98-79-EC体外诊断医疗器械指令
欧盟美国对体外诊断试剂的管理

第 一 类: 质 量 管 理 体 系 认 证 ISO 13485 第 二 类: 产 品 认 证
CE Mark
欧盟CE指令
Active implantable medical devices Medical devices (93/42/EEC)
(90/385/EEC)
In vitro diagnostic medical devices (98/79/EC)
quality system
Self-testing/professional use
自测 Self-testing examination performed by a layperson to evaluate an individual's health status NOTE 1 Typically performed in a home or other environment outside a healthcare institution without supervision by a healthcare professional.
体取得的样品、包括血液及组织供体的,无论单独使用 或是组合使用的人和医疗器械,包括试剂、试剂产品、 校准材料、控制材料、成套工具、仪表、装置、设备或 系统,其唯一或主要目的是提供以下信息:
- --
- --
有关生理学或病理学状态;或 有关先天性异常;或 用于确定安全性以及与可能接受治疗者的相容性;或 用于检查治疗措施
按照ISO13485& 98/79/EC附录4.3 运行质量体系QMS
认证机构(NB)进行审核 Auditing 符合性声明 DOC
CE XXXX
List A/清单A器械
欧盟指令法规及程序

N/A
附录 III
EC 符合性声明(制造商自我 声明)
N/A
公告机构是否介入 YES YES YES YES YES
YES NO
器械风险递增
6
注册和授权代表
98/79/EC 规定欧洲数据库的其中一个职责 是主管当局之间共享关键数据,但目前该 项目仍在进行中。一旦该项目执行,则制 造商只需在其所在国或在其授权代表 (AR) 所在国注册即可。目前,临时取代措施是 要求制造商在初始注册后,通知其他成员 国的主管当局。制造商如果确定了潜在的 目标市场,则需研究该国的具体实施要 求,以确定是否需要通报相关部门。
• “体外诊断医疗器械可以是试剂、试剂 产品、校准物、控制物、试剂盒、器 具、仪器、设备或系统等任何形式的医 疗器械,无论是单独使用或组合使用, 由制造商预期用于体外检测取自人体的 样本,包括血液和其他组织,且其主要 目的是提供如下信息:生理或病理状 态、或有关于先天性异常、或确定潜在 接受者的安全性和兼容性、或监测治疗 措施。”
对于 LIST A 中风险程度最高的器械,需要 有额外的措施来把控风险。在附录 IV 全面 质量保证的符合性评价程序下,每个 LIST A 的器械必须经过详细的技术文件评审, 即 EC 设计审查。无论是参照哪项程序, 在获得 CE 标志后,任一批 LIST A 的产 品在交付市场前,还需进行持续的逐批验 证,以确保器械性能的持续稳定。
98-79-EEC 协调标准列表——体外诊断试剂协调标准列表
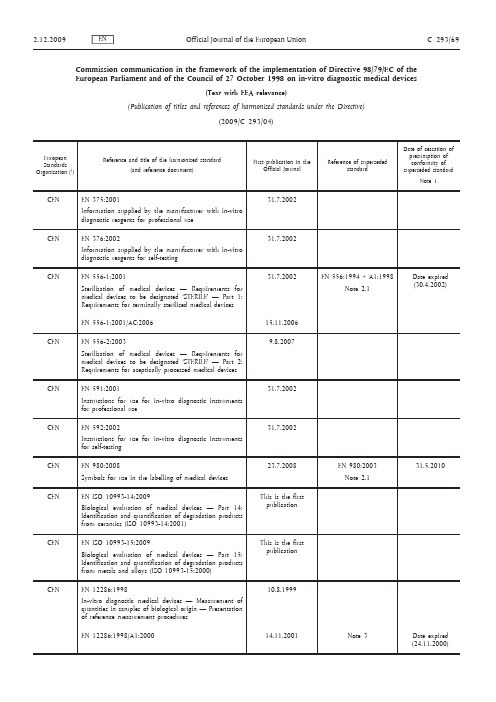
Commission communication in the framework of the implementation of Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in-vitro diagnostic medical devices(Text with EEA relevance)(Publication of titles and references of harmonised standards under the Directive)(2009/C 293/04)Cenelec: Avenue Marnix 17, 1000 Brussels, BELGIUM, tel. +32 25196871, fax: +32 25196919 (http://www.cenelec.eu).ETSI: 650, route des Lucioles, 06921 Sophia Antipolis, FRANCE, tel. +33 492944200, fax: +33 493654716 (http://www.etsi.eu).Note 1: Generally the date of cessation of presumption of conformity will be the date of withdrawal (dow), set by the European Standardisation Organisation, but attention of users of thesestandards is drawn to the fact that in certain exceptional cases this can be otherwise.Note 2.1: The new (or amended) standard has the same scope as the superseded standard. On the date stated, the superseded standard ceases to give presumption of conformity with the essentialrequirements of the Directive.Note 2.2: The new standard has a broader scope than the superseded standard. On the date stated the superseded standard ceases to give presumption of conformity with the essential requirements ofthe Directive.Note 2.3: The new standard has a narrower scope than the superseded standard. On the date stated the (partially) superseded standard ceases to give presumption of conformity with the essentialrequirements of the Directive for those products that fall within the scope of the newstandard. Presumption of conformity with the essential requirements of the Directive forproducts that still fall within the scope of the (partially) superseded standard, but that do notfall within the scope of the new standard, is unaffected.Note 3: In case of amendments, the referenced standard is EN CCCCC:YYYY, its previous amendments, if any, and the new, quoted amendment. The superseded standard (column 3) therefore consists ofEN CCCCC:YYYY and its previous amendments, if any, but without the new quoted amendment.On the date stated, the superseded standard ceases to give presumption of conformity with theessential requirements of the Directive.NOTE:— Any information concerning the availability of the standards can be obtained either from the European Standardisation Organisations or from the national standardisation bodies of which the list is annexed to the Directive 98/34/EC of the European Parliament and of the Council (1) amended by the Directive 98/48/EC (2).— Publication of the references in the Official Journal of the European Union does not imply that the standards are available in all the Community languages.— This list replaces all the previous lists published in the Official Journal of the European Union. The Commission ensures the updating of this list.— More information about harmonised standards on the Internet at http://ec.europa.eu/enterprise/ newapproach/standardization/harmstds/(1) OJ L 204, 21.7.1998, p. 37.(2) OJ L 217, 5.8.1998, p. 18.。
98-79-EC体外诊断医疗器械指令(中文版)(1998版)

欧洲议会和欧洲联盟理事会关于体外诊断医疗器械的98/79/EC指令1998年10月27日欧洲议会和欧洲联盟理事会考虑到建立欧洲经济共同体的条约,特别是该条约第100a条;考虑到欧洲联盟委员会提交的议案;考虑到经济与社会委员会的意见;依照《欧洲经济共同体条约》第189b条规定的程序采取行动;鉴于为顺利建立欧洲经济共同体内部市场应采取必要的措施;鉴于内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于成员国有关体外诊断医疗器械的安全性、健康保护和实施特性及核准程序的现行法律、法规和行政条款在内容和适用范围上是不同的;鉴于这些差异的存在形成了贸易壁垒;鉴于一项以欧洲联盟委员会的名义对各国立法所作的对比调查证明需要建立协调规则;鉴于对各国立法进行协调是消除此类自由贸易壁垒以及防止产生新的壁垒的唯一手段;鉴于这一目标无法由各成员国单独通过其他途径圆满地实现;鉴于本指令只规定为确保其适用的体外诊断医疗器械在最安全的条件下自由流通所需的此类要求;鉴于协调条款必须与成员国为管理直接或间接同此类器械有关的公共卫生和医疗保险计划资金筹措而采取的措施相区别;鉴于协调条款只要符合欧洲共同体法律就不影响成员国实施这些措施的能力;鉴于体外诊断医疗器械应向患者、使用者及第三方提供高水平的健康保护并达到制造商原先赋予它们的性能水准;鉴于因此,维持或提高各成员国所达到的健康保护水平是本指令的主要目的之一;鉴于按照理事会1985年5月7日关于技术协调与标准新方法的决议中所确定的原则,对有关产品的设计、制造和包装所制定的规则必须限于满足基本要求所必需的条款;鉴于因为这些要求是基本的,因此它们应当取代各国相应的条款;鉴于考虑到在设计时的技术和实际情况以及与高水平的健康保护与安全性相适应的技术、经济因素,在确定基本要求时,包括确定最大限度降低和减小危险的要求时,应当谨慎;鉴于大部分医疗器械已在理事会1990年6月20日关于使有源植入式医疗器械的法律趋于一致的90/385/EEC指令和理事会1993年6月14日关于体外医疗诊断器械除外的医疗器械的93/42/EEC指令中所涉及;鉴于本指令试图将协调扩展到体外诊断医疗器械,鉴于为了统一共同体规则,本指令在很大程度上是以上述两个指令的条款为基础的;鉴于预定用于研究而没有任何医疗目的的仪表、装置、器具、材料或其他物品,包括软件,均不被视为应进行性能评定的器械;鉴于虽然本指令并不适用于经过国际上认证的标准物质和用于外部质量评定计划的物质,但使用者用来确定或验证器械性能所需的校准和控制材料属于体外诊断医疗器械;鉴于考虑到辅助性原则,对于在卫生机构实验室内生产的并在该环境内使用的试剂以及不是以商业交易为目的的试剂,不属于本指令适用范围;鉴于,尽管如此,制造后预定用在专业和商业场合作医疗分析而非供出售的器械必须执行本指令条款;鉴于专门为体外诊断检查设计的机械实验室设备属于本指令适用范围,以及鉴于为了协调有关指令,对于欧洲议会及欧洲联盟理事会1998年6月22日关于使成员国有关机械设备的法律趋于一致的98/37/EC指令应作适当修改,以使其与本指令保持一致;鉴于本指令应当包括有关发射电离辐射的器械的设计和制造方面的要求;鉴于本指令并不影响欧洲联盟理事会1996年5月17日有关对保护工人和公众的健康免遭电离辐射危险的基本安全标准做出规定的96/29/Euratom指令得到实施;鉴于电磁兼容性问题已构成本指令基本要求的一个组成部分,因而理事会1989年5月2日关于使各成员国有关电磁兼容性的法律趋于一致的89/336/EEC指令不适用;鉴于为了有助于证明符合基本要求并能给予验证,需要协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于此类协调标准是由私法机构制定的,因此它们应当保持其非强制性文本的地位;鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)已根据它们与欧洲联盟委员会之间于1984年11月13日签署的合作总指导原则被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲联盟委员会的委托,由CEN或CENELEC或者由这两个机构共同按照1998年6月22日欧洲议会与欧洲联盟理事会关于在技术标准和法规领域提供信息程序的98/34/EC指令,依据上述总指导原则而批准的技术规范(欧洲协调文件标准);鉴于作为总原则的例外,制定通用技术规范时考虑了某些成员国目前的作法,在选择主要用于评定供血和器官捐赠的安全器械方面,应由公共当局来批准这类规范;鉴于这些特定规范应当用共同的技术规范来取代是适宜的;鉴于这些共同的技术规范可用于性能评定和再评定;鉴于来自各有关方面的科学专家可能参与制定通用技术规范和分析其他特殊或一般的问题;鉴于本指令所适用的产品制造还包括医疗器械的包装,因为此种包装与本指令的安全和性能方面有关;鉴于某些器械由于其性能随时间下降造成使用寿命有限,而这种性能下降是与它们的物理或化学特性,包括消毒和包装的完整性有关;鉴于制造商应当确定和注明器械将按预定用途运行的期限;鉴于标签上应当注明器械或其某个组成部分能够完全安全使用的有效日期;鉴于理事会在1993年7月22日关于用于技术协调指令的不同阶段合格评定程序模式及加贴和使用CE合格标志规则的93/465/EEC决定中,规定了协调的合格评定程序;鉴于对体外诊断医疗器械需要进行验证并且要求它们符合90/385/EEC指令及93/42/EEC指令,从而证明对这些模式增加细节是合理的;鉴于主要从合格评定程序考虑,有必要将体外诊断医疗器械分为两类主要产品;鉴于大部分此类器械对患者不会构成直接的危险,并且它们是由经过培训的专业人员使用的,而且获得的结果通常能采用其他手段予以确认,所以合格评定程序一般可由制造商单独负责进行;鉴于考虑到各成员国现有的法规及通报是按照98/34/EC指令规定的程序收到的,因此只有对那些在医疗工作中规定必须正确运行,并且其故障会对健康造成严重损害的器械才要求指定机构进行干预;鉴于在要求指定机构对其进行干预的体外诊断医疗器械中,用于输血和预防爱滋病及某些类型肝炎的批量产品从其设计制造目的出发,要求通过合格评定来保证最佳的安全性和可靠性水平;鉴于考虑到健康保护领域的技术进步和发展,要求不断更新需经第三方合格评定的体外诊断医疗器械清单;鉴于必须按照理事会1987年7月13日批准的关于委员会实施权程序的87/373/EEC决定中规定的程序Ⅲ(a)采取更新措施;鉴于欧洲议会、欧洲联盟理事会和欧洲联盟委员会已于1994年12月20日就按照《欧洲共同体条约》第189b条中规定的程序通过的法案实施措施达成了临时协议;鉴于医疗器械通常应加贴CE标志,表明其符合本指令的条款,能够在欧洲共同体内自由流通并按其预定用途投入使用;鉴于当要求指定机构干预时,制造商将能够从欧洲联盟委员会公布的机构名单中自行选择;鉴于虽然成员国没有义务确定此类指定机构,但它们必须保证被批准的指定机构遵守本指令规定的评定准则;鉴于指定机构的负责人和工作人员不得本人或通过中间人从接受评定和验证的企业获得任何利益,从而有损其独立性;鉴于负责市场监督的主管当局应能与制造商或其在欧洲共同体内的授权代表取得联系,以便采取必要的保护措施,尤其是在紧急情况下;鉴于为了统一实施本指令,特别是在市场监督方面,成员国之间进行信息合作和交流是必不可少的;鉴于为此有必要建立并管理一个数据库,这个数据库应包括有关制造商及其授权代表,投放市场的器械、证书的发放、中止或吊销,以及预警程序等方面的数据;鉴于建立一个有害事故报告(预警程序)体系是对市场监督,包括新器械性能进行监督的有效方法;鉴于从预警程序以及外部质量评定方案获得的信息有利于对器械分类做出决定;鉴于制造商必须向主管当局通报投放市场的“新产品”所用的技术和分析的物质或其他参数等情况;鉴于这种做法特别适用于基因甄别所使用的高密度DNA(脱氧核糖核酸)探查器械(通常称之为微片技术);鉴于当某个成员国认为,对于某个规定的产品或一批产品,为了保护健康和安全和/或保证履行对公众健康的责任,必须按照《欧洲共同体条约》第36条禁止或限制获得该产品或者对其规定特殊条件,该成员国可以采取任何必要的和合理的过渡措施;鉴于在此类情况下,欧洲联盟委员会可同有关方面和成员国磋商,如果各国的措施被证明是合理的,则应按照87/373/EEC决定中规定的程序Ⅲa 通过必要的欧洲共同体措施;鉴于本指令涉及到使用人体的组织、细胞或物质制造的体外诊断医疗器械;鉴于它并不涉及使用人体的物质制造的其他医疗器械;鉴于为了尽快地制定欧洲共同体法律,这方面的工作将继续进行下去;鉴于考虑到在采集和使用来自人体的物质的过程中必需保护人的完整性,实施欧洲联盟理事会公约中关于生物学和医学应用方面保护人权和人的尊严所规定的原则是合适的;鉴于各国有关伦理学的法规继续适用;鉴于为了使医疗器械指令保持一致,对本指令的某些条款需要作相应的修改;鉴于必须尽快就使用人体物质制造的医疗器械进行立法;兹通过本指令:第1条适用范围、定义1.本指令适用于体外诊断医疗器械及其附件。
临床检验的量值溯源问题

临床检验的量值溯源问题一、引言近年来,临床检验的量值溯源问题在国际上受到广泛重视。
欧洲议会和理事会1998年10月签署一项将于2003年12月生效的关于体外诊断器具的指令(Directive 98/79/EC)[1],该指令的一项关键内容是要求体外诊断器具的校准物质和/或质控物质定值的溯源性必须通过已有的高一级的参考方法和/或参考物质予以保证。
欧洲指令是法律文件,生效后有关各方必须执行。
为配合该欧洲指令的实施,国际标准化组织(ISO)于1999年起草了5个相关标准,其中与生产厂家关系比较密切的是ISO/DIS 17511“校准物质和质控物质定值的计量学溯源性”[2]和ISO/DIS 18153“酶催化浓度校准物质和质控物质定值的计量学溯源性”[3]。
以上指令和标准主要针对诊断试剂的生产。
对临床实验室检验来说,作为国际实验室认可依据的ISO/IEC 17025 “检测和校准实验室能力的通用要求”[4](我国国家标准GB/T 15481-2000和国家实验室认可委员会CNACL 201-2001“实验室认可准则”等同采用ISO/IEC 17025)和ISO/FDIS 15189 “医学实验室质量管理”[5]也都对临床检验结果的溯源性作出明确要求。
鉴于量值的溯源性将可能成为体外诊断试剂生产和使用中的重要质量指标,而我国试剂生产者和临床检验工作者对此计量学概念可能还不太熟悉,本文介绍临床检验量值溯源的基本概念、现状及有关问题。
二、量值的溯源性及溯源链的结构和工作原理ISO对溯源性的定义如下:测量结果或标准量值的属性,它使测量结果或标准量值通过连续的比较链与给定的参考标准联系起来,给定的参考标准通常是国家或国际标准,比较链中的每一步比较都有给定的不确定度。
不确定度是另一个计量学术语,ISO对它的定义为:与测量结果相关的参数,代表可能可合理地赋予被测量的值的分散性。
不确定度评定有A、B两类,A类评定基于测量结果,B类则基于经验或其它信息的概率分布, A类和B类合成为标准不确定度。
(CE认证用)欧盟体外诊断医疗器械指令 IVDD Directive 98_79_EC (2009年最新修订版-包括了所有的修订)

This document is meant purely as a documentation tool and the institutions do not assume any liability for its contents►B DIRECTIVE98/79/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCILof27October1998on in vitro diagnostic medical devices(OJ L331,7.12.1998,p.1)Amended by:Official JournalNo page dateL284131.10.2003►M1Regulation(EC)No1882/2003of the European Parliament and of theCouncil of29September2003L1881418.7.2009►M2Regulation(EC)No596/2009of the European Parliament and of theCouncil of18June2009Corrected by:►C1Corrigendum,OJ L22,29.1.1999,p.75(98/79/EC)►C2Corrigendum,OJ L6,10.1.2002,p.70(98/79/EC)DIRECTIVE98/79/EC OF THE EUROPEAN PARLIAMENTAND OF THE COUNCILof27October1998on in vitro diagnostic medical devicesTHE EUROPEAN PARLIAMENT AND THE COUNCIL OF THE EUROPEAN UNION,Having regard to the Treaty establishing the European Community,and in particular Article100a thereof,Having regard to the proposal from the Commission(1),Having regard to the opinion of the Economic and Social Committee(2),Acting in accordance with the procedure laid down in Article189b of the Treaty(3),(1)Whereas measures should be adopted for the smooth operation ofthe internal market;whereas the internal market is an area without internal frontiers in which the free movement of goods, persons,services and capital is ensured;(2)Whereas the content and scope of the laws,regulations andadministrative provisions in force in the Member States with regard to the safety,health protection and performance,charac-teristics and authorisation procedures for in vitro diagnostic medical devices are different;whereas the existence of such disparities creates barriers to trade,and whereas the need to establish harmonised rules has been confirmed by a comparative survey of national legislations carried out on behalf of the Commission;(3)Whereas the harmonisation of national legislation is the onlymeans of removing such barriers to free trade and of preventing new barriers from arising;whereas this objective cannot be achieved in a satisfactory manner by other means by the indi-vidual Member States;whereas this Directive lays down only such requirements as are necessary and sufficient to ensure, under the best safety conditions,free movement of the in vitro diagnostic medical devices to which it applies;(4)Whereas the harmonised provisions must be distinguished frommeasures adopted by the Member States to manage the funding of public health and sickness insurance schemes relating directly or indirectly to such devices;whereas,therefore,the harmonised provisions do not affect the ability of the Member States to implement such measures provided that they comply with Community law;(5)Whereas in vitro diagnostic medical devices should providepatients,users and third parties with a high level of health protection and attain the performance levels originally attributed to them by the manufacturer;whereas,therefore,maintenance or improvement of the level of health protection attained in the Member States is one of the main objectives of this Directive;(6)Whereas,in accordance with the principles set out in the Councilresolution of7May1985on a new approach to technical harmo-nisation and standards(4),rules regarding the design,manufacture(1)OJ C172,7.7.1995,p.21and OJ C87,18.3.1997,p.9.(2)OJ C18,22.1.1996,p.12.(3)Opinion of the European Parliament of12March1996(OJ C96,1.4.1996,p.31),Council common position of23March1998(OJ C178,10.6.1998, p.7)and Decision of the European Parliament of18June1998(OJ C210,6.7.1998).Council Decision of5October1998.(4)OJ C136,4.6.1985,p.1.and packaging of relevant products must be confined to the provisions required to meet the essential requirements;whereas, because they are essential,such requirements should replace the corresponding national provisions;whereas the essential requirements,including requirements to minimise and reduce risks,should be applied with discretion,taking into account the technology and practice at the time of design and technical and economic considerations compatible with a high level of protection of health and safety;(7)Whereas the major part of medical devices are covered by CouncilDirective90/385/EEC of20June1990on the approximation of laws relating to active implantable medical devices(1)and Council Directive93/42/EEC of14June1993concerning medical devices(2)with the exclusion of in vitro diagnostic medical devices;whereas this Directive seeks to extend the harmo-nisation to in vitro diagnostic medical devices and whereas,in the interest of uniform Community rules,this Directive is based largely on the provisions of the said two Directives;(8)Whereas instruments,apparatus,appliances,materials or otherarticles,including software,which are intended to be used for research purposes,without any medical objective,are not regarded as devices for performance evaluation;(9)Whereas,although internationally certified reference materialsand materials used for external quality assessment schemes are not covered by this Directive,calibrators and control materials needed by the user to establish or verify performances of devices are in vitro diagnostic medical devices;(10)Whereas,having regard to the principle of subsidiarity,reagentswhich are produced within health-institution laboratories for use in that environment and are not subject to commercialtransactions are not covered by this Directive;(11)Whereas,however,devices that are manufactured and intended tobe used in a professional and commercial context for purposes of medical analysis without being marketed are subject to this Directive;(12)Whereas mechanical laboratory equipment especially designed forin vitro diagnostic examinations falls within the scope of this Directive and whereas,therefore,in order to harmonise the relevant directives,Directive98/37/EC of the European Parliament and of the Council of22June1998on the approx-imation of the laws of the Member States relating to machinery(3),should be appropriately amended to bring it into line with this Directive;(13)Whereas this Directive should include requirements regarding thedesign and manufacture of devices emitting ionizing radiation;whereas this Directive does not affect the application of Council Directive96/29/Euratom of13May1996laying down basic safety standards for the protection of the health of workers and the general public against the dangers arising from ionising radiation(4); (14)Whereas,since electromagnetic compatibility aspects form anintegral part of the essential requirements of this Directive, Council Directive89/336/EEC of2May1989on the approxi-mation of the laws of the Member States relating to electro-magnetic compatibility(5)does not apply;(1)OJ L189,20.7.1990,p.17.Directive as last amended by Directive93/68/EEC(OJ L220,30.8.1993,p.1).(2)OJ L169,12.7.1993,p.1.(3)OJ L207,23.7.1998,p.1.(4)OJ L159,29.6.1996,p.1.(5)OJ L139,23.5.1989,p.19.Directive as last amended by Directive93/68/EEC(OJ L220,30.8.1993,p.1).(15)Whereas,in order to ease the task of proving conformity with theessential requirements and to enable conformity to be verified,it is desirable to have harmonised standards in respect of the prevention of risks associated with the design,manufacture and packaging of medical devices;whereas such harmonised standards are drawn up by private-law bodies and should retain their status as non-mandatory texts;whereas,to this end,the European Committee for Standardisation(CEN)and the European Committee for Electrotechnical Standardisation (Cenelec)are recognised as the competent bodies for the adoption ofharmonised standards in accordance with the general guidelines on cooperation between the Commission and those two bodies signed on13November1984;(16)Whereas,for the purpose of this Directive,a harmonised standardis a technical specification(European standard of harmonisation document)adopted,on a mandate from the Commission,by CEN or Cenelec or by both of those bodies in accordance with Directive98/34/EC of the European Parliament and of the Council of22June1998laying down a procedure for the provision of information in the field of technical standards and regulations(1),and pursuant to the abovementioned general guidelines;(17)Whereas,by way of exception to the general principles,thedrawing up of common technical specifications takes account of a current practice in some Member States whereby for selected devices mainly used for the evaluation of the safety of blood supply and of organ donation,such specifications are adopted by the public authorities;whereas it is appropriate that these particular specifications should be replaced by common technical specifications;whereas these common technical specifi-cations can be used for performance evaluation and reevaluation;(18)Whereas scientific experts from various interested parties couldbe involved in the drafting of common technical specifications and in the examination of other specific or general questions; (19)Whereas manufacturing,as covered by this Directive,alsoincludes the packaging of the medical device,insofar as such packaging is related to the safety and performance aspects of this device;(20)Whereas certain devices have a limited life owing to the declinein their performance over time,which is related,for example,to the deterioriation in their physical or chemical properties, including the sterility or integrity of the packaging;whereas the manufacturer should determine and indicate the period during which the device will perform as intended;whereas the labelling should indicate the date until which the device or one of its components can be used with complete safety;(21)Whereas,in Decision93/465/EEC of22July1993concerningthe modules for the various phases of the conformity assessment procedures and the rules for the affixing and use of the CE conformity marking,which are intended to be used in the technical harmonisation directives(2),the Council laid down harmonised conformity assessment procedures;whereas the details added to these modules are justified by the nature of the verification required for in vitro diagnostic medical devices and by the need for consistency with Directives90/385/EEC and 93/42/EEC;(22)Whereas it is necessary,essentially for the purpose of theconformity assessment procedures,to group in vitro diagnostic(1)OJ L204,21.7.1998,p.37.Directive as last amended by Directive98/48/EC(OJ L217,5.8.1998,p.18).(2)OJ L220,30.8.1993,p.23.medical devices into two main product classes;whereas,since the large majority of such devices do not constitute a direct risk to patients and are used by competently trained professionals,and the results obtained can often be confirmed by other means,the conformity assessment procedures can be carried out,as a general rule,under the sole responsibility of the manufacturer;whereas, taking account of existing national regulations and of notifi-cations received following the procedure laid down in Directive 98/34/EC,the intervention of notified bodies is needed only for defined devices,the correct performance of which is essential to medical practice and the failure of which can cause a serious risk to health;(23)Whereas,among the in vitro diagnostic medical devices forwhich intervention of a notified body is required,the groups of products used in blood transfusion and the prevention of AIDS and certain types of hepatitis require a conformity assessment guaranteeing,with a view to their design and manufacture,an optimum level of safety and reliability;(24)Whereas the list of in vitro diagnostic medical devices to besubjected to third-party conformity assessment needs updating, taking account of technological progress and of developments in the field of health protection;whereas such updating measures must be taken in line with procedure III(a)as laid down in Council Decision87/373/EEC of13July1987laying down the procedures for the exercise of implementing powers conferred on the Commission(1);(25)Whereas an agreement on a modus vivendi between the EuropeanParliament,the Council and the Commission concerning the implementing measures for acts adopted in accordance with the procedure laid down in Article189b of the Treaty was reached on20December1994(2);(26)Whereas medical devices should,as a general rule,bear the CEmarking indicating their conformity with the provisions of this Directive to enable them to move freely within the Community and to be put into service in accordance with their intended purpose;(27)Whereas manufacturers will be able,when the intervention of anotified body is required,to choose from a list of bodies published by the Commission;whereas,although Member States do not have an obligation to designate such notified bodies,they must ensure that bodies designated as notified bodies comply with the assessment criteria laid down in this Directive;(28)Whereas the director and staff of the notified bodies should not,themselves or through an intermediary,have any interest in the establishments subject to assessment and verification which is likely to compromise their independence;(29)Whereas the competent authorities in charge of marketsurveillance should be able,particularly in emergencies,to contact the manufacturer or his authorised representative estab-lished in the Community,in order to take any protection measures that should prove necessary;whereas cooperation and exchange of information between Member States are necessary with a view to uniform application of this Directive,in particular for the purpose of market surveillance;whereas to that end it is necessary to establish and manage a database containing data on manufacturers and their authorised representatives,on devices placed on the market,on certificates issued,suspended or withdrawn,and on the vigilance procedure;whereas a system(1)OJ L197,18.7.1987,p.33.(2)OJ C102,4.4.1996,p.1.of adverse incident reporting(vigilance procedure)constitutes a useful tool for surveillance of the market,including the performance of new devices;whereas information obtained from the vigilance procedure as well as from external quality assessment schemes is useful for decision-making on classifi-cation of devices;(30)Whereas it is essential that manufacturers notify the competentauthorities of the placing on the market of‘new products’with regard both to the technology used and the substances to be analysed or other parameters;whereas this is true in particular of high-density DNA probe devices(known as micro-chips)used in genetic screening;(31)Whereas,when a Member State considers that,as regards a givenproduct or group of products,it is necessary,in order to protect health and safety and/or ensure compliance with the imperatives of public health,in accordance with Article36of the Treaty,to prohibit or restrict their availability or to subject it to special conditions,it may take any transitional measures that are necessary and justified;whereas,in such cases,the Commission consults the interested parties and the Member States and,if the national measures are justified,adopts the necessary Community measures,in accordance with procedure III(a)as laid down in Decision87/373/EEC;(32)Whereas this Directive covers in vitro diagnostic medical devicesmanufactured from tissues,cells or substances of human origin;whereas it does not refer to the other medical devices manu-factured using substances of human origin;whereas,therefore, work will have to continue in this connection in order to produce Community legislation as soon as possible;(33)Whereas,in view of the need to protect the integrity of thehuman person during the sampling,collection and use of substances derived from the human body,it is appropriate to apply the principles laid down in the Convention of the Council of Europe for the protection of human rights and dignity of the human being with regard to the application of biology and medicine;whereas,furthermore,national regulations relating to ethics continue to apply;(34)Whereas,in the interests of overall consistency betweendirectives on medical devices,some of the provisions of this Directive should be incorporated into Directive93/42/EEC, which needs to be amended accordingly;(35)Whereas it is necessary to draw up as quickly as possible thelegislation which is lacking on medical devices manufactured using substances of human origin,HAVE ADOPTED THIS DIRECTIVE:Article1Scope,definitions1.This Directive shall apply to in vitro diagnostic medical devices and their accessories.For the purposes of this Directive,accessories shall be treated as in vitro diagnostic medical devices in their own right.Both in vitro diagnostic medical devices and accessories shall hereinafter be termed devices.2.For the purposes of this Directive,the following definitions shall apply:(a)‘medical device’means any instrument,apparatus,appliance,material or other article,whether used alone or in combination,including the software necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:—diagnosis,prevention,monitoring,treatment or alleviation of disease,—diagnosis,monitoring,treatment,alleviation or compensation for an injury or handicap,—investigation,replacement or modification of the anatomy or of a physiological process,—control of conception,and which does not achieve its principal intended action in or on the human body by pharmacological,immunological or metabolic means,but which may be assisted in its function by such means;(b)‘in vitro diagnostic medical device’means any medical devicewhich is a reagent,reagent product,calibrator,control material, kit,instrument,apparatus,equipment,or system,whether used alone or in combination,intended by the manufacturer to be used in vitro for the examination of specimens,including blood and tissue donations,derived from the human body,solely or principally for the purpose of providing information:—concerning a physiological or pathological state,or—concerning a congenital abnormality,or—to determine the safety and compatibility with potential reci-pients,or—to monitor therapeutic measures.Specimen receptacles are considered to be in vitro diagnostic medical devices.‘Specimen receptacles’are those devices, whether vacuum-type or not,specifically intended by their manu-facturers for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination.Products for general laboratory use are not in vitro diagnostic medical devices unless such products,in view of their character-istics,are specifically intended by their manufacturer to be used for in vitro diagnostic examination;(c)‘accessory’means an article which,whilst not being an in vitrodiagnostic medical device,is intended specifically by its manu-facturer to be used together with a device to enable that device to be used in accordance with its intended purpose.For the purposes of this definition,invasive sampling devices or those which are directly applied to the human body for the purpose of obtaining a specimen within the meaning of Directive93/42/EEC shall not be considered to be accessories to in vitro diagnostic medical devices;(d)‘device for self-testing’means any device intended by the manu-facturer to be able to be used by lay persons in a home envir-onment;(e)‘device for performance evaluation’means any device intendedby the manufacturer to be subject to one or more performance evaluation studies in laboratories for medical analyses or in other appropriate environments outside his own premises;(f)‘manufacturer’means the natural or legal person with responsi-bility for the design,manufacture,packaging and labelling of a device before it is placed on the market under his own name, regardless of whether these operations are carried out by that person himself or on his behalf by a third party.The obligations of this Directive to be met by manufacturers also apply to the natural or legal person who assembles,packages, processes,fully refurbishes and/or labels one or more ready-made products and/or assigns to them their intended purpose as devices with a view to their being placed on the market under his own name.This subparagraph does not apply to the person who,while not a manufacturer within the meaning of the first subparagraph, assembles or adapts devicesalready on the market to their intended purpose for an individual patient;(g)‘authorised representative’means any natural or legal personestablished in the Community who,explicitly designated by the manufacturer,acts and may be addressed by authorities and bodies in the Community instead of the manufacturer with regard to the latter's obligations under this Directive;(h)‘intended purpose’means the use for which the device isintended according to the data supplied by the manufacturer on the labelling,in the instructions for use and/or in promotional materials;(i)‘placing on the market’means the first making available in returnfor payment or free of charge of a device other than a device intended for performance evaluation with a view to distribution and/or use on the Community market,regardless of whether it is new or fully refurbished;(j)‘putting into service’means the stage at which a device has been made available to the final user as being ready for use on the Community market for the first time for its intended purpose.3.For the purposes of this Directive,calibration and control materials refer to any substance,material or article intended by their manufacturer either to establish measurement relationships or to verify the performance characteristics of a device in conjunction with the intended use of that device.4.For the purposes of this Directive,the removal,collection and use of tissues,cells and substances of human origin shall be governed,in relation to ethics,by the principles laid down in the Convention of the Council of Europe for the protection of human rights and dignity of the human being with regard to the application of biology and medicine and by any Member States regulations on this matter.5.This Directive shall not apply to devices manufactured and used only within the same health institution and on the premises of their manufacture or used on premises in the immediate vicinity without having been transferred to another legal entity.This does not affect the right of Member State to subject such activities to appropriate protection requirements.6.This Directive shall not affect national laws which provide for the supply of devices by a medical prescription.7.This Directive is a specific directive within the meaning of Article2(2)of Directive89/336/EEC,which shall cease to apply to devices which have complied with this Directive.Article2Placing on the market and putting into service Member States shall take all necessary steps to ensure that devices may be placed on the market and/or put into service only if they comply with the requirements laid down in this Directive when duly supplied and properly installed,maintained and used in accordance with their intended purpose.This involves the obligation of Member States to monitor the security and quality of these devices.This Article applies also to devices made available for performance evaluation.Article3Essential requirementsDevices must meet the essential requirements set out in Annex I which apply to them,taking account of the intended purpose of the devices concerned.Article4Free movement1.Member States shall not create any obstacle to the placing on the market or the putting into service within their territory of devices bearing the CE marking provided for in Article16if these devices have undergone conformity assessment in accordance with Article9.2.Member States shall not create any obstacle to devices intended for performance evaluation being made available for that purpose to the laboratories or other institutions listed in the statement referred to in Annex VIII if they meet the conditions laid down in Article9(4)and Annex VIII.3.At trade fairs,exhibitions,demonstrations,scientific or technical gatherings,etc.Member States shall not create any obstacle to the showing of devices which do not conform to this Directive,provided that such devices are not used on specimens taken from the participants and that a visible sign clearly indicates that such devicescannot be marketed or put into service until they have been made to comply.4.Member States may require the information to be supplied pursuant to Annex I,part B,section8to be in their official language (s)when a device reaches the final user.Provided that safe and correct use of the device is ensured,Member States may authorise the information referred to in the first subpar-agraph to be in one or more other official Community language(s).In the application of this provision,Member States shall take into account the principle of proportionality and,in particular:(a)whether the information can be supplied by harmonised symbols orrecognised codes or other measures;(b)the type of user anticipated for the device.5.Where the devices are subject to other directives concerning other aspects which also provide for the affixing of the CE marking,the latter shall indicate that the devices also fulfil the provisions of the other directives.However,should one or more of these directives allow the manu-facturer,during a transitional period,to choose which arrangements to apply,the CE marking shall indicate that the devices fulfil the provisions only of those directives applied by the manufacturer.In this case,the particulars of these directives,as published in the Official Journal of the European Communities,must be given in the documents,notices or instructions required by the directives and accom-panying such devices.Article5Reference to standards1.Member States shall presume compliance with the essential requirements referred to in Article3in respect of devices which are in conformity with the relevant national standards transposing the harmonised standards the reference numbers of which have been published in the Official Journal of the European Communities;Member States shall publish the reference numbers of such nationalstandards.2.If a Member State or the Commission considers that theharmonised standards do not entirely meet the essential requirementsreferred to in Article3,the measures to be taken by the MemberStates with regard to these standards and the publication referred to inparagraph1of this Article shall be adopted by the procedure defined inArticle6(2).3.Member States shall presume compliance with the essentialrequirements referred to in Article3in respect of devices designedand manufactured in conformity with common technical specificationsdrawn up for the devices in List A of Annex II and,where necessary,the devices in List B of Annex II.These specifications shall establishappropriate performance evaluation and re-evaluation criteria,batchrelease criteria,reference methods and reference materials.The common technical specifications shall be adopted in accordancewith the procedure mentioned in Article7(2)and be published in theOfficial Journal of the European Communities.Manufacturers shall as a general rule be required to comply with thecommon technical specifications;if for duly justified reasons manufac-turers do not comply with those specifications they must adopt solutionsof a level at least equivalent thereto.Where,in this Directive,reference is made to harmonised standards,thisis also meant to refer to the common technical specifications.▼M1Article6Committee on Standards and Technical Regulations1.The Commission shall be assisted by the Committee set up byArticle5of Directive98/34/EC(hereinafter referred to as‘theCommittee’).2.Where reference is made to this Article,Articles3and7ofDecision1999/468/EC(1)shall apply,having regard to the provisionsof Article8thereof.3.The Committee shall adopt its rules of procedure.▼M2Article71.The Commission shall be assisted by the Committee set up byArticle6(2)of Directive90/385/EEC.2.Where reference is made to this paragraph,Articles5and7ofCouncil Decision1999/468/EC(2)shall apply,having regard to theprovisions of Article8thereof.The period laid down in Article5(6)of Decision1999/468/EC shall beset at three months.3.Where reference is made to this paragraph,Article5a(1)to(4)andArticle7of Decision1999/468/EC shall apply,having regard to theprovisions of Article8thereof.4.Where reference is made to this paragraph,Article5a(1),(2),(4)and(6)and Article7of Decision1999/468/EC shall apply,havingregard to the provisions of Article8thereof.(1)Council Decision1999/468/EC of28June1999laying down the proceduresfor the exercise of implementing powers conferred on the Commission(OJL184,17.7.1999,p.23).(2)OJ L184,17.7.1999,p.23.。
欧洲议会和欧盟理事会指令(1998 年10 月27 日)

欧洲议会和欧盟理事会指令(1998年10月27日)关于协调各成员国有关体外医疗诊断设备法规的指令(98/79/EC)本指令适用范围:体外诊断医疗设备符号,信息—规范数据交换的术语本指令修订状况:(尚未对本指令进行修订)指令制修订依据及背景:考虑到建立欧共体的《条约》,尤其是第100a条款的规定,考虑到委员会的提案,考虑到经济及社会委员会的意见按照《条约》第189a款规定的程序,(1) 鉴于为使内部市场正常运作应采取一些措施;鉴于内部市场是一个确保商品、人员,服务及资本的自由流通的无内部边界的区域;(2) 鉴于成员国有关体外医疗诊断设备的安全,健康保护和性能特征及审批程序的法律,法规及管理规定的内容和范围是不同的;鉴于这种差异的存在给贸易带来阻碍,鉴于代表欧委会的国家立法进行一项对比调查证实了需要制订协调法规;(3) 鉴于为贸易自由排除壁垒,避免产生新的壁垒,唯一的手段是协调国家立法;鉴于各成员国单独采用其它的手段无法令人满意的达到该目的;鉴于本指令只规定的这些要求满足在最安全的条件下保证本指令适用的体外医疗诊断设备的自由流通;(4) 鉴于协调规定必须与成员国为管理直接或间接与这些设备有关的大众健康和医疗保险计划的基金筹措所采取的措施相区分;鉴于如这些措施符合欧共体的法律,则协调规定不会影响成员国予以实施的能力;(5) 鉴于体外医疗诊断设备应为患者、用户及第三者提供高水平的保护并达到制造商赋予产品的性能水准;鉴于,因此,在维持或改进各成员国已达到的保护水准是本指令的基本目标之一;(6) 鉴于根据1985年5月7日理事会决议对技术协调及标准的一种新方法的制定原则,有关相关产品的设计、制造及包装必须受到相关规定的限制,以满足基本要求;鉴于其基本性,这些要求应替代相应的国家规定;鉴于基本要求,包括降低及将风险最小化的要求,应考虑到设计当时的技术与应用及与保健及医疗安全水平相应的经济和技术因素,有区别地加以采用。
CE分类规则
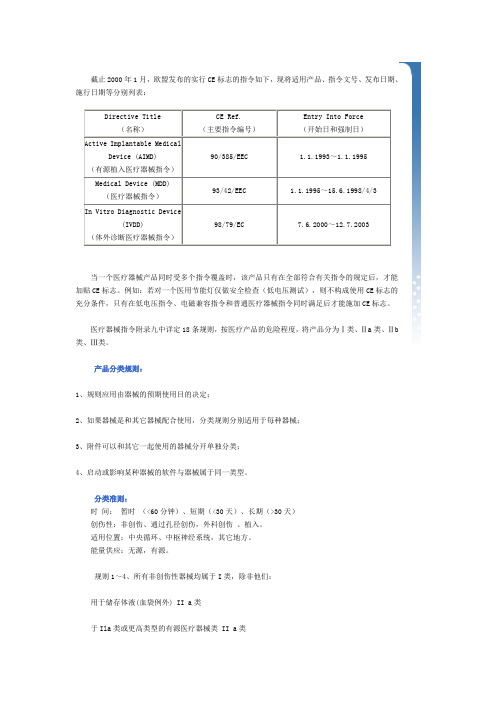
截止2000年1月,欧盟发布的实行CE标志的指令如下,现将适用产品、指令文号、发布日期、施行日期等分别列表:Directive Title (名称)CE Ref.(主要指令编号)Entry Into Force(开始日和强制日)Active Implantable MedicalDevice (AIMD)(有源植入医疗器械指令)90/385/EEC 1.1.1993~1.1.1995Medical Device (MDD)(医疗器械指令)93/42/EEC 1.1.1995~15.6.1998/4/3 In Vitro Diagnostic Device(IVDD)(体外诊断医疗器械指令)98/79/EC 7.6.2000~12.7.2003当一个医疗器械产品同时受多个指令覆盖时,该产品只有在全部符合有关指令的规定后,才能加贴CE标志。
例如:若对一个医用节能灯仅做安全检查(低电压测试),则不构成使用CE标志的充分条件,只有在低电压指令、电磁兼容指令和普通医疗器械指令同时满足后才能施加CE标志。
医疗器械指令附录九中详定18条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb 类、Ⅲ类。
产品分类规则:1、规则应用由器械的预期使用目的决定;2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;3、附件可以和其它一起使用的器械分开单独分类;4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:时间:暂时(<60分钟)、短期(<30天)、长期(>30天)创伤性:非创伤、通过孔径创伤,外科创伤、植入。
适用位置:中央循环、中枢神经系统,其它地方。
能量供应:无源,有源。
规则1~4、所有非创伤性器械均属于I类,除非他们:用于储存体液(血袋例外) II a类于Ila类或更高类型的有源医疗器械类 II a类改变体液成分 II a/II b类一些伤口敷料 II a/II b类规则5、侵入人体孔径的医疗器械暂时使用(牙科压缩材料、检查手套) I类短期使用(导管、隐形眼镜) II a类长期使用(正常牙线) II b类规则6-8、外科创伤性器械再使用的外科器械(钳子,斧子) I类暂时或短期使用(缝合针。
体外诊断试剂的政策法规

体外诊断试剂的政策法规全文共四篇示例,供读者参考第一篇示例:体外诊断试剂是医疗设备领域中一类应用广泛的产品,其在医疗诊断中扮演着至关重要的角色。
体外诊断试剂可以帮助医生诊断疾病、监测疗效、筛查疾病等,对于提高诊断的准确性和效率起着重要作用。
为了确保体外诊断试剂的质量和安全性,各国都制定了一系列的政策法规来规范和监管。
本文将重点介绍体外诊断试剂的政策法规及其相关内容。
一、FDA(美国食品药品监督管理局)对体外诊断试剂的监管在美国,FDA对体外诊断试剂的监管非常严格。
根据美国《药品和化妆品法案》,所有在美国市场销售的体外诊断试剂都必须经过FDA的审批。
FDA要求厂商在向FDA提交上市申请时,必须提供充分的临床数据和试验报告,以证明产品的有效性和安全性。
FDA还会对生产工艺、质量管理体系和产品标签等方面进行严格监督,确保体外诊断试剂的质量和安全性。
FDA还颁布了一系列的法规和指南,规定了体外诊断试剂在研发、生产、销售和使用过程中的要求和规范。
《体外诊断试剂法规》(21 CFR Part 809)、《体外诊断试剂注册和上市报告指南》等文件对体外诊断试剂的注册、标识、报告、质量管理等方面做出了详细规定,确保体外诊断试剂符合美国法律法规的要求。
二、欧盟对体外诊断试剂的监管在欧盟,体外诊断试剂的监管主要由欧盟委员会和各成员国的监管机构共同负责。
欧盟委员会根据《体外诊断试剂法规》(98/79/EC)对体外诊断试剂的注册、标识、质量管理、审评等方面做出了详细规定。
根据该法规,体外诊断试剂必须通过欧盟指定的评估机构进行审评,并获得CE认证方可在欧盟市场销售和使用。
在中国,体外诊断试剂的监管由国家药监局和各级地方药监部门共同负责。
中国的《医疗器械监督管理条例》、《医疗器械注册管理办法》等文件对体外诊断试剂的注册、生产、销售等方面做出了详细规定。
根据中国的相关法规,体外诊断试剂必须取得《医疗器械注册证》方可在中国市场销售和使用。
体外诊断医疗器械IVDD产品分类

体外诊断医疗器械IVDD产品分类根据98/79/EC(IVDD)指令附录2确定产品分类原则对有认证需求的产品进行分类。
分类的依据是产品所诊断的疾病。
常见产品的分类可参考下表:与上述诊断试剂配套使用的校准品、仪器、标本采集保存用具均属于体外诊断器械指令管理的范畴。
医疗器械FDA注册认证FDA对医疗器械的管理通过器械与放射健康中心(CDRH)进行的,中心监督医疗器械的生产、包装、经销商遵守法律下进行经营活动。
医疗器械范围很广,小到医用手套,大至心脏起博器,均在FDA监督之下,根据医疗用途和对人体可能的伤害,FDA将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,越高类别监督越多.如果产品是市场上不曾存在的新颖发明,FDA要求厂家进行严格的人体实验,并有令人信服的医学与统计学证据说明产品的有效性和安全性。
医疗器械的FDA认证,包括:厂家在FDA注册、产品的FDA登记、产品上市登记(510表登记)、产品上市审核批准(PMA审核) 医疗保健器械的标签与技术改造、通关、登记、上市前报告,须提交以下材料:(1)包装完整的产成品五份,(2)器械构造图及其文字说明,(3)器械的性能及工作原理;(4)器械的安全性论证或试验材料,(5)制造工艺简介,(6)临床试验总结,(7)产品说明书. 如该器械具有放射性能或释放放射性物质,必须详细描述.医疗器械的工厂和产品注册FDA对医疗器械有明确和严格的定义,其定义如下:“所谓医疗器械是指符合以下条件之仪器、装置、工具、机械、器具、插入管、体外试剂及其它相关物品,包括组件、零件或附件:明确列于National Formulary或the Unite States Pharmacopeia 或前述两者的附录中者;预期使用于动物或人类疾病,或其它身体状况之诊断,或用于疾病之治愈、减缓与治疗者;预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目的者”。
只有符合以上定义的产品方被看作医疗器械,在此定义下,不仅医院内各种仪器与工具,即使连消费者可在一般商店购买之眼镜框、眼镜片、牙刷与按摩器等健身器材等都属于FDA之管理范围。
体外诊断IVDD指令

体外诊断IVD D指令(CE认证主要步骤)2010-01-11 08:47欧洲委员会于1998年10月27日正式通过98/79/EC体外诊断医疗器材指令(In Vitro Diagnos tic Medical Devices Directi ve,以下简称IVD D指令),并公告于1998 年12月7日的第L331号欧盟公报上。
根据公报的内容,欧盟各成员国必须于2000年6月7日之前完成执行本指令所需要的相关法规命令修制定与公告,自2003年12月起,所有在欧盟各成员国销售的体外诊断医疗器材(In Vitro Diagnos tic Medical Devices,以下简称IVD)均须依照本指令完成符合性评价程序,贴上CE标记,才能在欧盟上市。
2004年5月,欧盟新增了十个成员国,加上原有的18个成员国,欧盟目前已有28个成员国,欧盟已是一个越来越大的单一市场。
国内的体外诊断医疗断器材生产厂家也意识到此点,但这个市场是有门槛的,这个门槛就是C E认证。
国内的生产厂家对此相对陌生,倍感困难,本文就申请CE认证主要步骤略作介绍。
第一步确定产品是否为IVD制造商首先要根据体外诊断医疗器材的定义,确定该产品是否为体外诊断医疗器材,根据欧盟体外诊断医疗器材指令98/79/EC的定义,体外诊断医疗器材的定义是指:任何试剂、校正物质、对照物质、仪器、装置、设备或系统的医疗器械,无论是单独或合并使用,由制造商指定其用途为用于体外检验采自人体的样本包括血液与组织,单独或主要用以提供以下相关信息者:·关于生理或病理状态·或关于先天异常·或决定与潜在接受移植人员之安全性与兼容性·或监控治疗效果。
样本容器也被认为是体外诊断医疗器材,无论是否为真空形式,由制造商指定用以储存采取自人体的样本,供体外诊断检验之用者皆属之。
CE认证指令
97/67/EC
Community postal services
邮政指令
76/769/EEC
Restrictions on marketing and use of certain dangerous substances and preparations
有毒有害物质管制指令(RoHS认证指令,化学指令)
热水锅炉指令
93/15/EEC
Explosives for civil uses/民用爆炸物指令
93/42/EEC
Medical devices
医疗器械指令
94/9/EC
Equipment explosive atmospheres (ATEX)
欧盟防爆指令
94/25/EC
Recreational craft
非自动衡器
90/384/EEC
1993年1月1日
主动性植入式医疗器械
90/385/EEC
1994年12月31日
燃气器具
90/396/EEC
1995年12月31日
锅炉
92/42/EEC
1998年1月1日
爆破性产品
93/15/EEC
2003年1月1日
通用医疗器械
93/42/EEC
1998年6月15日
低压电气安全
船用设备指令
2001/16/EC
Interoperability of trans-European conventional rail system
常规铁路系统的互通性指令
Directives based on some principles of the New Approach and the Global Approach
98-79-EC体外诊断医疗器械指令(中文版)

内部资料强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令 98/79/EC1998年10月27日强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令98/79/EC第1条范围·定义………………………………………………………………………第2条上市和投入使用………………………………………………………………………第3条基本要求………………………………………………………………………第4条自由流通………………………………………………………………………第5条标准………………………………………………………………………第6条标准和技术法规委员会……………………………………………………………第7条医疗器械委员会………………………………………………………………………第8条安全保障条款………………………………………………………………………第9条合格认证程序………………………………………………………………………第10条制造者和器械的注册登记……………………………………………………………第11条防范程序………………………………………………………………………第12条欧洲资料库………………………………………………………………………第13条特别卫生监督措施………………………………………………………………………第14条对附录II的修订和降低条款……………………………………………………………第15条指定认证部门………………………………………………………………………第16条C E标志………………………………………………………………………第17条C E标志使用不当………………………………………………………………………第18条拒绝或限制的规定………………………………………………………………………第19条保密规定………………………………………………………………………第20条成员国之间的合作………………………………………………………………………第21条指令的修订………………………………………………………………………第22条执行,过渡规定………………………………………………………………………第23条生效日期………………………………………………………………………第24条发给成员国………………………………………………………………………附录I 基本要求…………………………………………………………………………………附录II 和第9(2)及9(3)条有关的器械清单…………………………………………………附录II I E C合格声明……………………………………………………………………………附录IV E C 合格声明(全面质量保证体系)……………………………………………………附录V E C样品检查…………………………………………………………………………附录V I E C验证…………………………………………………………………………………附录VI I合格声明(生产质量保证)……………………………………………………………附录V I I I性能评价器械的报告书和程序………………………………………………………附录I X指定认证部门的选派准则……………………………………………………………附录X C E合格标志……………………………………………………………………………强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令98/79/EC1998年10月27日欧洲议会和欧盟理事会根据欧共体成立条约第100a条的规定和欧盟委员会的建议,征求了经济和社会委员会的意见,按照条约第189b条所规定的程序,并鉴于下列各点,发布此指令。
EN13612:2002中文翻译

98/79/EC指令,关于体外诊断医疗设备(IVD MDs)requires in Annex III, section 3,indent 11 and section 6.1, in Annex IV, section 3.2 c) and in Annex V, section 3,生产商在他的技术文档里提供证据,表明该IVD MD象所声明的一样表现,是否这些声明是由技术,分析或诊断支持。
这些证据可以由生产商已有数据显示,或科学文献支持,或由临床或其它适用环境下收集来的相应于使用用途的性能评估数据。
若一个性能评估研究是必要的,合适的,来支持该IVD MD的性能声明,本标准描述生产商如何可以满足他的责任来引导一个科学性的性能评估研究。
评估计划适合于该IVD MD的属性和预期用途,同时考虑到在科学文献和标准中给出的不同的推荐建议。
考虑到IVD MD的由指令98/79/EC指定的宽广的范围,并考虑到,到目前为止,没有统一的应用文档,这就是本标准的目的,表述一个性能评估的通用元素。
所描述的许多项目的适应性由IVD MD 复杂程度所决定。
在起草本标准时,设想到欧盟会出版一些通用技术描述(CTS),相关于指令98/79/EC,关于体外诊断医疗设备。
进一步预测到,这些会由欧盟的官方杂志所引用。
特别是这些CTS会应用到附录II 的列表A中的体外诊断器中,及附录II的列表B中。
生产商因此应在指令98/79/EC 条款5“参考标准”中,考虑这些CTS。
范围本欧盟标准针对IVD MD的性能评估,包括自我测试的IVD MD。
本标准描述了生产商对性能评估研究的责任和一般要求,以及研究的计划,实行,评估和记录。
这不针对某些IVD MD的特殊评估计划或特别使用。
注对出版物的选择,见文献录。
当一个生产商维持一个质量系统,本标准解决了设计验证和设计变化的顺应性,如EN ISO 9001,EN46001和EN928所描述,尤其考虑到IVD MD的性质和使用。
关于体外诊断医疗器械的9879EC指令

第13条 特殊的健康监督措施
对于某一产品,如果成员国认为,为了确保健康与安全和/或 确保依照《欧洲共同体条约》第36条遵守公共卫生要求,应 禁止、限制获得这类产品或使这类产品必须遵守特殊要求。 则该成员国可以采取合理的过渡性措施,并应通知欧洲联盟 委员会及其他成员国,并说明理由。欧洲联盟委员会应与有 关各方磋商,如果该国采取的措施经证明是合理的,即应根 据程序批准必要的共同体措施。
eg:尿杯
第1条 适用范围、定义
(c) “附件”是指虽然不是体外诊断医疗器械,但其制造商明 确规定与某种器械一起使用、使该器械能够按照其预定目的 应用的物品。在本定义中,取样器械或那些为获取 93/42/EEC指令含义内的样品而直接施于人体的器械不应认 为是体外诊断医疗器械的义
CE标示是制造商的符合标志,表示符合所有现行的指令。对于大多数销 售到欧盟的产品而言,CE标示的使用和声明产品的符合性 ,是强制性 的法令条文。有此符合标志,产品可自由在会员国流通。CE标示必须标 示在产品装置上,或是在包装上显示。然而此CE标示并不能免除国家执 法单位对于未符合标示的产品所采取的行动。
质保部 王娟
关于体外诊断医疗器械的98/79/EC指令
欧盟与EC指令
欧盟成立后,实施一套制度以保护消费者与工作者 的健康,商品的状态与环境。在这套新制度之下, 欧盟与一些欧洲自由贸易协会的国家,制定了EC (European Community)指令,其中强制执行的 指令称为EEC(European Economic Community) 指令 。
(d) “自我检测器械”是指制造商规定能够由非专业 人员在家庭环境中使用的任何器械;
eg:易知欣、易知盖
第1条 适用范围、定义
关于体外诊断的医疗器械通用技术规范1

理事会理事会指令2002年5月7日关于体外诊断的医疗器械通用技术规范(通过文件号C(2002)1344之公告)文章与EEA相关(2002/364/EC)欧洲共同体理事会考虑到建立欧洲经济共同体的条约。
考虑到欧洲国会和理事会1998年10月27日体外诊断医疗器械指令 98/79/EC,特别是其第5(3)条第二段,鉴于:(1)指令98/79/EC 陈述了体外诊断医疗器械上市时必须满足基本要求,并把与协调标准保持一致作为与相关基本要求一致的前提。
(2)除了利用这些基本条例,通用技术规范的提出应考虑到一些成员国当前措施,这些针对于指定器械的措施选择主要应用于对供血和器官捐赠的安全性评价,公众当局也接受这些规范。
这些通用技术规范可以用于性能评估和再评估。
(3)来自各个有关方面的科学专家参加了此次通用技术规范的起草。
(4)指令98/79/EC指出成员国应默认为最高风险类别中特定器械的器械设计和制造应符合为其制定的通用技术规范,这些规范与基本要求相关。
这些规范建立了适当的性能评估和再评估标准,批放行标准,参考方法和参考物质。
(5)根据通用准则,要求制造商满足通用技术规范。
如果,在一些得到证实的适当理由下,制造商可以不依从这些规范,但必须采用与此规范水平至少相当的解决方法。
(6)为这个决议所提供的方法应符合指令90/385/EEC(2)的第6(2)条建立的委员们的意见。
兹通过本指令:第1条本决议附录中所提到的技术规范被采用作为通用技术规范,适用于指令98/79/EC附录2列表A中的体外诊断的医疗器械。
第2条本决议向各成员国提出。
完成于布鲁塞尔,2002年5月7日。
给委员会Erkki LIIKANEN委员会委员附录CTS-体外诊断医疗器械通用技术规范1.范围这些通用技术规范适用于附录2,列表A中列出的器械:——试剂和试剂产品,包括相关的校准和控制材料,用于决定以下血型:ABO系统,凝血(C,c,D,E,e)和抗Kell型。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令 98/79/EC1998年10月27日强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令98/79/EC第1条范围·定义………………………………………………………………………第2条上市和投入使用………………………………………………………………………第3条基本要求………………………………………………………………………第4条自由流通………………………………………………………………………第5条标准………………………………………………………………………第6条标准和技术法规委员会……………………………………………………………第7条医疗器械委员会………………………………………………………………………第8条安全保障条款………………………………………………………………………第9条合格认证程序………………………………………………………………………第10条制造者和器械的注册登记……………………………………………………………第11条防范程序………………………………………………………………………第12条欧洲资料库………………………………………………………………………第13条特别卫生监督措施………………………………………………………………………第14条对附录II的修订和降低条款……………………………………………………………第15条指定认证部门………………………………………………………………………第16条C E标志………………………………………………………………………第17条CE标志使用不当………………………………………………………………………第18条拒绝或限制的规定………………………………………………………………………第19条保密规定………………………………………………………………………第20条成员国之间的合作………………………………………………………………………第21条指令的修订………………………………………………………………………第22条执行,过渡规定………………………………………………………………………第23条生效日期………………………………………………………………………第24条发给成员国………………………………………………………………………附录I 基本要求…………………………………………………………………………………附录II 和第9(2)及9(3)条有关的器械清单…………………………………………………附录III EC合格声明……………………………………………………………………………附录IV EC 合格声明(全面质量保证体系)……………………………………………………附录V EC 样品检查…………………………………………………………………………附录VI EC 验证…………………………………………………………………………………附录VII 合格声明(生产质量保证)……………………………………………………………附录VIII 性能评价器械的报告书和程序………………………………………………………附录IX 指定认证部门的选派准则……………………………………………………………附录X CE合格标志……………………………………………………………………………强制实行法规欧洲议会和欧盟理事会体外诊断医疗器械指令98/79/EC1998年10月27日欧洲议会和欧盟理事会根据欧共体成立条约第100a条的规定和欧盟委员会的建议,征求了经济和社会委员会的意见,按照条约第189b条所规定的程序,并鉴于下列各点,发布此指令。
1、欧盟内部市场是一个没有内部边境的市场。
在市场内要保障商品、人员、劳务和资本的自由流通。
为此必须采取各项措施使市场顺利运转。
2、各成员国在体外诊断医疗器械的安全性、卫生保健、性能、特性和核准程序方面,现行法律、法规和行政规章规定的内容和范围各有不同;这些差别的存在造成了贸易壁垒。
代表欧盟委员会实施的国家立法的广泛调查已经证实:需要建立相互协调的法规。
3、国家立法的协调是消除壁垒走向自由贸易并阻止新壁垒产生的唯一手段。
然而,单一的成员国用其它方法不可能满意地达到这一目标;本指令就制定了这样一些必须的要求以成功地保障体外诊断医疗器械在其应用地区自由流通。
4、协调各国的法规应同各成员国采取的与这些医疗器械有直接或间接关系的公共卫生和医疗保险制度的管理措施区别开来,以免影响各成员国实施上述管理措施的能力,只要这些措施不违反共同体法律。
5、体外诊断医疗器械应向患者、用户和第三方提供高水平的健康保障,并达到生产者原来赋予的使用性能,本指令的主要目的之一就是要保持和提高各成员国已经达到的保健水平。
6、按照1985年5月7日理事会对技术协调和标准化新政策所作决议原则,各成员国关于相关产品设计、生产、包装方面的规定必须同满足基本要求的条款相一致。
并且,因为是基本要求,它应当取代各成员国的相应规定。
要慎重地实施基本要求,包括减少和降低风险的要求。
应考虑设计时的技术和实践,使技术和经济方面的考虑与高水平的安全和健康保障相协调。
7、1990年6月20日理事会指令90/385/EEC协调了成员国关于有源植入性医疗器械的法规,1993年6月14日关于医疗器械的理事会指令未包括体外诊断医疗器械,这两个指令涵盖了医疗器械的主要部分。
为此,本指令拟对体外诊断医疗器械进行协调。
为使欧共体的规定协调一致,本指令的内容大部分基于上述两指令的规定。
8、用于研究目的而无任何医疗目的的仪器、设备、器具、材料或其他物件,包括软件在内,均不作为医疗器械进行评估。
9、虽然,国际上认定的基准材料和用于外部质量鉴定的材料不属本指令所包括的范围,但用户用于确认或验证器械性能的校准设备和对照材料也属于体外诊断医疗器械。
10、根据补充规定的原则,在卫生机构实验室生产并在该环境下应用的试剂,且不用于商业处理的目的时,不属本指令所规定的范围。
11、然而,生产出并为医学分析目的用于专业和商业方面而上市的器械应执行本指令。
12、特别为体外诊断检查而设计的机械实验室的设备应属本指令所包括的范围。
因此,为协调相关的指令,1998年6月22日对成员国与机械有关的法律进行协调的理事会和欧洲议会指令98/37/EC,应予以适当修订以使其和本指令相一致。
13、本指令应包括具有电离辐射的器械的设计和生产要求,本指令并不影响1996年5月13日的理事会指令96/29/Euratom的应用,该指令规定了由电离辐射引起的工作人员健康保障和通用公众危险防护基本安全标准。
14、由于电磁兼容要求构成了本指令基本要求的组成部分,1998年5月2日用于协调成员国关于电磁兼容的理事会指令89/336/EEC不再使用。
15、为了简化保证符合基本要求的工作并使这一符合性得到验证,希望制定和医疗器械设计、生产、包装有关的风险预防协调标准。
这些协调标准由民间法律机构起草,属于非强制性规范。
为此,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)根据欧盟委员会同这两个机构于1984年11月13日签订的合作通用指南被作为采用协调标准的主管单位。
16、按本指令的目的,协调标准时根据欧盟委员会(以下简称欧委会)委托,由CEN和CENELEC分别发布或联合发布的技术规范(协调文件的欧洲标准),发布协调标准应按照欧洲议会和理事会1998年6月22日指令98/34/EC关于技术和法规发布程序的规定并应符合上述合作通用指南。
17、作为通用原则的例外,共同技术规范的拟定要考虑某些成员国的实际情况,对主要用于供血和捐献组织安全评估的选定的器械,这种规范是由政府机构发布的,用共同技术规范代替特殊规范较为适宜,这些共同技术规范可用于性能的评估和再评估。
18、在起草共同技术规范和检查其它特殊和一般问题时可将代表不同利益部门的科学专家包括在内。
19、本指令内所说的制造也包括了医疗器械的包装,因为包装也和器械的安全性和使用性能有关。
20、由于性能随着时间增长向下降,某些器械的寿命是有限的。
举例说,这与其物理和化学特性的变质有关,包括其包装的灭菌与完整性;制造者应决定并指明器械具备预期性能的期限;标签上应注明器械或其某一部组成部分可以完全安全使用的日期期限。
21、1993年7月22日的决定(93/465/EEC)涉及了合格认证程序在不同阶段的模式和CE合格标志的使用与附加规则;这些内容拟应用于技术协调指令中;在上述决定中,理事会规定了协调的合格认证程序,而体外诊断器械所需验证的性质及与指令90/385/EEC 保持一致性的需要,证明模式的附加细节是有道理的。
22、为了合格认证程序的需要,应将体外诊断医疗器械分为两个基本类别,这些器械的绝大部分并不对患者构成直接风险,并由能胜任的经培训的专业人员使用,其得到的结果常常可由其他手段确认,按通常惯例,由制造者单独负责即可实施合格认证程序;然而,考虑到成员国法规的存在和指令98/34/EC中规定的程序所附带的通告,在某些特定医疗器械其正确使用对医疗实践非常重要,并且其失效将对健康造成严重风险时,指定认证部门的介入就是需要的。
23、在需要指定部门介入的那些体外诊断医疗器械当中,用于输血且要预防爱滋病和某些类型肝炎的产品则需要一种从设计和生产角度使安全性、可靠性处于最佳水平的合格评定程序。
24、考虑到技术进步和保健领域的发展,由第三方面进行合格判定的体外诊断医疗器械的清单需要更新;此项更新措施的采取必须和1987年7月13日理事会决定87/373/EEC 所规定的程序III(a)相一致,该决定规定了授予欧盟委员会的执行权的运行程序。
25、对于按条约第1896条规定的程序发布的执行措施条例,在欧洲议会、理事会和欧委会间于1994年12月20日达成了一项暂行协定。
26、按惯例,医疗器械应带有CE标志,以表明其符合本指令规定,能在共同体内自由流通,并能按其预定目的投入使用。
27、当需指定部门介入时,制造者能够自委员会公布的部门名单中进行选择;虽然成员国没有责任来选派此类指定部门,但他们必须保证:一旦选派的部门作为指定认证部门,必须符合本指令规定的合格认证规则。
28、指定部门的工作人员和负责人,不论是他们自己或通过中间人,都不应由于在认证和验证项目中有任何利益关系,以致损害了他们的独立性。
29、负责市场监督的主管机关,特别是在紧急情况下,应能和制造者或他们在共同体内设立的授权代理人进行接触,以便采取需要的任何保护措施;从本指令应用的一致生角度出发,多成员国之间的使用和情报交流是需要的,特别是在市场监督方面;因此,需要建立和管理一个资料库,该资料库应包括:制造者和他们的授权代理人、上市的器械、认证证书的发出、终止或撤消、防范程序;因此,有害事件报告制度(防范程序)就成了市场监督(包括新器械使用性能监督)的有用工具;从防范程序中和外部质量评定系统中获取的信息对器械分类的决策是有用的。