出现时表明其全称。
配送中心上情况小结
配送中心2018年上情况小结 增强全员安全意识。不断深入开展安全文化建设,四是抓宣传教育。加强警示教育。形成人人重视安全、关注安全、防范事故的良好氛围。 中心紧紧围绕相关文件指示精神,2018年上半年的工作当中。主要做到一下工作: 做到四个到位”不断强化目标管理抓安全一完善机制。 为进一步提升配送中心安全管理水平,1组织机构到位。今年上半年。建立了配送中心主任为安全生产的一级管理者,分管安全管理工作的副主任为安全生产的二级管理者,各部门负责人为安全生产的三级管理者,基层员工为安全生产的四级管理员的安全管理网络,完善安全管理机构。 并将该项工作和他各项工作同规划、同布置、同落实、同考核。认真落实《安全生产法》湖北省消防条例》等相关法律法规。上半年来,2安全意识到位。配送中心始终高度重视安全工作。召开了多次以抓安全稳定工作为主题的办公会,贯彻落实国家局、省局电视电话会议精神,对各项工作进行具体的研究和部署,为全面确保安全生产奠定了良好的思想基础。 年初,4考核兑现到位。为进一步完善安全目标考核机制。针对不同职能部门和各岗位的安全职责,突出针对性、约束性和可操作性,修订完善了配送中心各部门、各岗位安全工作考核细则,并将其列入考核与评比中予以兑现。基本形成了月度有检查、有考核,年度有评比、有表彰机制,做到安全目标考核的系统化。开展“三项活动”不断加强安全教育培训促安全二精心组织。 特别是把安全工作作为优秀车组评选的重要指标。通过树立典型,1积极开展优
秀车组评选活动。坚持把优秀车组评选活动作为一项基础性工作来抓。表彰先进,充分调动各车组的安全争先意识。根据《配送中心优秀车组评选办法》每季度开展一次优秀车组评选活动,截至目前,累计表彰人次,取得了较好效果。 指定专人及时向市局上报整治工作情况;二是严格落实,2积极开展安全专项整治活动。一是广泛动员、统一思想。把安全专项整治行动作为一项重要工作抓实、抓好。同时。齐抓共管。建立各部门联动机制,落实中高度事故隐患督办制度;三是全面总结,及时上报。对专项整治工作中发现的问题,进行全面整改,并工作过程进行全面总结,确保行动取得实效。 召开专题会议落实活动要求,3积极开展安全生产月活动。一是统一部署。保证“安全生产月”正常开展。二是精心组织、广泛开展宣传教育活动。组织全体干部职工学习《湖北省消防条例》充分利用led显示屏,广泛开展《安全生产月》宣传,提高员工的安全防范意识。三是认真组织检查,注重隐患整改。以开展“安全生产月”活动为契机,对车辆、办公区、仓库等重点部位进行一次安全大检查。抓好“两项建设”不断夯实安全管理基础防安全三立足实际。 以安全促发展”安全文化理念,1抓好安全文化建设。根据省局“以文化促安全。单位积极探索安全文化建设,主要抓了以下几个方面:一是积极推行7s现场管理。通过运用科学的管理方法,促使全员把安全工作从小处做起,从自我做起;二是积极推行安全目视化管理。上半年,单位制作“有电危险”库区禁止吸烟”您已进入监控区域”等安全警示牌块,营造了实时的全覆盖的安全警示网络;三是抓好安全动态宣传。大院安装了led显示屏,每周为全体员工发送安全提示短信,使全体员工随时都能了解各类安全动态和天气信息,让安全宣传“动”起来。四是编写了安全教育短片,让安全警示教育“活”起来,起到良好效果。
临床试验项目总结报告分中心小结表盖章申请表
临床试验项目总结报告/分中心小结表盖章申请表 申请人上交申请表及总结报告后7个工作日联系机构办公室取回,联系人孟老师
27557209
倾听,岁月吹奏长笛,叮咚流韵,悠悠靡音。午夜乘鸾云游,虔念飘洋过海,痴缠驻守天籁,心灵驿站舒馨。 静看月色,锦瑟未央,娇羞情窦初开,醉饮一阙诗经蒹葭。蝶舞翩翩,漫渡凌云,不胜凌波。踏波无浪,携手聚集灵气,惠风庭前守望。 月光盈照,心池微醉,诗意芳华,缠绕穹空,悠然蜗居栖息。一弯柔情似水,秋水无痕。素风掠过心苑,犹如怡景,纤尘不染竹篱。 心事平静安享,唏嘘红尘踏歌,恬淡悠远,听一天马行空。信鸽滑翔,风翅逍遥,音律穿越,坐拥不老青春。芳菲西楼浣韵,丝织金色笑语,笛箫琴瑟,植于心谷漾溢,盎然灵仙之气。 风舞天涯极光,月下袅娜,醉醒守望蔓坨。心路溪谷,一簇芬芳妖娆,跫音,捕捉千里之外。杳杳讯息,铺展羁旅,心境驾舟,红尘泅渡,渡口歌者,匆匆驶过天籁。 盈盈月盘,金枝玉叶为媒,凌波旅途迢迢,轻摇浅阶相遇。杯盏,斟下一弯月镰,拂去忧愁烦恼,与云雾朦胧醉卧,与影对饮,翠幽。 独坐春夜,静观夏夜,旖旎秋夜,缱绻冬夜,夜夜,坐拥与你,衣袂飘飘。醉花荫处,一笺风语痴迷,沉香,沁脾。那一弯春溪泉涌,吟唱过往清风,心情澄澈释放,思绪执手填词。聆听青春嫣然。 梦中一道霓虹,化为亘古琴音。相约午夜,缱绻风景,旖旎浪漫。扯下一苑芳草,萋萋蔓绿,感怀飘然轮回。登程人生,若即若离,云水禅心,未央别具一格。 岁月如斯,了然于胸,与你秉烛执手,问情生命。编辑记忆,跋涉心灵轨迹,如梦鼾声,柔感情怀。走出尘世喧嚣,缘结地老天荒。待,小荷风情初窦,与天地之生灵,吟咏。 卷壁苍夜,浅笑一弯碧月。谱一阙红尘神曲,载渡。寄语晨时星光,盈照旭日,小鸟伊人,浮光霓裳,晓风过隙,情泻千里,凌霄悄然寄挂。 袅袅炊烟,一宛悠然,馨暖徜徉,花前月下徘徊,天涯无痕。阡陌碧波,幸福瑶池,花枝摇曳,奏一曲天籁吟韵。诗书充盈,短笛横吹,歌赋旖旎,坐拥,永恒岁月…… 倾听,岁月吹奏长笛,叮咚流韵,悠悠靡音。午夜乘鸾云游,虔念飘洋过海,痴缠驻守天籁,心灵驿站舒馨。 静看月色,锦瑟未央,娇羞情窦初开,醉饮一阙诗经蒹葭。蝶舞翩翩,漫渡凌云,不胜凌波。踏波无浪,携手聚集灵气,惠风庭前守望。 月光盈照,心池微醉,诗意芳华,缠绕穹空,悠然蜗居栖息。一弯柔情似水,秋水无痕。素风掠过心苑,犹如怡景,纤尘不染竹篱。 心事平静安享,唏嘘红尘踏歌,恬淡悠远,听一天马行空。信鸽滑翔,风翅逍遥,音律穿越,坐拥不老青春。芳菲西楼浣韵,丝织金色笑语,笛箫琴瑟,植于心谷漾溢,盎然灵仙之气。 风舞天涯极光,月下袅娜,醉醒守望蔓坨。心路溪谷,一簇芬芳妖娆,跫音,捕捉千里之外。杳杳讯息,铺展羁旅,心境驾舟,红尘泅渡,渡口歌者,匆匆驶过天籁。 盈盈月盘,金枝玉叶为媒,凌波旅途迢迢,轻摇浅阶相遇。杯盏,斟下一弯月镰,拂去忧愁烦恼,与云雾朦胧醉卧,与影对饮,翠幽。 独坐春夜,静观夏夜,旖旎秋夜,缱绻冬夜,夜夜,坐拥与你,衣袂飘飘。醉花荫处,一笺风语痴迷,沉香,沁脾。那一弯春溪泉涌,吟唱过往清风,心情澄澈释放,思绪执手填词。聆听青春嫣然。
临床试验总结报告的设计与撰写
一、临床试验方案设计 临床试验方案由研究者或申办者拟订,应符合GCP要求。研究者和申办者均应在已制定的临床试验方案上签名并签署日期。 临床试验设计的基本原则:①代表性:受试者样本符合总体规律;②重复:结果经得起重复验证;③随机:受试者随机分配入组;④对照与盲法:避免条件误差与主观因素。 试验方案的格式包括:①封页:包括题目、申办者和临床试验机构的名称与地址,拟订日期;②正文:GCP要求的23项;③封底:各参与的临床试验机构与主要研究者、申办者的名称与联系方式;④主要参考文献。 临床试验方案设计主要内容(GCP第四章第十七条)有以下23条: (一)试验题目: (二)试验目的,试验背景,临床前研究中有临床意义的发现和与该试验有关得临床试验结果、已知对人体的可能危险与受益,及试验药物存在人种差异的可能; (三)申办者的名称和地址,进行试验的场所,研究者的姓名、资格和地址; (四)试验设计的类型,随机化分组方法及设盲的水平; (五)受试者的入选标准,排除标准和剔除标准,选择受试者的步骤,受试者分配的方法; (六)根据统计学原理计算要达到试验预期目的所需的病例数; (七)试验用药品的剂型、剂量、给药途径、给药方法、给药次数、疗程和有关合并用药的规定,以及对包装和标签的说明; (八)拟进行临床和实验室检查的项目、测定的次数和药代动力学分析等; (九)试验用药品的登记与使用记录、递送、分发方式及储存条件; (十)临床观察、随访和保证受试者依从性的措施; (十一)中止临床试验的标准,结束临床试验的规定; (十二)疗效评定标准,包括评定参数的方法、观察时间、记录和分析; (十三)受试者的编码、随机数字表及病例报告表的保存手续; (十四)不良事件的记录要求和严重不良事件的报告方法、处理措施、随访的方式、时间和转归; (十五)试验用药品编码的建立和保存,揭盲方法和紧急情况下破盲的规定; (十六)统计分析计划,统计分析数据集的定义和选择; (十七)数据管理和数据可溯源性的规定; (十八)临床试验的质量控制和质量保证; (十九)试验相关的论理学; (二十)临床试验预期的进度和完成日期;
多中心临床试验的各中心小结表
药物临床试验结题申请表项目名称 研究药物名称 临床试验批件号 本中心伦理委员会批件号 负责专业 研究时间年月——年月 试验计划入组 受试者数 筛选人数 入组人数完成试验人数 SAE发生有□无□发生SAE的药物编号 本中心药研经费编 号(经费核查用) 附经费说明 提交资料清单 (附表1) 有□无□总结报告/分中心小结有□无□ 药物管理情况药物管理员签字: 日期: 项目监查情况监查员签字:CRO 名称: 日期: 专业/项目质控情况质控员签字: 日期: 项目负责人审核意见签字:日期: 专业负责人审核意见签字:日期: 伦理委员会意见签字:日期: 机构质控意见签字:日期:
附表1 药物临床试验存档资料清单 项目名称: 申办单位: 负责科室: 移交人签名: 接受部门:药研机构办公室 联系电话: 接收人签名: 移交日期: 接受日期: 序号 移交文件 有/无 序号 移交文件 有/无 1 临床试验申请表 22 监查报告 2 临床试验备案表 23 研究者致申办者、药品监督管理局、 3 国家食品药品监督管理局批件 伦理委员会的严重不良事件报告 4 企业资质 24 已签名的知情同意书 5 试验药物的药检证明 25 原始医疗文件 6 药物标签 26 病例报告表(已填写、签名、注明日期) 7 说明书样稿 27 试验药物登记表 8 研究者手册 28 试验药物回收表 9 试验方案及其修正案(已签名) 29 试验药物销毁证明 10 病例报告表、研究病例、日记卡(样表) 30 生物样品留存记录 11 知情同意书(样稿) 31 揭盲记录(组长单位) 12 伦理委员会批件 32 质控检查记录 13 协议书或合同书 33 统计报告 14 研究者履历及相关文件 34 分中心小结 15 临床试验有关的实验室检测正常值范围 35 总结报告(组长单位:签名、盖章) 16 医学或实验室操作的质控证明 36 17 各种文件更新版本 37 18 启动培训记录 38 19 受试者筛选表与入选表 39 20 受试者鉴认代码表 40 21 试验药物与试验相关物资的接收记录 41
药物临床试验总结报告的设计规范
药物临床试验总结报告的设计规范 版本号 1.0 页数11页 起草人起草日期年月日审核人审核日期年月日批准人批准日期年月日颁布日期年月日起效日期年月日 威海市立医院 药物临床试验机构
药物临床试验总结报告的设计规范 一目的 为真实地反应反映该试验的实施过程和客观地表述药物的有效性和安全性及其应用价值。 二范围 所有由本院牵头或协助参加的新药临床试验。 三内容 1报告封面 参照国家食品药品监督管理总局有关药品注册申报资料的形式要求。 2签名页 1)报告题目 2)执笔者签名。 3)主要研究者对研究试验报告的声明。 申明已阅读了该报告,确认该报告准确描述了试验过程和结果。 4)主要研究者签名和日期。(包括统计学工作者) 3报告目录 每个章节、附件、附表的页码。 4缩略语 正文中首次出现的缩略语应规范拼写,并在括号内注明中文全称。应以列表形式提供在报告中所使用的缩略语、特殊或不常用的术语定义或度量单位。 5伦理学问题 1)确认试验实施符合赫尔辛基宣言及伦理学原则。 2)伦理委员会组成及批准临床试验方案情况说明,并在附件中提供独立伦理委员会成 员表。 3)描述如何及何时获得与受试者入选相关的知情同意书,并在附件中提供原稿。 6报告摘要 报告摘要应当简洁、清晰的说明以下要点,通常不超过1500字。 1)试验题目。 2)临床批件文号。 3)主要研究者和临床试验单位。 4)试验的起止日期(第一例受试者第一次访视至最后一名受试者最后一次访视日期)。
5)试验目的及观察指标。 6)对研究药物的作用类别和主治功能的描述。 7)对试验设计作简短描述,包括试验设计类型(平行、交叉、成组序贯等)、设盲水 平(双盲、单盲或开放)、随机分组方法、对照的形式(安慰剂、阳性药对照、剂 量对照)、疗程。 8)试验人群。 9)给药方案(包括对照组) 10)评价标准(有效性和安全性评价指标)。 11)统计分析方法或模型(包括基线评价、组间比较、协变量分析、综合比较等)。 12)受试者入组情况及各组人口学资料。 13)各组疗效结果(主要和次要疗效指标)。 14)各组安全性结果(不良事件及严重不良事件)。 15)结论(有效性和安全性结论)。 7报告正文 1)前言 一般包括:受试药品研究背景;研究单位和研究者;目标适应症和受试人群、 治疗措施;受试者样本量;试验的起止日期;国家食品药品监督管理局批准临床试验的文号;制定试验方案时所遵循的原则、设计依据;申报者与临床研究单位之间有关特定试验的协议或会议等予以说明或描述。简要说明临床试验经过及结果。 2)试验目的 应提供对特定试验目的的陈述(包括主要、次要目的)。注意具体说明本项试验的受试因素、受试对象、研究效应,明确试验要回答的主要问题,明确药品的临床定位。 3)试验方法 (1)试验设计 a)总体研究设计和计划的描述(包括临床试验的流程图)。如试验过程中方案有 修正,应说明原因、更改内容及依据。 b)对试验总体设计的依据、合理性进行讨论,具体内容应视设计特点进行有针对 性的阐述。如采用单盲或开放试验设计,应说明理由。 c)提供样本含量的具体计算方法、计算过程以及计算过程中所用到的统计量的估 计值及其来源依据。 d)描述期中分析计划。
最新药物临床试验工作总结-(1)
药物临床试验机构工作总结 根据江苏省三级综合医院医疗技术水平标准(2017版)和江苏省省级临床重点专科评分标准的相关要求,我院自2017年1月份启动创建国家药物临床试验机构。依照《药物临床试验质量管理规范》(2003版)的标准有序开展各项工作。以质量控制为抓手,注重内涵建设;加强机构办管理人员及各专业科室相关人员的培训;强化临床试验相关的伦理审查;规范伦理委员会的管理;大大提高了临床试验质量和管理水平。现对2017年的工作总结如下: 一、建立健全组织管理机构 我院自2017年1月份启动国家药物临床试验机构创建工作,3月份召开了创建动员大会。先后成立了国家药物临床试验工作领导小组、组织管理机构、机构伦理委员会和机构工作实施方案。机构办公室配置机构药房,资料档案室、质控小组。根据各临床专业的综合实力及科研技术水平遴选了17个临床专业研究团队。 二、制定规章制度、标准操作规程 机构办公室严格按照《药物临床试验质量管理规范》(2003版)
和《药物临床试验机构资格认定标准》的具体要求,起草制定了药物临床试验管理制度20条、各类人员职责14条、药物临床试验标准操作规程89条、药物临床试验设计规范5条并汇编成书100本,汇编印发应知应会手册300册。 三、强化药物临床试验培训,保证试验过程规范,结果科学可靠 积极组织专业人员参加院、内外GCP及相关技术规范培训:2017年的3-6月份有关院领导带领办公室成员先后到省、市级具有国家药物临床试验机构资质的6家医院调研参观学习;4-5月份组织两次38人次参加的江苏省药物临床试验法规、技术及实务操作和伦理审查技术高级培训班;7月份邀请有关药物临床试验知名专家,在我院举办由中国药理学会药物临床试验专业委员会主办的国家级药物临床试验质量管理规范培训班,约240人参加。目前共有129人次取得了国家级、省级药物临床试验培训证书。在创建期间组织院内培训8次约300人次,涉及内容包括对17个临床专业科室的研究者、质量管理员、药品管理员的专题培训及药物临床试验安全性评价与不良事件处理、进行心肺复苏、呼吸机、除颤仪等设备使用的SOP进行培训。四、 五、完成试验设施设备的配备 2017年8月份完成了机构办公室、伦理委员会办公室及各申报专
临床试验总结报告的体例格式和内容要求
临床试验总结报告体例和内容要求 1.题目封面 封面题页应包括如下内容: ——试验题目 ——试验药物/研究产品的名称 ——试验用药品的适应症 ——如不能在题目中表明、则简要描述(以1~2句话)设计(平行、交叉、双盲、随机)比较(安慰剂、活性成分、剂量/反应),间隔,剂量和病例数。 ——申办者姓名 ——原始规划与计划的确认(编码或数目,确认日期) ——试验预期进度与进程安排 ——研究开始数据(受试的第一位病人,或任何其它数据) ——后期研究终末数据(最终一个受试者的名称、或终末研究者完成的数据) ——试验研究完整的数据(最后的受试者) ——参加者或合作者或主办者责任医务官等人士的名称和职责。 ——药业公司名称/主办方签名(在公司/主办者中负责研究报告的人,公司/主办方联络者的姓名,电话和传真号码,以备在回顾研究报告提出问题时,在此页码或应用的字母有所表明)。 ——表明该项研究是否在优良临床试验管理规范(GCP)条件下进行的包括应达到的基本文件、研究设施、人员知识结构,统一培训等。 ——报告的资料(通过题目和资料,确定与该项研究相同的任何其它较早报告) 2.内容概述 应提供—个临床试验的主要内容提要(通常限于3页内容),该提要应包括试验题目、研究人员、研究出版物名称、试验时间、试验目的、试验方法、试验研究样本、诊断及进入研究的主要标准、试验的产品、剂量、给药方式及批号、治疗持续时间、参考的治疗、剂量、给药方式及批号、评价标准(有效性、安全性)、统计方法、总结——结论(效能结论、安全性结论、结论)及报告日期。该提要应包括表明结果的数字资料,而不仅仅是文字和P 值(见附件l,研究内容概述)。 3.内容目录 内容表格应包括: ——页码或其它每一个部分的局部资料,包括归纳性表格、图和图表。 ——附录、列表和任何能提供的病例报告形式的汇总和定位。 4.名词、术语缩写和定义 应提供报告中使用的—系列缩写符、特殊的或不常用的术语,或测量单位定义,应拼写
临床试验分中心小结表的设计规范
临床试验分中心小结表的设计规范 版本号 1.0 页数4页 起草人起草日期年月日审核人审核日期年月日批准人批准日期年月日颁布日期年月日起效日期年月日 威海市立医院 药物临床试验机构
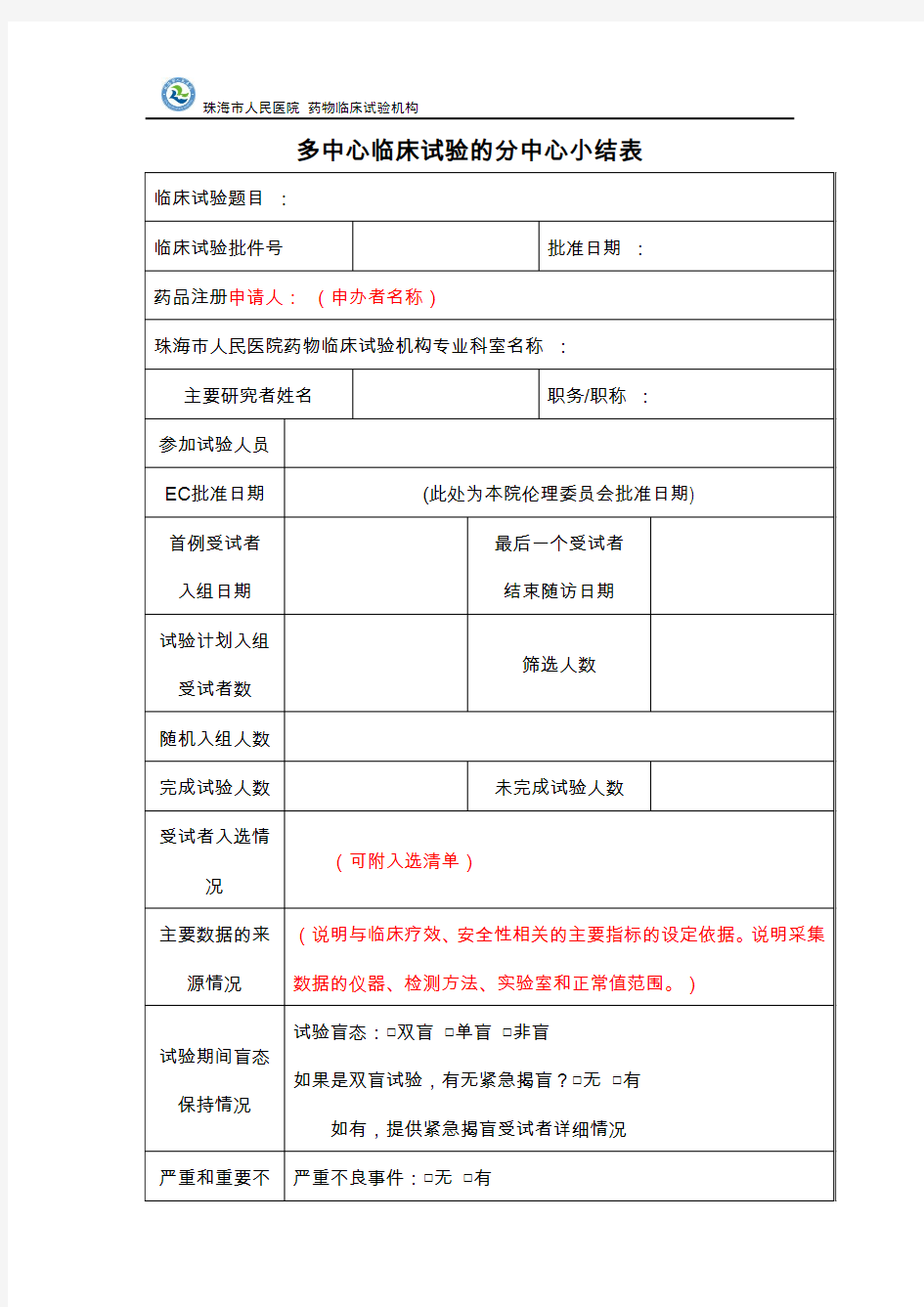
多中心临床试验的分中心小结表 临床试验题目临床试验题目应明确临床试验的分期和项目临床试验批件号批准日期 药品注册申请人 临床试验机构及专业名称 本中心试验负责人姓名职务/职称 参加试验人员(可提供附表)参加试验人员姓名、职称、所在科室、研究中分工等信息 参见附表1 伦理委员会名称伦理委员会批准日期 第一个受试者入组日期最后一个受试者结束随访日期 试验计划入组受试者数筛选人数入组(随机化)人数完成试验人数未完成试验人数 受试者入选情况一览表(可提供附表)需提供所有签署知情同意书的受试者编号(或姓名缩写)、知情同意日期、筛选失败原因、入组日期、药物编号、未完成试验者的中止原因与日期。详见:附表2 《受试者筛选/入选情况表》 主要数据的来源情况说明与临床疗效、安全性相关的主要指标的设定依据。 说明采集数据的仪器、检测方法、实验室和正常值范围。 附表3 《实验室采集数据仪器、检测方法正常值范围一览表》 试验期间盲态保持情况试验盲态:□双盲□单盲□非盲 如果是双盲试验,有无紧急揭盲?□无□有如有,提供紧急揭盲受试者详细情况 严重和重要不良事件 发生情况严重不良事件:□无□有 重要不良事件:□无□有 如有,提供发生严重和重要不良事件受试者情况及与试验药物的关系判断。 临床研究监查情况委派临床试验监查员单位:□申请人□CRO 监查次数:次监查质量评价: 主要研究者的评论 本中心主要研究者对本项临床试验的质量控制和试验情况作出评论,并对试验结果的真实性作出声明。 本中心主要研究者签名:日期: 本中心临床试验机构 管理部门审核意见盖章:日期:
临床试验观察表讲解
阿美宁病例报告表 (Case Report Form) 受试者姓名: 家庭地址: 联系电话: 试验中心名称: 申办单位:江苏豪森药业股份有限公司 在正式填表前请认真阅读下列填表说明 病例报告表填写说明: 1.筛选合格者填写病例报告表。 2.病例报告表填写务必准确、清晰,不得任意涂改,错误之处纠正时需用横线居中化出,并签署修改者姓名缩写及修改时间。 3.填写记录一律用钢笔或碳素签字笔。 4.患者姓名拼音缩写四个需填满,两字姓名填写两字拼音前两个字母;三字姓名填写三字首字母及第三字第二字母;四字姓名填写每个字的首字母。 5.表中凡有“□”的项,请在符合的条目上划“√”。表格中所有栏目均应填写相应的文字或数字,不得留空。 6.所有检验项目因故未查或漏查,请填写ND;具体合并用药剂量和时间不明,请先写NK。 7.试验期间应如实填写合并用药记录表、不良事件记录表。记录不良事件的发生时间、严重程度、持续时间、采取的措施和转归。如有严重不良事件发生(包括临床验证过程中发生需住院治疗、延长住院时间、伤残、影响工作能力危及生命或死亡、导致先天畸形等事件),必须立即通知主要研究单位伦理委员会及申办单位。
8.临床试验应严格按照临床试验方案要求进行。试验不同时期需完成的检查和需记录的项目,请对照临床流程图执行。
受试者知情同意书 敬爱的患者: 我们现在正在进行一项临床研究,该项临床研究是经吉林省药品监督管理局备案的,研究的目的是评价物理治疗的高科技产品----温热电位治疗仪的疗效和安全性。 温热磁疗仪是XXX医疗器械有限公司根据韩国专利技术研制生产的医疗器械,本治疗仪是通过XXXXXXXXXXXXXXXXXXXXXXXXXXXXXX机理,主要用于XXXXXXXXXXXXX治疗。具有平衡机体阴阳,增强脏腑机能,促进新陈代谢,调节植物神经,改善心脑血管血液供应,促进血液循环,调节血管张力,降低血液粘稠度,防治动脉粥样硬化,促进组织的再生修复功能,增强机体免疫功能及消炎止痛等作用。其注册标准已经XX省食品药品监督管理局备案,其技术指标经XX省医疗器械检验所检测合格。现拟进行临床验证。 在临床观察过程中,请您在可能的前提下积极配合临床医生按方案要求进行临床试验观察。本仪器是安全的,无任何风险和副作用的,医生和使用单位将会尽力防止由于本试验可能带来的伤害,如果产生由于本次试验因素给您带来任何不良影响,您将会得到及时、必要的治疗,所需要的费用由生产该仪器的企业承担,并将会按有关规定得到相应的补偿。 绝大多数患者能从本研究中获益。您将有权在任何时间询问有关本研究的任何问题。您参加本研究完全是自愿的,您将有权决定在任何时间退出本研究,将不会再任何方面影响医生对您的治疗,您的权利将得到充分的保障。医生和研究单位将尽力防止由于本研究可能带来的伤害。 本研究的所有资料将是保密的,有关您的个人资料不会出现在总结报告或发表文献中。伦理委员会将公正此项研究是安全和合乎道德的,并在赫尔辛基宣言指导下进行。 志愿受试者声明: 作为受试者,我已了解以上情况,同意参加本研究,按照临床试验方案的要求,按时用药,按时复诊,及时报告出现的不良反应。我有权随时退出该项试验。但在无特殊情况下,尽可能完整的接受本次临床试验研究。 受试者签字:研究者签字: 日期:日期:
临床试验总结报告的撰写.doc
临床试验总结报告的撰写 定义:是反映药物临床研究设计、实施过程,并对试验结果 作出分析、评价的总结性文件,是正确评价药物是否 具有临床实用价值(有效性和安全性)的重要依据, 是药品注册所需的重要技术资料。 基本准则:真实、完整地描述事实 科学、准确地分析数据 客观、全面地评价结局 结构与内容:药品名称:资料项目编号: 33- Ⅱ ****II期临床试验研究报——以 *** 为对照药评价 *** 治疗 *** 安全性有效性的分层区组 随机、双盲双模拟、平行对照、多中心临床研究 研究机构名称: *** (负责单位)(盖章) ***(参加单位)(盖章)研究机构地址及电话:** 省** 市******** 主要研究者:***主任医师(签名): 试验起止日期: **** 年** 月- **** 年** 月 原始资料保存地点: *** 医院 联系人姓名: *** 联系人电话: **** 申报单位: *** (盖章) 报告签名 报告题目: 主要研究者声明及签名 我已详细阅读了该报告,该报告客观、准确描述了 试验过程和结果。 ***医院 ***医师(签名):年月日 研究负责人签名 ***医院 ***医师(签名):年月日
统计分析负责人签名 ***医院 ***医师(签名):年月日 申办者声明及签名 我们对该临床试验的全过程进行了监查,试验按临床试方案进行,我们已详细阅读了该报告,该报告客观、准确描述了试验过程和结果。 ***公司 负责人: *** (签名):年月日 监查员: *** (签名):年月日 执笔者签名 ***医院 ***医师(签名):年月日 报告目录 缩略语 论理学声明 报告摘要 试验目的 试验方法 讨论 结论 参考文件 附件 缩略语 缩写中文全称英文全称 ALT 丙氨酸氨基转换酶alannine transaminase RBC 红细胞red blood cell WBC 白细胞white blood cell N 中性粒细胞neutrophilic granulocyte L 淋巴细胞lymphocyte PLT 血小板blood platelet
I期临床试验总结报告样本
SFDA临床试验批准号:2002XXXXXX号 XXX注射液 Ⅰ期临床人体耐受性试验总结报告 试验单位:XXXXXXXX医院 试验负责:XXX 试验设计:XXX,XXXX 试验日期:2003年X月X日至X月X日 申办单位:XXXXX有限公司 (以下仅提供报告格式,其数据和文字均为虚构,如有巧合,纯属偶然;数据前后有矛盾之处,也请原谅) XXX注射Ⅰ期临床人体耐受性试验总结报告 摘要 单次给药的耐受性试验:30名健康受试者,根据体重和性别随机分配到7个剂量组(组1、组2、组3、组4、组5、组6、组7)。组1、组2每组2人;组3、组4、组5每组6人;组6、组7每组4人。参照费氏递增法(改良Fibonacci法)递增给药,各组分别静脉滴注XXX注射液5、10、20、30、40、50、60ml,滴速20-30滴/min。每个受试者只接受一个相应的剂量,从小剂量开始,每个剂量应用后未见不良反应,才可用下
一个剂量。观察给药后健康人体对XXX注射液的反应和耐受性。 多次(累积)给药的耐受性试验:12名健康受试者根据体重和性别随机分配到甲、乙两个剂量组,每组6人。分别静脉滴注XXX注射液20、10 ml,滴速20-30滴/min,连续5d。 结论:(1)每次按10 ml/次给药,有可能引血胆红素轻度增高或尿胆原阳性,但受试者无明显不适,因此推荐II期临床试验为10 ml/次;15 ml/次有可能引起血胆红素轻度增高或尿胆原阳性,但风险不会太大,可在严密监护下尝试。20 ml/次虽然引起的不良反应尚属轻度,但发生的频度较高,慎用。(2)10 ml/次就有可能引起轻度不良反应,主要为血胆红素轻度增高或尿胆原阳性,但受试者无自觉症状。20 ml/次可引起的不良反应有所增加。这种血胆红素轻度增高或尿胆原阳性,且两次重复结果相近,推测是药物引起的轻度溶血所致。其他主要不良反应为口干(2例)、头晕(1例)、皮疹(1例)。 根据国家药品监督管理局XXX号批文的要求,按照《新药审批办法》,《药品临床试验管理规范》(GCP),《中药新药临床研究的技术要求》,以及《中药新药临床研究指导原则》和XXX的化学组成、功能主治、药效学、毒理学研究资料,于2003年7月17日~2003年9月20日,对XXX有限公司申报的XXX注射液中药二类新药,进行Ⅰ期临床人体耐受性试验,总结报告如下。 1 研究目的 选择健康人为受试者,从安全的初始剂量开始,观察人体对XXX注射液的耐受性,为制定本品的Ⅱ期临床试验给药方案提供依据。 2 临床资料与研究方法 病例选择 入选标准 1.健康志愿者。 2.年龄在18~50岁,男女各半。 3.体重在标准体重的±10%范围内[标准体重(kg)=×(身高cm-80)]。 4.无烟酒嗜好。 5.心、肝、肾、血液等检查指标均在正常范围。 6.知情同意,志愿受试。 排除标准 1.妊娠期、哺乳期妇女。 2.具有原发性心、肝、肾、血液学疾病者。 3.精神或法律上的残疾患者。 4.有酒精、药物滥用病史。 5.过敏体质(对两种或两种以上药物或食物、花粉过敏史者)。 6.3个月内参加过其他药物临床试验者。 7.正在应用其他预防和治疗的药物者。 8.研究者认为不能入组的其他原因。 剔除标准 对已被选入本临床研究,属于以下情况之一者,作为剔除病例。 1.不符合纳入标准,或符合排除标准者
医疗器械临床试验报告范本
附件5 医疗器械临床试验报告本报告编号: ×××临床试验报告 试验用医疗器械名称: 型号规格: 试验用医疗器械的管理类别: 需进行临床试验审批的第三类医疗器械是□否□ 中国境同类产品有□无□ 临床试验机构: 临床试验开始时间: 临床试验结束时间: 方案编号: 方案版本号和日期: 研究者: 申办者: 代理人: 监查员:
年月日 填写说明: 1.临床试验机构和研究者应当本着认真负责的态度,严格按照临床试验方案实施临床试验,公正、客观地编制临床试验报告。 2.临床试验机构和研究者应当对试验报告的真实性和科学性负责。 3.本报告应当由研究者签名和注明日期,临床试验机构临床试验管理部门出具意见、签章和注明日期。
一、一般信息 二、摘要 三、简介 简单介绍试验用医疗器械的相关研发背景(例如原因、目的、目标人群、治疗、时间、主要终点等)。 四、临床试验目的 五、临床试验方法 六、临床试验容 七、临床一般资料 (一)试验围(病种) (二)病例的选择 1.入选标准 2. 排除标准 (三)样本量的计算 (四)病例数 入组情况 八、试验用医疗器械和对照用医疗器械/对照诊疗方法 (一)试验用医疗器械 (二)对照用医疗器械/对照诊疗方法 九、所采用的统计分析方法及评价方法 (一)统计分析方法 1.分析人群
2.统计分析方法 (二)统计评价方法 1.有效性终点 2.安全性终点 (三)缺失值和异常值的处理 十、临床评价标准 (一)有效性评价标准 1.主要指标 2.次要指标 (二)安全性评价标准 1.主要指标 2.次要指标 十一、临床试验的组织结构 十二、伦理情况说明 十三、临床试验结果 十四、临床试验中发现的不良事件及其处理情况 (一)不良事件定义 (二)不良事件严重程度判定 (三)不良事件与试验用医疗器械及操作关系的判定 (四)严重不良事件定义 (五)本试验发现的不良事件、可能导致严重不良事件的器械缺陷
产前诊断分中心总结
*******院 三年产前诊断分中心 工作总结 *******院是我市唯一一家担负着妇幼保健、医疗、科研、教学、计划生育、健康教育和信息管理的三级妇幼保健机构,是全市22个县(市)、区妇幼保健机构的技术指导中心和业务管理机构,负责全市的产前筛查、产前诊断的信息统计,有完整的妇幼保健三级监测网。产前诊断技术作为出生缺陷的二级预防措施,在出生缺陷防治工作中起着重要作用。我院又是石家庄市产前筛查中心及新生儿疾病筛查诊治中心,这样就形成了产前筛查与产前诊断有机结合的技术服务网络和质量控制体系,可大大提高此项工作的顺利进行。我院还注重保健宣传工作,利用出生缺陷防治宣传日举行大型义诊活动,举办第五届全国产房内危重新生儿急救学习班暨第七届石家庄市围产医学年会,为孕前、孕期妇女普及如何做到优生优育,宣传如何预防出生缺陷和残疾儿出生的科学知识,让广大的育龄妇女了解我国出生缺陷的现况,宣传了我国实施的以婚前保健、产前诊断、新生儿疾病筛查为主的三级综合性防治策略,让群众自觉做到优生优育,提高我国的人口素质。 为了提高产前诊断技术水平,我院每年都会派出相关专业技术人员参加国家及省卫生厅举办的产前诊断技术培训班,并派出实验室人员分别到美国、湖南、广州和河北省级医院产前诊断中心进修,这样都为提高我院产前诊断技术提供了良好的技术基础。产前诊断中心自
2014年省卫计委进行周期校验后,三年来的工作情况总结如下: 一、组织管理 我院产前诊断分中心经河北省卫生厅批准于2008年10月成立,医院成立了产前诊断分中心组织机构,由主管副院长莫中福担任组长,主管副院长吴荣芹、主管副院长苏学艳担任副组长的领导小组,下设产前诊断办公室、产前诊断质控组及产前诊断技术服务组。产前诊断办公室设在医务科;产前诊断技术服务组由我院围产保健、妇科、产科、新生儿科、功能科、遗传咨询科等科室联合组成;产前诊断质控组由我院质控办、医务科、护理部、感控科、产前诊断门诊、遗传咨询科、功能科等相关科室组成。产前诊断分中心设有遗传咨询门诊、细胞遗传诊断室、产前筛查超声室、细胞遗传诊断室、生化免疫遗传诊断室、分子遗传诊断室、产前诊断资料室等,开展遗传咨询、细胞遗传、分子遗传、产前筛查、产前诊断及新生儿疾病筛查、诊断和治疗、43种遗传代谢病筛查、产前及新生儿耳聋基因检测、叶酸代谢能力基因检测等,真正起到预防三级出生缺陷的目的。 二、人员配置 我院产前诊断分中心拥有一支紧密协作、技术精湛、态度严谨的技术骨干组成的团队: 1、产前诊断门诊占地约100平米,现有医护人员5人,其中高级职称3人,中级职称2人,均接受过全国及河北省卫计委组织的产前诊断相关培训,并同时取得大型医疗设备上岗证,一名副主任护师负责病案管理及随访、统计工作,一名主管护师负责羊膜腔穿刺手术
药物临床试验机构筹建工作总结及整改措施
姓名:XXX 部门: XX部YOUR LOGO Your company name 2 0 X X 药物临床试验机构筹建工作总结 及整改措施
药物临床试验机构筹建工作总结及整改措施 XXXX年XX月XX日国家药监局专家专家对我院药物临床试验机构xx专业资格认定进行了现场检查,对我院的工作给予充分的肯定,并给出了综合评定意见,认为我院:专业技术力量较强,医疗设备齐全,专业病原病种充足,能满足药物临床试验的需要,医院领导重视药物临床试验机构的建设,成立了管理机构并制定了相关的管理制度、设计规范和SOP,机构人员接受过不同层次的GCP和药物临床试验技术培训,基本具备开展药物临床试验的条件。 但SFDA药品认证管理中心专家组也指出了检查中我们尚存在的一些问题。针对这些问题,医院领导组织有关人员进行了认真的讨论和研究,并做出如下整改: 1. 机构选派部分研究和管理人员到XX临床试验基地进行进修学习,学习结束后,重新制定和完善了部分管理制度、技术设计规范和SOP,使其符合本专业的特点及GCP要求并具有可操作性。 2. 机构和专业分别选派具有相应的专业技术职称,参见过GCP 培训,并有参加过药物临床试验的经历并掌握临床试验相关技术的及相关法律的人员,设专业质控员和机构办公室质控员,分别从专业和实验全过程对药物临床实验进行监督检查,实现并强化药物临床实验的三级质控,完善药物临床试验质量保证体系与制度,确保我院承担的国家药物临床试验过程规范,结果真实可靠,保证受试者和申办单位的合法权益。 3. 选派机构主要研究和管理人员参加了国家级的GCP培训并取得了培训证书。同时在院内继续进行GCP知识和试验技术知识培训,并从认定工作结束后开始定期派机构管理人员对GCP知识和试验技 术知识掌握情况进行抽查和督促,使所有试验相关人员熟悉GCP知识 第2 页共4 页
客户服务分中心工作总结
客户服务分中心2012年工作总结 来 w .5 K https://www.360docs.net/doc/b911849325.html, 3 客户服务分中心2012年工作总结 2012年中心针对新的管理的模式,不断调整和适应新的营销形势,引导职工在端正思想和规范工作思路的同时,树立信心,在工作管理和经营管理方面取得新的进步。一年来,中心各项经营指标在全体员工不懈的努力下都得到了完成。 下面从几个方面阐述今年的工作。 一、经营工作 1-10份经营指标: 售电量:22167.17万kwh(其中有损,12622.02,无损,4280.87,工业园,5264.28);线损率:4.49%;售电均价:有损634.02元/kkwh,无损713.05元/kkwh。 (一)、电费回收 电费的回收是落实经营目标的终极目的,保证当月电费的回收到位是衡量我们工作的重要的砝码。当月未应收电费全部结零。
(二)、增供扩容 保持锐志进取,努力拓展电力营销业务,多渠道、全方位服务电力市场,是增供扩销的必由之策;2012年中心的供、售电量较之以往增大许多。当年新增专变用户60户,容量KVA。 (三)、线损 线损是保障各项经济指标完成的重要参数,设备、人员管理制度上的完善措施又是保障和控制这一重要参数得以实现的重要手段。2012年填补亏空后综合线损率顺利完成公司下达的年度指标,8.1%。 (四)、用电普查与反窃电管理 用电普查的目的是理顺和掌握辖区内各类用电户的情况,并及时纠正用电不规范行为和规范用电秩序。定期或不定期对辖区内不同的用电性质的单位和个体分类巡视检查。今年结合公司布置的自查自纠和低压集抄工作,对辖区内所有低压进行了普查,近40000户。对专用用户的计量装置全面清理和核实,建立真实、准确的资料档案。这次按照公司要求,再次对所有专变终端进行清理登记。 (五)、工程资料及基础资料 低压集抄全覆盖,中心是第一个实施的供电单位。因此,基础资料的翔实准确是保证该系统初始化的重要依据。低压配变台区图的绘制和修正工作在本月底无条件执行完成。分管主任和营业班专责负责监督工程全过程。截至目前为止,已完成39000户,266个台区。(六)、优质服务与行风建设
临床试验中期监查报告样本
临床试验终期监查报告 监查目的:(标记所有需要监查的项目) □全面监查 □法规卷宗(Ⅰ、Ⅱ) □知情同意(Ⅲ、Ⅳ) □方案依从性和病例报告表(Ⅴ、Ⅵ、Ⅶ) □药品资料(Ⅳ、Ⅶ) □研究条件(Ⅸ、Ⅹ) □其他 是否不适合未访视一、研究卷宗/法规资料监查 (在研究卷宗中是否有如下资料) 1 附有签名的方案和方案修正案□□□□ 2 伦理委员会通过的知情同意书样本□□□□ 3 所有病例报告表样本□□□□ 4 研究者手册□□□□
5 研究人员履历/资格说明□□□□ 6 研究人员相关培训文件□□□□ 7 伦理委员会通过的研究相关资料□□□□ 8 实验室资格及正常值范围□□□□ 二、中心监察是否不适合未访视 1 所有需要的文件是否都有□□□□ 2 对开展临床试验至关重要的设施是否都正常 □□□□运转 3 研究者和其工作人员保留着和开展试验相关 □□□□的记录 三、知情同意/入选情况是否不适合未访视 1 已签署的原始知情同意书都在受试者档案里□□□□ 2 是否有筛选日志□□□□ 3 是否有入选日志□□□□ 四、研究药物是否不适合未访视 1 试验药物是否保存在安全的地方□□□□ 2 试验药物是否由专人分发□□□□ 3 药物运送记录及时准确并有日期及签名□□□□ 4 药物分发和数量记录是否及时准确□□□□ 5 药物是否按照方案分发□□□□ 五、方案依从性是否不适合未访视 1 筛选过程正确□□□□ 2 所有受试者都符合入选/排除标准□□□□ 3 不良事件是否正确报告,记录,评价和解决□□□□ 4 受试者到诊及过程是否按照方案规定顺序完成□□□□
5 是否按照方案给予研究干预□□□□ 6 是否有严重不良事件(SAEs)□□□□ 六、病例报告表(CRFs)和源文件是否不适合未访视 1 能否提供CRFs和相应的源文件查阅□□□□ 2 源文件是否足以供CRFs核查□□□□ 3 CRFs是否完整,清晰,准确□□□□ 4 CRFs是否及时完成和递交(如数据录入)□□□□ 七、违背方案是否不适合未访视 1 以前没有注意到的违背方案之处,监查是否 □□□□发现以及正确记录 八、中心实验室规程是否不适合未访视 1 样本是否按照方案收集和保存□□□□ 2 医务人员是否对实验室数据的临床意义作出 □□□□合适的评价和记录 九、研究设施/人员招募情况是否不适合未访视 1 研究中心的设施是否还合适□□□□ 2 工作人员是否还合适□□□□ 3 目前受试者入选率是否符合要求□□□□ 十、中心现状是否不适合未访视 1 中心是否有可以影响开展临床试验的变化□□□□