palbociclib的全部数据
帕博西尼(Ibrance,Palbociclib)FDA 官方说明书

帕博西尼(Ibrance,Palbociclib)FDA官方说明书1 适应症和用途IBRANCE是适用与来曲唑联用对有雌激素受体(ER)-阳性,人表皮生长因子受体2(HER2)-阴性晚期乳癌绝经后妇女作为初始基于内分泌治疗对其转移疾病的治疗。
这个适应症是根据无进展生存(PFS)在加速批准下被批准的[见临床研究(14)]。
对此适应症的继续批准可能取决于在验证性试验中临床获益的证明和描述。
2 剂量和给药方法2.1 一般给药信息IBRANCE的推荐剂量是一粒125 mg胶囊口服服用每天一次共21天,接着不用治疗7天组成一个28天完整疗程。
IBRANCE应与食物服用[见临床药理学(12.3)]与来曲唑2.5 mg每天一次联用连续28-天疗程自始至终给予。
应鼓励患者在每天接近相同时间服用他们的剂量。
如患者呕吐或丢失一剂,在那天不应服用另外剂量。
在寻常的时间服用下一次处方剂量。
IBRANCE胶囊应被整吞(在吞咽前不要咀嚼,压碎或打开胶囊)。
如破碎,压碎或不完整时不应摄入胶囊。
2.2 剂量调整建议根据个体安全性和耐受性调整IBRANCE剂量[见警告和注意事项(5)]。
某些不良反应的处理[见警告和注意事项(5)]可能需要暂时中断剂量/延迟和/或减低剂量,或永久终止如同表1,2和3提供每种剂量减低计划[见警告和注意事项(5),不良反应(6)和临床研究(14)]。
见制造商处方资料对共同给药产品,来曲唑,在毒性事件中剂量调整指导原则和其他相关安全性资料或禁忌证。
为与强CYP3A抑制剂使用剂量调整避免强CYP3A抑制剂的同时使用和考虑没有或小CYP3A抑制作用另外同时药物。
如患者必须用强CYP3A抑制剂共同给药,减低IBRANC剂量至75 mg每天一次。
如强抑制剂被终止,增加IBRANCE剂量(抑制剂的3–5个半衰期后)至强CYP3A抑制剂使用前剂量[见药物相互作用(7.1)和临床药理学(12.3)]。
3 剂型和规格125 mg胶囊:不透明硬明胶胶囊,大小0,有焦糖帽和体,帽上用白墨汁印,体上“PBC 125”。
药物帕布昔利布(palbociclib)合成检索总结报告
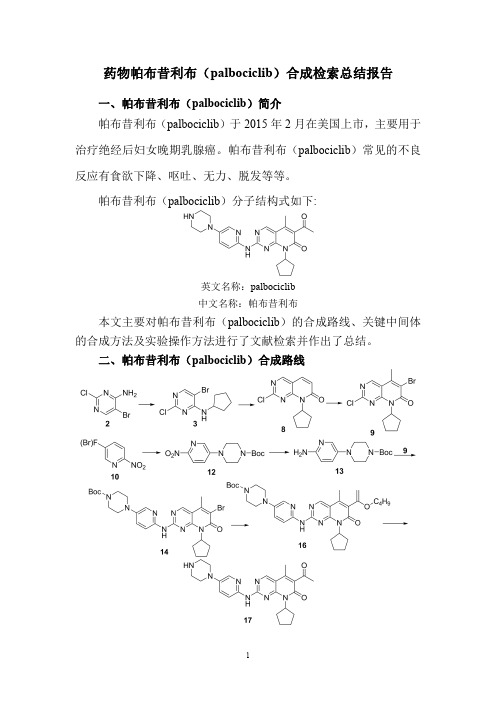
药物帕布昔利布(palbociclib)合成检索总结报告一、帕布昔利布(palbociclib)简介帕布昔利布(palbociclib)于2015年2月在美国上市,主要用于治疗绝经后妇女晚期乳腺癌。
帕布昔利布(palbociclib)常见的不良反应有食欲下降、呕吐、无力、脱发等等。
帕布昔利布(palbociclib)分子结构式如下:英文名称:palbociclib中文名称:帕布昔利布本文主要对帕布昔利布(palbociclib)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、帕布昔利布(palbociclib)合成路线三、帕布昔利布(palbociclib )合成检索总结报告(一)帕布昔利布(palbociclib )中间体3的合成方法一合成方法实验步骤参考文献操作方法一Compound 2(208.4g,1mol)was added to the 2L three-necked flask,Copper trifluoroacetate (2.8g,0.01mol)1,4-dioxane (833.6g),Sodium hydroxide (60g,1.5mol),cyclopentyl bromide 1(223.6g,1.5mol),incubate at 80o C for 6hours,the reaction is over,Add saturated ammonium chloride solution (832g)and stir well,extract with ethyl acetate (300g),the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure,the obtained solid was collected in a 2L three-necked flask,and isopropyl alcohol (832g)was added,activated carbon (10g),stirred at 60°C for 1hour,filtered while hot,The filtrate was naturally cooled to 10°C to precipitate solids and filtered,The filter cake was vacuum dried at 40°C to obtain Compound 3,a white solid 249g,and a yield of 90%.The HPLC purity was 99.5%.CN108299311;(2018);(A)Chinese(二)帕布昔利布(palbociclib )中间体3的合成方法二合成方法实验步骤参考文献操作方法一Compound 2(208.4g,1mol)was added to the 2L three-necked flask,Copper acetate (2g,0.01mol),dichloroethane (625g),sodium hydroxide (100g,2.5mol),cyclopentyl chloride 4(261.5g,2.5mol),incubate at 80o C for 10hours,After the reaction,add saturated ammonium chloride solution (832g)and stir well.Extracted with ethyl acetate (300g),the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure,The obtained solid was collected in a 2L three-necked flask,ethyl acetate (832g)was added,activated carbon (10g),stirred at 60°C for 1hour,filtered while hot,The filtrate was naturally cooled to 10°C to precipitate solids and filtered,The filter cake was vacuum dried at 40°C to obtain Compound 3,white solid 213g,yield 77%,The HPLC purity was 98.5%.CN108299311;(2018);(A)Chinese(三)帕布昔利布(palbociclib )中间体3的合成方法三合成方法实验步骤参考文献操作方法一To a solution of 5-bromo-2,4-dichloropyrimidine 6(45.6g,200mmol)in dioxane (400mL)was added N-cyclopentylamine 5(20.4g,240mmol)at room temperature.The mixture thus obtained was stirred at room temperature for 6h.The reaction mixture was then diluted with ethyl acetate and washed with brine and dried over MgSO 4.The solvent was evaporated to give the title compound as a light yellow solid 3(56g,100%)which was used in next step without further purification.WO2009/85185;(2009);(A1)English 操作方法二2,4-dichloro-5-bromopyrimidine 6(200g,888mmol)and 1.4L of dichloromethane were added to a reaction flask,and sodium hydrogencarbonate (372g,4440mmol)was added.At room temperature,cyclopentylamine 5(90.7g,1066mmol)diluted with 0.6L of dichloromethane was slowly added dropwise,and reacted at room temperature for 13h.The sodium hydrogencarbonate and the resulting inorganic salt were removed by suction filtration,the mother liquor was evaporated to remove dichloromethane,and 1.0L of n-heptane was added to recrystallize.236.7g of a white solid were obtained,the yield of 3was 97.5%,the purity was >99.8%,and the impurity 5-bromo-4-chloro-2-cyclopentyl-Aminopyrimidine.Melting point:94-96°109206373;(2019);(A)Chinese 操作方法三5-Bromo-2,4-dichloropyrimidine 6(227.9g,1mol)was sequentially added to a 2L three-necked flask.Isopropyl alcohol (360g,6mol),diisopropylethylamine (167.7g,1.3mol)was added at -15°C.A solution of cyclopentylamine 5(106.2g,1.3mol)in isopropanol (120g,2mol)was added dropwise.The reaction was carried out for 2to 3hours,and the solid was filtered.The filter cake was taken in a 2L reaction flask,petroleum ether (600g)was added thereto,stirred for 1hour,filtered again,and the filter cake was washed with petroleum ether (100g).The filter cake was collected and dried under vacuum at 40°C to give 260.0g of Intermediate II,white solid,yield 94%.CN108299422;(2018);(A)Chinese A solution of 5-bromo-2,4-dichloropyrimidine (6)(5g,22mmol)and N,N-diisopropylethylamine (5.7g,44mmol)in ethanol (30mL)was cooled to 10°C under nitrogenCombinatorial Chemistry and操作方法四atmosphere.Charged cyclopentylamine 5(2.1g,24.1mmol)and stirred for 5hours at 40°C.Progress of the reaction wasmonitored by TLC and solvent was evaporated undervacuum.Resulting residue was stirred with hexane (20mL)for 2hours at 0°C.Precipitate was filtered and washed withhexane to obtain compound (3).Off-white crystalline solid,Yield:90%,mp.95-97°C.High Throughput Screening ;vol.20;nb.8;(2017);p.703–712.操作方法五In a large sealed tube is added 5-bromo-2,4-dichloropyri-midine 6(3g,13.2mmol)in 100mL of EtOH.Then cyclopentyl amine 5(1.95mL,19.75mmol)and N,N'-diisopropylethylamine (3.36mL,19.8mmol)are added to the solution at rt.The solution is then stirred rt overnight.Solvent is evaporated and the crude is purified using silica gel chromatography (15%ethyl acetate/85%hexane)to give (5-bromo-2-chloro-pyrimidin-4-yl)-cyclopentyl-amine 3as a white solid (3.25g,89%).WO2010/20675;(2010);(A1)English 操作方法六To a vessel was added absolute ethanol (3000mL,3.0vol)followed by 5-bromo-2,4-dichloropyrimidine 6(mw 227.87;1000g,1.0equiv.).Triethylamine (612mL,1.0equiv.)was added,and then cyclopentylamine 5(mw 85.15;520mL,1.2equiv.)was added slowly over 2hours to control the mild exotherm.After completion of cyclopentylamine addition,the reaction was seeded with 5-bromo-2-chloro-6-cyclopentylamino-pyrimidine 3(5g,0.5wt%)to induce crystallization,if needed.The reaction was stirred at 25°C for 2hours.Water (2500mL,2.5vol)was added to the vessel at 20-25°C at a rate of 30mL/min.The mixture was cooled to 8-12°C at 2°C/min.The slurry was kept at 8-12°C for 1hour and then filtered onto a 2Whatman paper filter.The cake was rinsed with n-heptane (2000mL).The cake was reslurried with n-heptane on the filter drier (2000mL).The material was dried overnight in the vacuum oven at 50-55°C to give 5-bromo-2-chloro-6-cyclopentylamino-pyrimidine 3(1020g;84%)as a white solid.WO2014/128588;(2014);(A1)English(四)帕布昔利布(palbociclib )中间体8的合成合成方法实验步骤参考文献To a vessel was added 5-bromo-2-chloro-6-cyclopentyl-amino-pyridimidine 3(10.0g,1.0equiv.)along with。
palbociclib英文说明书

6 7 7
FULL PRESCRIBING INFORMATION 1 INDICATIONS AND USAGE
IBRANCE is indicated in combination with letrozole for the treatment of postmenopausal women with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer as initial endocrine-based therapy for their metastatic disease. This indication is approved under accelerated approval based on progression-free survival (PFS) [see Clinical Studies (14)] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. 2 2.1 DOSAGE AND ADMINISTRATION General Dosing Information
----------------------- WARNINGS AND PRECAUTIONS ---------------------- • Hematologic: Neutropenia may occur. Monitor complete blood count prior to start of IBRANCE therapy and at the beginning of each cycle, as well as on Day 14 of the first two cycles, and as clinically indicated. (5.1) • Infections: Monitor for signs and symptoms and withhold dosing as appropriate. (5.2) • Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.4, 8.1, 8.3) ------------------------------ ADVERSE REACTIONS ----------------------------- Most common adverse reactions (incidence ≥10%) were neutropenia, leukopenia, fatigue, anemia, upper respiratory infection, nausea, stomatitis, alopecia, diarrhea, thrombocytopenia, decreased appetite, vomiting, asthenia, peripheral neuropathy, and epistaxis. (6) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer, Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or /medwatch. ------------------------------ DRUG INTERACTIONS------------------------------ • CYP3A Inhibitors: Avoid concurrent use of IBRANCE with strong CYP3A inhibitors. If the strong inhibitor cannot be avoided, reduce the IBRANCE dose. (2.2, 7.1) • CYP3A Inducers: Avoid concurrent use of IBRANCE with strong and moderate CYP3A inducers. (7.2) • CYP3A Substrates: The dose of sensitive CYP3A4 substrates with narrow therapeutic indices may need to be reduced when given concurrently with IBRANCE. (7.3) See 17 for PATIENT COUNSELING INFORMATION and FDAapproved patient labeling. Revised: [02/2015]
爱博新(哌柏西利胶囊)说明书

核准日期:哌柏西利胶囊说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称:哌柏西利胶囊商品名称:爱博新® / IBRANCE®英文名称:Palbociclib Capsules汉语拼音:Paiboxili Jiaonang【成份】本品主要成份为哌柏西利其化学名称为:6-乙酰基-8-环戊基-5-甲基-2-[[5-(1-哌嗪基)-2-吡啶基]氨基]吡啶[2,3-d]嘧啶-7(8H)-酮化学结构式:分子式:C24H29N7O2分子量:447.53辅料名称:微晶纤维素、单水乳糖、羧甲基淀粉钠、胶态二氧化硅、硬脂酸镁【性状】本品为胶囊剂,内容物为类白色至黄色粉末。
【适应症】本品适用于激素受体(HR)阳性、人表皮生长因子受体2(HER2)阴性的局部晚期或转移性乳腺癌,应与芳香化酶抑制剂联合使用作为绝经后女性患者的初始内分泌治疗。
【规格】(1) 75 mg; (2) 100 mg; (3) 125 mg【用法用量】应由具抗癌药物使用经验的医生开始并监督本品治疗。
推荐剂量哌柏西利的推荐剂量为125 mg,每天一次,连续服用21 天,之后停药7 天(3/1 给药方案),28 天为一个治疗周期。
治疗应当持续进行,除非患者不再有临床获益或出现不可接受的毒性。
当与来曲唑联用时,来曲唑的推荐剂量为 2.5 mg,口服,每天一次,在整个28 天治疗周期连续服药。
具体请参见来曲唑批准的说明书。
给药方法口服。
应与食物同服,最好随餐服药以确保哌柏西利暴露量一致(见【药代动力学】)。
哌柏西利不得与葡萄柚或葡萄柚汁同服(见【药物相互作用】)。
哌柏西利胶囊应整粒吞服(吞服前不得咀嚼、压碎或打开胶囊)。
如果胶囊出现破损、裂纹或其他不完整的情况,则不得服用。
应鼓励患者在每天大约相同的时间服药。
如果患者呕吐或者漏服,当天不得补服。
应照常进行下次服药。
剂量调整建议根据个体安全性和耐受性调整哌柏西利的剂量。
出现某些不良反应时可能需要暂时中断/延迟给药和/或减低剂量,或永久停药来进行控制,请参照表1、2 和 3 中提供的方案进行剂量调整(见【注意事项】和【不良反应】)。
Ibrance中文说明书

【药物名】Palbociclib【商品名】Ibrance【美国上市时间】乳腺癌,2015年2月【类别】抑制剂【靶点】CDK4,CDK6【分子结构】分子式:C24H29N7O2化学名:6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one结构式为:分子量:447.54 Da【生产公司】Pfizer 辉瑞公司【购买地】美国【剂型和规格】口服胶囊,规格为:125 mg、100 mg和75 mg。
125mg胶囊:不透明硬明胶胶囊,大小0,在胶囊的焦糖帽上用白墨汁印有“Pfizer”,在浅橙色体上印有“PBC 125”。
100mg胶囊:不透明硬明胶胶囊,大小1,在胶囊的焦糖帽上用白墨汁印有“Pfizer”,在浅橙色胶囊体上印有“PBC 100”。
75mg胶囊:不透明硬明胶胶囊,大小2,在浅橙色胶囊帽上用白墨汁印有“Pfizer”,在浅橙色胶囊体上印有“PBC 75”。
【处方】Palbociclib是一种黄色至橙色粉有pKa为7.4(第二哌嗪氮)和3.9(吡啶氮)。
在或低于pH 4,Palbociclib行为如同高溶解度化合物。
高于pH 4,药物物质溶解度显著减低。
无活性成分:微晶纤维素,一水乳糖,羟基乙酸淀粉钠,胶体二氧化硅,硬脂酸镁,和硬明胶胶囊壳。
浅橙色,浅橙色/焦糖和焦糖不透明胶囊壳含明胶,红色氧化钛,黄色氧化铁,和二氧化钛;和印刷油墨含虫胶,二氧化钛,氢氧化铵,丙二醇和二甲基硅油。
【作用机理】Palbociclib是一种周期蛋白-依赖激酶(CDK)4和6的抑制剂。
周期蛋白D1和CDK4/6是导致细胞增殖信号通路的下游。
在体外,Palbociclib雌激素受体(ER)-阳性乳癌细胞株通过阻断细胞从细胞周期G1进入S期的进展减低细胞增殖。
用palbociclib和抗雌激素的联合处理与单独各个药物处理比较,乳癌细胞株导致视网膜母细胞瘤蛋白(Rb)磷酸化的减低导致减低E2F表达和信号和阻止增加生长。
Cancer Cell:CDK46抑制剂强大的临床应用仅仅是因为阻滞细胞周期?不,癌症研究不该局限于此

Cancer Cell:CDK4/6抑制剂强大的临床应用仅仅是因为阻滞细胞周期?不,癌症研究不该局限于此原创2018-05-04 订阅号APExBIO两种密切相关的细胞周期蛋白依赖性激酶CDK4和CDK6以及它们的结合伴侣cyclin D1、D2和D3是细胞周期进程中的关键调控因子。
Rb(视网膜母细胞瘤蛋白)是CDK4 / 6的主要细胞周期靶标。
图1 CDK4 / 6被抑制后细胞周期停滞Palbociclib(帕博西尼),Abemaciclib(玻玛西尼)和Ribociclib是目前三种针对CDK4 / 6的特异性抑制剂。
这些药物的毒性低,可广泛用于治疗各种肿瘤,在乳腺癌中应用最广泛。
CDK4 / 6抑制剂既可单独使用,也可以与其他药物一起联用,其临床潜力巨大。
因此,了解它们发挥抗肿瘤作用的机制显得尤为重要。
如图2中列举了Palbociclib与其他药物联用治疗不同癌症的临床试验结果。
图2 Palbociclib与其他药物联用治疗多种癌症(这里只列举部分,更多见原始文献)CDK4 / 6抑制剂属于新一代治疗药物。
Palbociclib是第一个显示临床疗效的CDK4 / 6抑制剂,Ribociclib在结构上与Palbociclib非常相似。
很多体外研究确定,Abemaciclib和Ribociclib对CDK4比对CDK6更有效,Palbociclib具有相似的效力。
Palbociclib,Abemaciclib和Ribociclib这三种药物均可口服,但各自具有不同的药代动力学和临床毒性,如图3所示。
图3 三种药物对比自CDK4 / 6抑制剂与激素受体拮抗剂来曲唑的联合使用在乳腺癌中取得显著成功后,近来更多其他组合已进入多种疾病的临床试验。
许多早期临床试验的结果显示CDK4 / 6抑制剂可以增强其他信号通路靶点抑制剂的效力(图2)。
CDK4 /6抑制剂与ER、PI3K/mTOR、MEK、aromatase、FGFR、BTK、RTK等靶点的抑制剂联合使用,具有很好的临床效果。
Palbociclib_isethionate_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Palbociclib (isethionate) is a highly selective inhibitor of CDK4/6 with IC 50s of 11 nM/16 nM, and shows no activity against CDK1/2/5,EGFR, FGFR, PDGFR, InsR, etc.IC50 & Target: IC50: 11 nM (CDK4), 16 nM (CDK6)In Vitro: Palbociclib exhibits absolute selectivity for CDK4/6 with little or no activity against other CDKs. Palbociclib is effective at reducing Rb phosphorylation at Ser 780 and Ser 795 in MDA–MB–435 breast carcinoma cells with IC 50 of 66 nM and 63 nM,respectively. Palbociclib is a potent inhibitor of cell growth and suppresses DNA replication by preventing cells from entering Sphase. Palbociclib inhibits thymidine incorporation into the DNA of Rb–positive human breast (such as MDA–MB–435, MCF–7), colon (H1299), and lung carcinomas (Colo–205) as well as human leukemias (CRRF–CEM and K562), with IC 50 values rangingfrom 0.04–0.17 μM. Palbociclib significant increases the percentage of MDA–MB–453 in G1 period [1]. Palbociclib inhibitsphosphorylation of Rb in cycling CD138+ primary bone marrow myeloma cells, nontransformed primary B cells, MM1.S and CAG HMCLs cells line with IC 50 of <0.1 μM, 0.05 μM, and 60–70 nM, respectively. Palbociclib treatment also induces G1 arrest of CD138+primary bone marrow myeloma and nontransformed primary B cells. Palbociclib induces G1 arrest in MM1.S with IC 50 ofappr 0.05 μM [2]. Palbociclib preferentially inhibits proliferation of luminal estrogen receptor–positive (including HER2–positive)human breast cancer cell lines. Palbociclib increases gene expression of pRb and cyclin D1 and decreases gene expression of CDKN2A (p16) in most sensitive lines. Palbociclib enhances sensitivity to tamoxifen in cell lines with conditioned resistance to ER blockade [3].In Vivo: Palbociclib(150 mg/kg. p.o.) produces rapid Colo–205 colon carcinoma xenografts regressions and acorresponding tumor growth delay. Palbociclib (150 mg/kg, p.o.) induces complete tumor stasis and cell kill inMDA–MB–435 breast carcinoma. Palbociclib (150 mg/kg) also induces significant tumor regression in micebearing the SF–295 glioblastoma xenografts, and in ZR–75–1 breast and PC–3 prostate tumor models (completesuppression of tumor growth). Palbociclib (150 mg/kg) suppresses Rb Ser 780 phosphorylation in MDA–MB–435 breastcarcinoma over the full 24–hour period. Palbociclib (150 mg/kg) down–regulates expression of four E2F–regulatedgenes CDC2, CCNE2, TK1, and TOP2A in Colo–205 carcinoma xenografts [1]. Palbociclib also rapidly inhibits myeloma tumor growth [2]. PROTOCOL (Extracted from published papers and Only for reference)Cell Assay:[3]Cells are seeded in duplicate at 5,000 to 10,000 cells per well in 24–well plates. The day after plating, differentconcentrations of Palbociclib are added. Control wells without drug are also seeded. At the end of incubation, cells are trypsinizated and placed in Isotone solution and counted immediately using a Coulter Z2 particle counter.Animal Administration:[1]Mice (18–22 g) are randomized and then implanted s.c. with tumor fragments (appr 30 mg) into the region of the right axilla. Treatment is initiated when tumors reach 100 to 150 mg. Palbociclib is given according to the schedule and doseProduct Name:Palbociclib (isethionate)Cat. No.:HY-A0065CAS No.:827022-33-3Molecular Formula:C 26H 35N 7O 6S Molecular Weight:573.66Target:CDK Pathway:Cell Cycle/DNA Damage Solubility:H 2O: ≥ 66.66 mg/mLindicated in the table and figure legends by gavage as a solution in sodium lactate buffer (50 mM, pH 4.0) based on mean group body weight. In all experiments, there are 12 mice in the control group and 8 mice each in the treated groups. Additional details for each experiment are given in the table legends.References:[1]. Fry DW, et al. Specific inhibition of cyclin–dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther, 2004, 3(11), 1427–1438.[2]. Baughn LB, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin–dependent kinase 4/6. Cancer Res. 2006 Aug 1;66(15):7661–7.[3]. Finn RS, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor–positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
帕博西尼说明书翻译

Ibrance(palbociclib)使用说明书2015年第一版批准日期:2015年2月3日;公司:PfizerInc.加速批准,突破性治疗指定和优先审评FDA药品评价和研究中心血液学和肿瘤产品室主任说:“palbociclib添加至来曲唑对被诊断有转移乳癌妇女提供一种新颖治疗选择,”“FDA承诺通过我们的加快批准监管加快癌症药物的上市批准。
”完整处方资料。
IBRANCE人表皮生剂量和给药方法?IBRANCE胶囊是与食物与来曲唑联用口服。
(2)?⑴推荐开始剂量:125mg每天一次与食物服用共21天接着7天不治疗。
(2.1)?⑵建议根据个体安全性和耐受性中断和/或剂量减低给药。
(2.2)?剂型和规格胶囊:125mg,100mg,和75mg(3)?禁忌证无(4)?警告和注意事项⑴血液学:可能发生中性粒细胞减少。
监视完全血细胞计数IBRANCE治疗开始前和在每个疗程开始,以及在头两个疗程第14天,和当临床指示时。
(5.1)?(5.4,8.1,不良反应CYP3ACYP3ACYP3A底物:有狭窄治疗指数敏感CYP3A4底物当与IBRANCE同时给予时剂量可能需要减低。
(7.3)?完整处方资料1适应证和用途?IBRANCE是适用与来曲唑联用对有雌激素受体(ER)-阳性,人表皮生长因子受体2(HER2)-阴性晚期乳癌绝经后妇女作为初始基于内分泌治疗对其转移疾病的治疗.? 这个适应证是根据无进展生存(PFS)在加速批准下被批准的[见临床研究(14)]。
对此适应证的继续批准可能取决于在验证性试验中临床获益的证明和描述。
2剂量和给药方法?2.1一般给药信息?IBRANCE疗7与来曲唑剂量。
2.2/或减低剂(5),不良反应(6)和临床研究(14)]。
见制造商处方资料对共同给药产品,来曲唑,在毒性事件中剂量调整指导原则和其他相关安全性资料或禁忌证。
为与强避免强如抑制3125mg“PBC125”100mg体上75mg“PBC75”。
Palbociclib (PD-0332991)高度选择性的CDK4和6抑制剂生物活性CAS号827022-32-2

Cdk4/6 Inhibition Induces Epithelial-Mesenchymal Transition and Enhances Invasiveness in Pancreatic Cancer Cells. Liu F et al. Mol Cancer Ther. 2012 Aug 6. PMID: 22869556.
实验操作 来自于公开的文献,仅供参考
细胞实验
细胞系 方法
HCT116, SW480, Lovo and LS174T cells
Cell proliferation and colony formation assays Cells were seeded in a 96-well plate at 2000–6000 cells/well, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cell proliferation was determined using MTS solution (Promega), and formal assessment for synergy performed per the Chou Talalay method [64, 65] using CompuSyn (ComboSyn, Inc, Paramus, NJ). For colony formation assay, cells were seeded in a 6-well plate at 8000– 80,000 cells/well, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 2–3 weeks. Cell colonies were fixed with ice-cold methanol and stained with 1% crystal violet. The density of colonies over the plate area was quantified by ImageJ (NIH) [66]. For detection of exposed phosphatidylserine residues reflective of apoptosis, Annexin V-FITC apoptosis assay kit (BD Biosciences) was used. Cells were seeded at 500,000 cells/ well in 6-well plates, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cells were washed and stained with annexin V-FITC and propidium iodide, and flow cytometry was performed using the Gallios flow cytometer (Beckman Coulter) and analyzed with Kaluza flow analysis software (Beckman Coulter). To measure cell senescence, treated cells were stained for senescence-associated beta-galactosidase (Chemicon). Cells were seeded at 500,000 cells/well in 6-well plates, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cells were washed with PBS, fixed with glutaraldehyde and methanol, washed twice more, and then incubated overnight in the dark at 37°C under ambient atmospheric conditions with X-Gal. Subsequently, cells were washed, and 10 high-powered light microscopy images of each well were captured, and cells with blue staining were manually counted.
乳腺癌用药 哌柏西利 palbociclib

乳腺癌用药哌柏西利 palbociclib制剂与规格:胶囊:75mg、100mg、125mg适应证:本品适用于激素受体(HR)阳性、HER2阴性的局部晚期或转移性乳腺癌:(1)与芳香化酶抑制剂联合使用,作为绝经后女性患者的初始内分泌治疗。
鉴于芳香化酶抑制剂的作用机制,绝经前/围绝经期女性接受哌柏西利与芳香化酶抑制剂联合治疗时,必须进行卵巢切除或使用促黄体生成激素释放激素(LHRH)激动剂抑制卵巢功能。
(2)与氟维司群联合使用治疗内分泌治疗后进展的转移性乳腺癌女性患者。
哌柏西利联合氟维司群用于绝经前/围绝经期女性,需要与LHRH激动剂联合用药。
(3)男性乳腺癌:2019年4月4日,FDA批准了美国哌柏西利HR阳性、HER2阴性晚期男性乳腺癌的适应证。
合理用药要点:1.在接受哌柏西利治疗前,应在有资质的病理实验室进行检测证实HR阳性、HER2阴性患者方可使用。
HR阳性的定义为雌激素受体免疫组化染色显示超过1%的肿瘤细胞核染色阳性。
HER2阴性的定义为IHCO-1+或FISH阴性。
12.本品起始剂量是125mg/d,4周为一个用药周期:服药3周后需停药1周。
应与食物同服,不得与葡萄柚或葡萄柚汁同服,最好随餐服药以确保哌柏西利暴露量一致。
3.常见副作用为骨髓抑制,因此建议在使用本品前行血常规检查,在中性粒细胞绝对计数≥1.0×109/L且血小板计数≥50×109/L时开始接受治疗。
在第15天检测血常规,中性粒细胞绝对计数0.5×109/L至≤1.0×109/L时,可以继续服药至21天。
如果第15天中性粒细胞绝对计数≤0.5×109/L 时,需暂停服用哌柏西利,直至恢复至≥1.0×109/L,再以降低一个剂量级开始下一疗程治疗。
如果下个疗程前一天检测血常规,中性粒细胞恢复至≥1.0×109/L,可以原剂量开始下一疗程,但如果延迟恢复,则需要降低一个剂量级开始下一疗程。
Palbociclib_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Palbociclib is a highly specific inhibitor of Cdk4 (IC 50=11 nM) and Cdk6 (IC 50=16 nM), having no activity against a panel of 36additional protein kinases.IC50 & Target: IC50: 11 nM (Cdk4), 16 nM (Cdk6)[1]In Vitro: The IC 50 of Palbociclib (PD 0332991) for reduction of retinoblastoma (Rb) phosphorylation at Ser 780 in MDA–MB–435breast carcinoma cells is 66 nM. Palbociclib is equally effective at reducing Rb phosphorylation at Ser 795 in this tumor with an IC 50of 63 nM, and similar effects on both Ser 780 and Ser 795 phosphorylation are obtained in the Colo–205 colon carcinoma [1]. The MP–MRT–AN (AN), KP–MRT–RY (RY), G401, and KP–MRT–NS (NS) cell lines are effectively inhibited by Palbociclib (PD) in aconcentration–dependent manner in a WST–8 assay. The IC 50s are 0.01 μM, 0.01 μM, 0.06 μM, and 0.6 μM, respectively. Incontrast, the KP–MRT–YM (YM) cell line is resistant to Palbociclib (IC 50>10 μM). The flow cytometry results show that Palbociclib at concentrations between 0 to 1.0 μM induces G1 arrest in the AN, RY, G401 and NS cell lines in a concentration–dependent manner,but has no effect on YM cells. The BrdU incorporation results are consistent with the WST–8 and flow cytometry results: PD reduces BrdU incorporation (indicating G1 arrest) in the AN, RY, G401 and NS cell lines, but not in the YM cell line. Palbociclib, even at a concentration of 0.05 μM, significantly reduces BrdU incorporation in the AN, RY, and G401 cell lines (p<0.05)[2].In Vivo: Palbociclib (PD 0332991) exhibits significant antitumor efficacy against multiple human tumor xenograft models. In mice bearing Colo–205 colon carcinoma xenografts (p16 deleted), daily p.o. dosing for 14 days with Palbociclib (150 or 75 mg/kg)produces rapid tumor regressions and a corresponding tumor growth delay of ~50 days with >1 log of tumor cell kill at the highest dose tested. At 37.5 mg/kg, the tumor slowly regressed during treatment. Even at doses as low as 12.5 mg/kg, a 13–day growth delay is obtained indicating a 90% inhibition of tumor growth rate. Likewise, robust antitumor activity is seen in the MDA–MB–435breast carcinoma (p16 deleted) where complete tumor stasis is apparent at 150 mg/kg and some cell kill is evident at the highest dose [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]CDK assays are performed in 96–well filter plates. All CDK–cyclin kinase complexes are expressed in insect cells through baculovirus infection and purified. The substrate for the assays is a fragment (amino acids 792–928) of pRb fused to GST (GST.RB–Cterm). The total volume in each well is 0.1 mL containing a final concentration of 20 mM Tris–HCl, pH 7.4, 50 mM NaCl, 1mM dithiothreitol, 10 mM MgCl 2, 25 μM ATP (for CDK4–cyclin D1, CDK6–cyclin D2, and CDK6–cyclin D3) or 12 μM ATP (for CDK2–cyclin E, CDK2–cyclin A, and CDC2–cyclin B) containing 0.25 μCi of [γ–32P]ATP, 20 ng of enzyme, 1 μg of GST.RB–Cterm,and Palbociclib (0.001–0.1μM). All components except the [γ–32P]ATP are added to the wells, and the plate is placed on a plate mixer for 2 min. The reaction is started by adding the [γ–32P]ATP and the plate is incubated at 25°C for 15 min. The reaction is terminated by addition of 0.1 mL of 20% trichloroacetic acid and the plate is kept at 4°C for at least 1 hour to allow the substrate toProduct Name:Palbociclib Cat. No.:HY-50767CAS No.:571190-30-2Molecular Formula:C 24H 29N 7O 2Molecular Weight:447.53Target:CDK Pathway:Cell Cycle/DNA Damage Solubility:DMSO: 0.2 mg/mL (Need ultrasonic or warming)precipitate. The wells are then washed 5 times with 0.2 mL of 10% trichloroacetic acid and radioactive incorporation is determined with a β plate counter.Cell Assay: Palbociclib (PD) is prepared in DMSO and stored (-80°C), and then diluted with appropriate media before use[1].[1]MRT cell lines, G401, MP–MRT–AN (AN), KP–MRT–RY (RY), KP–MRT–NS (NS), and KP–MRT–YM (YM) cell lines are seeded in normal growth medium into 96–well cell plates. After 24 h, the culture medium is replaced with culture medium containing Palbociclib (0.05 or 1 μM) or DMSO. Cells are cultured and treated in triplicate. Cell proliferation is determined 8 days after the treatment by WST–8 assay using a Cell Counting Kit–8.Animal Administration: Palbociclib (PD 0332991) is prepared as a solution in sodium lactate buffer (50 mM, pH 4.0) (Mice)[1].[1] Mice (18–22 g) are randomized and then implanted s.c. with tumor fragments (30 mg) into the region of the right axilla. Treatment is initiated when tumors reach 100 to 150 mg. PD 0332991 (150 or 75 mg/kg, p.o.) is given according to the schedule and dose indicated in the table and figure legends by gavage as a solution in sodium lactate buffer (50 mM, pH 4.0) based on mean group body weight. In all experiments, there are 12 mice in the control group and 8 mice each in the treated groups. Additional details for each experiment are given in the table legends.References:[1]. Fry DW, et al. Specific inhibition of cyclin–dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427–38.[2]. Katsumi Y, et al. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun, 2011, 413(1), 62–68.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
KRAS突变非小细胞肺癌的预后和疗效

KRAS突变⾮⼩细胞肺癌的预后和疗效RAS癌基因参予细胞⽣长和分化的调控,参与多种肿瘤的形成与发展。
与⼈类肿瘤相关的RAS基因有三种,即HRAS、KRAS和NRAS,它们分别定位于11、12、1号染⾊体,见图1。
KRAS突变已被明确为NSCLC发⽣的驱动基因,在肺鳞癌中⽐例极少,在东亚国家肺腺癌中约占8-10%,在欧美国家肺腺癌中约占20-30%。
中国肺腺癌中KRAS突变⽐例为8.3%,优势⼈群为男性、吸烟史、浸润性黏液型腺癌和实体型腺癌,G12C最常见(33.6%),G12D(23.9%)和G12V(22.1%)其次,见图2。
疾病预后KRAS突变⾮⼩细胞肺癌患者与EGFR/ALK/ROS1突变患者相⽐,有较短的⽣存期。
⽽且KRAS突变患者有显著的临床异质性,与KRAS突变亚型⽆关,与其共存基因突变有关。
美国纪念斯隆-凯特琳癌症中⼼应⽤⼆代测序⽅法筛选出330例KRAS突变的晚期⾮⼩细胞肺癌患者,评估共存的最常见的基因组改变。
见图2,TP53、STK11(LKB1)和KEAP1/NFE2L2是最常见的共存基因突变。
STK11(LKB1)或KEAP1/NFE2L2共存突变患者总⽣存期明显缩短,⽽TP53共存突变对总⽣存期没有影响,见图3。
多变量分析显⽰,KEAP1/NFE2L2突变是独⽴的不良预后预测因⼦,⽽TP53或STK11(LKB1)突变与预后⽆关*。
*STK11(LKB1)突变患者中63%同时也有KEAP1/NFE2L2突变。
临床前研究显⽰,KEAP1/NFE2L2突变对铂类耐药,导致含铂化疗⽅案疗效较差。
KEAP1/NFE2L2共存突变的KRAS⾮⼩细胞肺癌患者对PD1/PD-L1免疫治疗响应也较差。
化疗疗效荷兰的⼀个回顾性研究显⽰,晚期KRAS突变NSCLC⼀线化疗采⽤紫杉醇⽅案的疗效最佳,总有效率50%,联合贝伐单抗时更⾼达62%,培美曲赛⽅案的总有效率为21%,吉西他滨⽅案的总有效率为25%,具体数据见表1。
国内第2款CDK46抑制剂获批,乳腺癌患者又有治疗新选择!

国内第2款CDK46抑制剂获批,乳腺癌患者⼜有治疗新选择!好消息!2021年1⽉5⽇,礼来制药正式宣布阿贝西利⽚于2020年12⽉29⽇获得国家药品监督管理局(N M P A)的批准于国内上市。
阿贝西利的适应症为:1、与芳⾹化酶抑制剂联⽤,作为激素受体(H R)阳性、H E R2阴性局部晚期或转移性绝经后⼥性乳腺癌患者初始内分泌治疗;2、与氟维司群联⽤,治疗初始内分泌治疗失败疾病进展的晚期或转移性H R+/H E R2-⼥性乳腺癌患者。
阿贝西利是继哌柏西利后国内上市的第2款C D K4/6抑制剂。
阿贝西利的上市同时也意味着中国⼴⼤H R+/H E R2-晚期乳腺癌患者⼜拥有了⼀项治疗新选择。
⼤家可能对C D K4/6抑制剂的认识并不多,那就跟着科普君⼀起了解下这类药物吧。
1C D K4 /6抑制剂如何发挥作⽤?C D K4 /6是细胞周期的关键调节因⼦,通过与细胞周期蛋⽩形成复合物。
对于H R+乳腺癌患者⽽⾔,体内的C D K4 /6过度表达最终可导致细胞增殖失控,从⽽演变成恶性肿瘤。
⽽C D K4 /6抑制剂可阻断C D K4 /6激酶活性,降低R b蛋⽩磷酸化⽔平,使其⽆法释放转录因⼦,抑制癌细胞分裂增殖,恢复细胞周期控制,阻断肿瘤细胞增殖,抑制乳腺癌细胞⽣长。
疗效如何呢?Palbociclib(哌柏西利,商品名:爱博新)Abemaciclib(阿贝西利,商品名:唯择)Ribociclib(玻马西尼)M O N A R C H pl us是第⼀个、同时也是唯⼀的在以中国患者为主的H R+,H E R2-晚期乳腺癌⼥性中证实C D K4和C D K6抑制剂临床获益的国际多中⼼I I I期临床研究。
M O N A R C H pl us研究结果证实,阿贝西利联合⾮甾体芳⾹化酶抑制剂使⽆疾病进展⽣存期(P F S)得到显著改善;阿贝西利联合氟维司群相⽐单药氟维司群治疗延长中位P F S 5.88个⽉。
辉瑞乳腺癌药物Palbociclib的合成

辉瑞乳腺癌药物Palbociclib获FDA优先审评资格背景介绍:2014年10月13日,辉瑞宣布FDA授予该公司Palbociclib上市申请优先审评资格,这款药物与诺华来曲唑合并用药作为一线疗法用于先前未接受过系统治疗的雌激素受体阳性(ER+)、HER2阴性晚期乳腺癌绝经后女性。
于今年8月份提交的该上市申请的审评期限到2015年4月13日结束。
辉瑞公司最近发布了PALOMA-1研究的2期临床试验结果,结果非常振奋人心,对乳腺癌患者来说可能是一个好消息。
该研究发现,palbociclib联合来曲唑对绝经后的局部浸润性乳腺癌患者或新近诊断的雌激素受体(ER)阳性,HER-2阴性的转移性乳腺癌患者非常有效。
Palbociclib(正式名PD-0332991)最早进入人们视野的是在2012年圣安东尼奥乳腺癌会议上(SABCS),一经发布就引起行业广泛关注。
Palbociclib是一种口服的细胞周期素依赖性激酶4、6的抑制药物,主要通过调节细胞周期发挥作用。
Palbociclib 主要通过抑制CDK4/6活性来阻止细胞由G1期到S期进而抑制DNA的合成。
来自加州大学洛杉矶分校Jonsson 综合癌症中心的Richard S. Finn医生在此次会议上公布了PALOMA-1研究的中期研究结果:Palbociclib联合来曲唑可将乳腺癌患者的中位无疾病生存期(PFS)提高到26.1个月,而单用来曲唑的PFS只有7.5个月。
基于这一中期研究结果,2013年4月FDA授予Palbociclib治疗转移性乳腺癌突破性治疗药物。
辉瑞临床肿瘤部医疗事务高级副总裁、首席医疗官Mace Rothenberg医生称:“对于绝经后ER阳性、HER2阴性的晚期乳腺癌患者,这可能是一个非常振奋人心的消息。
”palbociclib可能会改变绝经后ER阳性、HER2阴性晚期乳腺癌患者传统的治疗方案。
我们不久将和FDA及其他医疗监管机构讨论该试验的一系列结果以决定接下来的研究方向,希望该药能尽快上市。
palbociclib 说明书

处方信息要点---------------------- 警告和注意事项 --------------------------这些要点并未包含安全和有效使用 IBRANCE 所需的所有信息。
请参见 IBRANCE 的完整处方信息。
• 血液方面:可能发生中性粒细胞减少症。
在IBRANCE 治疗开始前、每个周期开始时以及前两个周期的第 14 天监测全血细胞计数。
(5.1) IBRANCE ®(Palbociclib )口服胶囊 美国首次批准时间:2015 年 • 感染:监测体征和症状并酌情暂停给药。
(5.2)------------------------ 适应症及用法 ------------------------------------•胚胎-胎仔毒性:可导致胎仔伤害。
向患者告知对胎儿的毒性并使用有效避孕措施。
(5.4, 8.1, 8.3)IBRANCE 是一种激酶抑制剂,适用于与来曲唑联合用药,治疗雌激素受体(ER )阳性、人体表皮生长因子受体 2(HER2)阴性的晚期乳腺癌绝经后女性患者,以作为针对转移性疾病的初始内分泌疗法。
该适应症根据无进展生存期(PFS )获得加速批准。
该适应症的继续批准可能取决于在一项验证性试验中的临床获益验证和描述。
(1)--------------------------- 不良反应 -----------------------------最常见的不良反应(发生率 ≥10%)包括中性粒细胞减少症、白细胞减少症、疲劳、贫血、上呼吸道感染、恶心、口腔炎、脱发、腹泻、血小板减少症、食欲减退、呕吐、衰弱、周围神经病变和鼻衄。
(6) -------------------- 用法用量 -------------------------------------------如要报告疑似不良反应,请通过以下方式联系辉瑞公司:与来曲唑合用时,IBRANCE 胶囊与食物同服。
收藏贴!乳腺癌靶向药物:帕博西尼Ibrance(palbociclib)显奇效

收藏贴!乳腺癌靶向药物:帕博西尼Ibrance(palbociclib)显奇效全球肿瘤医生网 second_opinion全球肿瘤医生网,国内最专业的综合抗癌网站,肿瘤国内顶尖专家会诊、国际顶尖专家会诊、国际癌症中心就医、出国就医、顶尖癌症基因检测个性化治疗平台。
癌症治疗进展可能令人沮丧, 然而在加州大学洛杉矶分校(UCLA)肿瘤学家Richard Finn报告说,在乳腺癌的标准治疗中增加一种名为帕博西尼Ibrance(palbociclib)的药物,较来曲唑可使乳腺癌患者无进展生存期,从平均10个月增加到20个月。
因为其良好的治疗效果,2015年4月16日,辉瑞(Pfizer)突破性乳腺癌药物Ibrance (palbociclib)在2015年2月凭借一项II期研究的无进展生存期(PFS)数据获得了FDA的加速批准,用作乳腺癌的一线治疗。
IBRANCE(palbociclib) capsules 帕博西尼IBRANCE(palbociclib) capsules 帕博西尼, palbociclib是一种实验性、口服、靶向性制剂,能够选择性抑制细胞周期蛋白依赖性激酶4和6(CDK4/6),恢复细胞周期控制,阻断肿瘤细胞增殖。
细胞周期失控是癌症的一个标志性特征,CDK4/6在许多癌症中均过度活跃,导致细胞增殖失控。
CDK4/6是细胞周期的关键调节因子,能够触发细胞周期从生长期(G1期)向DNA复制期(S1期)转变。
在雌激素受体阳性(ER+)乳腺癌中,CDK4/6的过度活跃非常频繁,而CDK4/6是ER信号的关键下游靶标。
临床前数据表明,CDK4/6和ER 信号双重抑制具有协同作用,并能够抑制G1期ER+乳腺癌细胞的生长。
IBRANCE是一种口服胶囊,一般与来曲唑联用。
推荐剂量:125mg每天一次与食物服用共21天接着7天不治疗。
FDA于2013年4月授予palbociclib治疗晚期或转移性ER+/HER2-乳腺癌的突破性疗法认定。
爱博新(哌柏西利胶囊)说明书

核准日期:之南宫帮珍创作哌柏西利胶囊说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称:哌柏西利胶囊商品名称:爱博新® / IBRANCE®英文名称:Palbociclib Capsules汉语拼音:Paiboxili Jiaonang【成分】本品主要成分为哌柏西利其化学名称为:6-乙酰基-8-环戊基-5-甲基-2-[[5-(1-哌嗪基)-2-吡啶基]氨基]吡啶[2,3-d]嘧啶-7(8H)-酮化学结构式:分子式:C24H29N7O2分子量:辅料名称:微晶纤维素、单水乳糖、羧甲基淀粉钠、胶态二氧化硅、硬脂酸镁【性状】本品为胶囊剂, 内容物为类白色至黄色粉末.【适应症】本品适用于激素受体(HR)阳性、人表皮生长因子受体2(HER2)阴性的局部晚期或转移性乳腺癌, 应与芳香化酶抑制剂联合使用作为绝经后女性患者的初始内分泌治疗.【规格】(1)75mg; (2) 100 mg; (3) 125mg【用法用量】应由具抗癌药物使用经验的医生开始并监督本品治疗.推荐剂量哌柏西利的推荐剂量为 125 mg, 每天一次, 连续服用 21 天, 之后停药 7 天(3/1 给药方案), 28 天为一个治疗周期.治疗应当继续进行, 除非患者不再有临床获益或呈现不成接受的毒性.当与来曲唑联用时, 来曲唑的推荐剂量为 2.5 mg, 口服, 每天一次, 在整个 28 天治疗周期连续服药.具体请拜会来曲唑批准的说明书.给药方法口服.应与食物同服, 最好随餐服药以确保哌柏西利流露量一致(见【药代动力学】).哌柏西利不得与葡萄柚或葡萄柚汁同服(见【药物相互作用】).哌柏西利胶囊应整粒吞服(吞服前不得咀嚼、压碎或翻开胶囊).如果胶囊呈现破损、裂纹或其他不完整的情况, 则不得服用.应鼓励患者在每天年夜约相同的时间服药.如果患者呕吐或者漏服,当天不得补服.应照常进行下次服药.剂量调整建议根据个体平安性和耐受性调整哌柏西利的剂量.呈现某些不良反应时可能需要暂时中断/延迟给药和/或减低剂量, 或永久停药来进行控制, 请参照表 1、2 和 3 中提供的方案进行剂量调整(见【注意事项】和【不良反应】).表 1.呈现不良反应时哌柏西利剂量调整的建议在开始哌柏西利治疗前、每个治疗周期开始时、前 2 个治疗周期的第 15天以及有临床指征时应监测全血细胞计数.对前6个治疗周期内发生最高严重水平为1或2级中性粒细胞减少症的患者, 其后续周期的全血细胞计数监测时间应为每3个月一次、各周期开始之前以及有临床指征时.建议在中性粒细胞绝对计数(Absolute Neutrophil Count, ANC)≥1,000/mm3 且血小板计数≥50,000/mm3时接受哌柏西利.的剂量调整和管理—血液学毒性ANC = 中性粒细胞绝对计数;CTCAE = 不良事件通用术语标准;LLN = 正常值下限.a 表格适用于除淋巴细胞减少症以外的所有血液学不良事件(除非陪伴临床事件, 如机会性感染)bANC :1 级:ANC < LLN - 1500/mm 3; 2 级:ANC 1000 - <1500/mm 3; 3 级:ANC 500 <1000/mm 3; 4 级:ANC <500/mm 3.的剂量调整和管理—非血液学毒性CTCAE = 不良事件通用术语标准.特殊人群老年人≥65 岁的患者无需调整哌柏西利的剂量(见【药代动力学】).儿科人群尚未确定哌柏西利在≤18 岁儿童和青少年患者中的平安性和疗效.没有数据可用.肝损伤轻度或中度肝损伤患者(Child-Pugh A 级和B 级)无需调整哌柏西利的剂量.重度肝损伤(Child-Pugh C 级)患者的推荐剂量为75 mg, 每天一次, 采纳3/1给药方案(见【注意事项】和【药代动力学】).肾损伤轻度、中度或重度肾损伤患者(肌酐清除率[Creatinine Clearance, CrCl] ≥15mL/min)无需调整哌柏西利的剂量.需要血液透析患者的数据不充沛, 无法对该人群提供任何剂量调整建议(见【注意事项】和【药代动力学】).与 CYP3A 强效抑制剂合用时的剂量调整防止陪伴使用 CYP3A 强效抑制剂, 考虑替换为没有或只有微弱CYP3A 抑制作用的其他陪伴用药.如果患者必需合用 CYP3A 强效抑制剂, 则将哌柏西利的剂量减少至 75 mg, 每天一次.如果停用强效抑制剂, 则将哌柏西利的剂量增加至开始使用 CYP3A 强效抑制剂之前的剂量(在抑制剂的 3 至 5 个半衰期后)[拜会【药物相互作用】和【药代动力学】]【不良反应】本说明书描述了在临床试验中观察到的判断为可能由哌柏西利引起的不良反应及其近似的发生率.由于每项临床试验的条件各不相同, 在一个临床试验中观察到的不良反应的发生率不能与另一个临床试验观察到的不良反应发生率直接比力, 也可能不能反映临床实践中的实际发生率.平安性特征概要哌柏西利的总体平安性特征评估来自在HR 阳性、HER2 阴性晚期或转移性乳腺癌随机研究中接受哌柏西利与内分泌疗法联合治疗(527 例与来曲唑联用和 345例与氟维司群联用)的 872 例患者的合并数据 [包括研究PALOMA-1 (A5481003), 研究PALOMA-2(A5481008), 研究PALOMA-3 (A5481023)].临床研究中, 接受哌柏西利治疗的患者陈说的最罕见(≥20%)的任何级另外不良反应为中性粒细胞减少症、感染、白细胞减少症、疲乏、恶心、口腔炎、贫血、脱发和腹泻.哌柏西利的最罕见(≥2%)的≥3 级不良反应为中性粒细胞减少症、白细胞减少症、贫血、疲乏和感染.在研究 PALOMA-2中评估了哌柏西利(125 mg/天)联合来曲唑(2.5 mg/天)治疗对比抚慰剂联合来曲唑治疗的平安性.哌柏西利联合来曲唑的中位治疗继续时间为 19.8 个月, 而抚慰剂联合来曲唑的中位治疗继续时间为 13.8 个月.在接受哌柏西利联合来曲唑治疗的患者中, 有 36% 的患者因任何级另外不良反应而减量.43/444(9.7%)例接受哌柏西利联合来曲唑治疗的患者以及13/222(5.9%)例接受抚慰剂联合来曲唑治疗的患者发生了与不良反应相关的永久停药.招致接受哌柏西利联合曲唑治疗的患者永久停药的不良反应包括中性粒细胞减少症(1.1%)和丙氨酸转氨酶升高(0.7%).不良反应列表表 4 陈说了 3 项随机研究 [研究PALOMA-1 (A5481003), 研究PALOMA-2 (A5481008), 研究PALOMA-3 (A5481023) ] 的合并数据集中的不良反应.合并数据集中哌柏西利治疗的中位继续时间为12.7 个月.表5陈说了3项随机研究的合并数据集中的实验室检查异常.按系统器官分类和发生频率列出不良反应.发生频率界说为:十分罕见(≥1/10)、罕见(≥1/100 至 <1/10)和偶见(≥1/1,000 至 <1/100).a依照 17.1 版 MedDRA 列出首选术语(Preferred Term, PT).b感染包括系统器官分类感染和侵染类疾病部份的所有 PT.c中性粒细胞减少症包括以下 PT:中性粒细胞减少症、中性粒细胞计数降低.d白细胞减少症包括以下 PT:白细胞减少症、白细胞计数降低.e贫血包括以下 PT:贫血、血红卵白降低、血细胞比容降低.f血小板减少症包括以下 PT:血小板减少症、血小板计数降低.g口腔炎包括以下PT:阿弗他性口腔黏膜炎、唇炎、舌炎、舌痛、口腔溃疡、粘膜炎症、口腔疼痛、口咽不适感、口咽疼痛、口腔黏膜炎.h皮疹包括以下PT:皮疹、斑丘疹、皮疹瘙痒、红斑皮疹、丘疹样皮疹、皮炎、痤疮样皮炎、毒性斑疹.表 5. 基于3项随机研究(N=872)合并数据集的实验室检查异常WBC-白细胞;AST-天冬氨酸氨基转移酶;ALT-丙氨酸氨基转移酶;N-患者人数;N/A-不适用.注:实验室检查结果依照NCI CTCAE版本4.0严重水平级别分级.* 来曲唑或氟维司群在PALOMA-2和PALOMA-3两项研究中, 纳入了200例亚裔患者.在接受哌柏西利的亚裔患者中陈说的 3 级或 4 级中性粒细胞减少症和白细胞减少症发生率高于非亚裔患者, 因而在亚裔患者中的剂量中断、剂量减少和周期延迟发生频率也略高于非亚裔患者, 但通过方案规定的剂量调整可控制总体平安性, 亚裔患者与非亚裔患者具有相似的中位治疗继续时间.根据对已有的哌柏西利剂量流露、平安性和疗效数据进行的累积分析, 将125mg每日一次作为亚裔患者的起始剂量是恰当的.需根据患者个体的平安性和耐受性, 严格遵循说明书调整哌柏西利剂量.特定不良反应的描述总体而言, 3项随机研究中有703 例(80.6%)无论以何种联用方案接受哌柏西利治疗的患者陈说了任何级另外中性粒细胞减少症, 其中分别有 482 例(55.3%)和 88 例(10.1 %)患者陈说 3 级和 4 级中性粒细胞减少症(见表 4).3 项随机临床研究中, 至首次发生任何级别中性粒细胞减少症的中位时间为 15 天(12-700天), ≥3 级中性粒细胞减少症的中位继续时间为 7 天.0.9% 接受哌柏西利与氟维司群联用和 2.1% 接受哌柏西利与来曲唑联用的患者陈说了发热性中性粒细胞减少症.在总体临床研究中, 年夜约 2% 接受哌柏西利治疗的患者曾陈说过发热性中性粒细胞减少症.【禁忌】对活性成分或章节【成分】项下所列的任一辅料过敏者禁用.禁止使用含圣约翰草的制品(见【药代相互作用】).【注意事项】绝经前/围绝经期女性鉴于芳香化酶抑制剂的作用机制, 绝经前/围绝经期女性接受哌柏西利与芳香化酶抑制剂联合治疗时, 必需进行卵巢切除或使用促黄体生成激素释放激素(Luteinizing Hormone Releasing Hormone, LHRH)激动剂抑制卵巢功能.哌柏西利联合氟维司群用于绝经前/围绝经期女性的研究中, 仅与LHRH激动剂联合用药.危重内脏疾病(转移)尚未在危重的有内脏疾病(转移)患者中研究哌柏西利的疗效和平安性(见【临床试验】).血液学毒性中性粒细胞减少症是临床研究中最常陈说的不良反应, 临床研究中年夜约有2%的接受哌柏西利治疗的患者曾陈说过发热性中性粒细胞减少症, 并陈说了 1 例中性粒细胞减少性败血症引起的死亡.应在哌柏西利治疗开始前、每个周期开始时、前两个周期的第 15 天以及呈现临床指征时监测全血细胞计数.对呈现 3 或 4 级中性粒细胞减少症的患者, 建议中断给药、减少剂量或延迟开始治疗周期, 并进行密切监测.(见【用法用量】和【不良反应】).医生应告知患者立即陈说任何发热事件.感染因为哌柏西利具有骨髓抑制特性, 其可使患者易于呈现感染.多项随机研究报道了哌柏西利组患者的感染率高于各自的对比组患者.分别有 4.5% 和 0.7% 的接受哌柏西利任何联用方案治疗的患者发生了 3 级和 4 级感染(见【不良反应】).应监测患者的感染体征和症状而且适那时应给予治疗(见【用法用量】).患者在呈现任何骨髓抑制或感染体征或症状时立即陈说, 例如发热、寒战、头晕、气短、无力或出血和/或瘀伤倾向加重.肝损伤中度或重度肝损伤患者应慎用哌柏西利, 并密切监测毒性体征(见【用法用量】和【药代动力学】).肾损伤中度或重度肾损伤患者应慎用哌柏西利, 并密切监测毒性体征(见【用法用量】和【药代动力学】).与 CYP3A4 抑制剂或诱导剂联合治疗强效 CYP3A4 抑制剂可招致毒性增加(见【药物相互作用】).哌柏西利治疗期间应防止与强效 CYP3A 抑制剂合用.仅在认真评估潜在获益和风险后才可考虑同时使用.如不能防止与强效 CYP3A 抑制剂同时使用, 应将哌柏西利的剂量降至 75 mg 每天一次.停止使用强效抑制剂时, 应将哌柏西利的剂量(抑制剂的 3-5 个半衰期后)增加至开始使用强效 CYP3A 抑制剂前的剂量(见【药物相互作用】).与 CYP3A 诱导剂同时使用可招致哌柏西利的流露量降低, 所以有缺乏疗效的风险.因此, 应防止哌柏西利与强效 CYP3A4 诱导剂合用.哌柏西利与中效 CYP3A 诱导剂同时使用时无需调整剂量(见【药物相互作用】).有生育能力的女性或其配偶有生育能力的女性或其男性配偶在使用哌柏西利治疗期间必需使用一种高效的避孕方法(见【孕妇及哺乳期妇女用药】).乳糖哌柏西利含乳糖.存在半乳糖不耐症、Lapp 乳糖酶缺乏症或葡萄糖-半乳糖吸收不良症等罕见遗传疾病的患者不得服用哌柏西利.对驾驶和把持机器能力的影响哌柏西利对驾驶和把持机器能力的影响很小.可是, 哌柏西利可能引起疲乏, 患者在驾驶或把持机器时应谨慎.【孕妇及哺乳期妇女用药】有生育能力的女性/避孕接受本药品治疗的有生育能力的女性或其男性配偶, 应在治疗期间以及完成治疗后分别至少 3 周(女性)或 14 周(男性)内采用充沛的避孕办法(如, 双重屏障避孕)(见【药物相关作用】).妊娠尚缺乏关于孕妇使用哌柏西利的数据或数据有限.植物研究显示哌柏西利具有生殖毒性(见【药理毒理】).不建议孕妇和未采用避孕办法的有生育能力的女性使用哌柏西利.哺乳尚未在人体或植物中进行相关研究以评价哌柏西利对乳汁生成的影响、是否存在于母乳中或对母乳喂养婴儿的影响.尚不清楚哌柏西利是否会分泌至人类乳汁中.接受哌柏西利治疗的患者不应哺乳.生育力在非临床生殖毒性研究中, 未发现对年夜鼠的发情周期(雌性)或交配和生育力(雄性和雌性)有影响.尚未获得对人类生育力影响的临床数据.根据非临床平安性研究中雄性生殖器官的变动(睾丸曲细精管变性、附睾精子减少、精子活力和密度降低以及前列腺分泌减少), 哌柏西利治疗可能会损害男性的生育力(见【药理毒理】).因此, 男性在开始哌柏西利治疗前应考虑保管精液.【儿童用药】尚未确定哌柏西利在18 岁及以下的儿童和青少年患者中的平安性和疗效.尚无相关数据.【老年用药】在PALOMA-2研究中接受哌柏西利治疗的 444 例患者中, 181(41%)例患者≥65 岁, 48(11%)例患者≥75 岁.未发现上述患者与年轻患者在哌柏西利的平安性或有效性方面存在不同.65 岁及以上患者无需调整哌柏西利的剂量(见【药代动力学】).【药物相互作用】哌柏西利主要被CYP3A 和磺基转移酶(Sulphotransferase, SULT)SULT2A1代谢.在体内, 哌柏西利是CYP3A 的时间-依赖性弱抑制剂.其它药品对哌柏西利药代动力学的影响CYP3A 抑制剂的影响同时给予多剂量 200 mg 伊曲康唑与单剂量 125 mg 哌柏西利, 相对独自给予单剂量 125 mg 哌柏西利, 哌柏西利的全身流露量(AUC inf)和峰浓度(C max)分别增加了约 87% 和 34%.应防止与强效 CYP3A 抑制剂合用, 包括但不限于:克拉霉素、茚地那韦、伊曲康唑、酮康唑、洛匹那韦/利托那韦、奈法唑酮、奈非那韦、泊沙康唑、沙奎那韦、特拉匹韦、泰利霉素、伏立康唑和葡萄柚或葡萄柚汁(见【用法用量】和【注意事项】).与轻度和中度 CYP3A 抑制剂合用时无需调整剂量.CYP3A 诱导剂的影响同时给予多剂量 600 mg 利福平与单剂量 125 mg 哌柏西利, 相对独自给予单剂量 125 mg 哌柏西利, 哌柏西利 AUC inf和 C max分别降低了约 85% 和 70%.应防止与强效 CYP3A 诱导剂合用, 包括但不限于:卡马西平、恩杂鲁胺、苯妥英、利福平和圣约翰草(见【禁忌】和【注意事项】).同时给予多剂量每天 400 mg 莫达非尼(一种中效 CYP3A 诱导剂)与单剂量 125 mg 哌柏西利, 相对独自给予单剂量 125 mg 哌柏西利, 哌柏西利 AUC inf和 C max分别降低了约 32% 和 11%.与中效 CYP3A 诱导剂合用时无需调整剂量(见【注意事项】).抗酸药的影响餐后(摄入中脂餐)同时给予多剂量质子泵抑制剂(Proton Pump Inhibitor, PPI)雷贝拉唑与单剂量 125 mg 哌柏西利, 相对独自给予单剂量 125 mg 哌柏西利, 哌柏西利 C max降低了 41%, 但对 AUC inf的影响有限(降低了 13%).空腹条件下同时给予多剂量质子泵抑制剂(PPI)雷贝拉唑与单剂量 125 mg 哌柏西利, 哌柏西利 AUC inf和 C max分别降低了 62% 和 80%.因此, 哌柏西利应与食物同服, 最好随餐服用(见【用法用量】和【药代动力学】).鉴于 H2 受体拮抗剂和局部抗酸剂与 PPI 相比对胃内 pH 的影响较小, 哌柏西利与食物同服时, 预期 H2 受体拮抗剂或局部抗酸剂对哌柏西利的流露量无临床相关影响.哌柏西利对其它药品药代动力学的影响在每天给予 125 mg 到达稳态后, 哌柏西利是一种弱的时间-依赖性 CYP3A 抑制剂.与咪达唑仑独自给药相比, 多剂量哌柏西利与咪达唑仑同时给药时, 咪达唑仑 AUC inf和 C max值分别增加了 61% 和 37%.治疗指数狭窄的敏感 CYP3A4 底物(如, 阿芬太尼、环孢素、双氢麦角胺、麦角胺、依维莫司、芬太尼、匹莫齐特、奎尼丁、西罗莫司和他克莫司)与哌柏西利同时使用时可能需要降低剂量, 因为哌柏西利可增加它们的流露量.哌柏西利与来曲唑之间的药物相互作用一项乳腺癌患者临床研究的药物相互作用(Drug-Drug Interaction, DDI)评价部份的数据标明, 哌柏西利与来曲唑联用时, 两种药品之间无药物相互作用.他莫昔芬对哌柏西利流露量的影响在健康男性受试者中进行的一项DDI 研究的数据标明, 单剂量哌柏西利与多剂量他莫昔芬同时给药, 与哌柏西利独自给药时的流露量相当.哌柏西利与氟维司群之间的药物相互作用在乳腺癌患者中进行的一项临床研究的数据标明, 哌柏西利与氟维司群联用时, 两种药品之间无临床相关药物相互作用.哌柏西利与口服避孕药之间的药物相互作用尚未对哌柏西利与口服避孕药之间的DDI 进行研究(见【孕妇和哺乳期妇女用药】).与转运卵白的体外研究根据体外研究数据, 预计哌柏西利抑制肠道P-糖卵白(P-Glycoprotein, P-gp)和乳腺癌耐药卵白质(Breast Cancer Resistance Protein, BCRP)介导的转运.因此, 哌柏西利与 P-gp (如, 地高辛、达比加群、秋水仙碱)或 BCRP(如, 普伐他汀、瑞舒伐他汀、柳氮磺胺吡啶)的底物类药品合并用药可增加它们的治疗作用和不良反应.根据体外研究数据, 哌柏西利可抑制摄取转运体有机阳离子转运卵白 OCT1, 因此可增加该转运卵白的底物类药品(如, 二甲双胍)的流露量.【药物过量】尚无针对哌柏西利的特效解毒药.如果哌柏西利用药过量, 可能呈现胃肠道(如, 恶心、呕吐)和血液学(如, 中性粒细胞减少症)毒性, 应给予一般的支持性治疗.【临床试验】随机Ⅲ期研究 PALOMA-2:哌柏西利与来曲唑联用作为雌激素受体(ER)阳性、HER2 阴性的晚期或转移性乳腺癌患者初始内分泌治疗在 ER 阳性、HER2 阴性的不能通过手术切除或放疗治愈的局部晚期乳腺癌患者或既往未接受过针对转移灶的全身治疗的晚期乳腺癌患者中进行了一项随机、双盲、抚慰剂对比、国际多中心研究, 评价了哌柏西利与来曲唑联用和来曲唑与抚慰剂联用的疗效.共计 666 例绝经后妇女以 2:1 的比例随机分配至哌柏西利 + 来曲唑组或抚慰剂 + 来曲唑组, 并按病灶部位(内脏、非内脏)、从完成(新)辅助治疗至疾病复发的无病间期(新发转移、12 个月、>12 个月)、既往(新)辅助抗肿瘤治疗的类型(既往激素治疗、无既往激素治疗)分层.研究排除存在晚期、症状性、内脏转移, 短时间内可能呈现危及生命的并发症(包括年夜量积液无法控制 [胸膜积液、心包液、腹膜积液]、肺淋巴管炎以及肝脏受累面积超越 50%)的患者.患者继续接受分配的治疗, 直到发生客观疾病进展、症状恶化、不成接受的毒性、死亡或裁撤同意书, 以先发生者为准.不允许治疗组间交叉治疗.哌柏西利 + 来曲唑组与抚慰剂 + 来曲唑组之间患者的基线人口统计学以及预后特征具有可比性.入组本研究的患者的中位年龄为62 岁(范围:28-89岁), 大都患者为碧眼儿(78%), 且大都患者的美国东部肿瘤协作组(ECOG)体力状态(PS)为 0 或 1(98%).在诊断为晚期乳腺癌前, 48.3% 患者接受过化疗和56.3% 患者接受过抗激素治疗, 37.2% 的患者既往未接受过全身治疗.年夜大都患者(97.4%)在基线时有转移病灶, 23.6% 的患者只有骨转移, 49.2% 的患者有内脏转移.研究的主要终点是由研究者依照实体瘤疗效评价标准(Response Evaluation Criteria in Solid Tumors, RECIST) v1.1 评估的无进展生存期(Progression-Free Survival, PFS).主要疗效终点包括客观缓解率(Objective Response Rate, ORR)、临床获益缓解(Clinical Benefit Response, CBR)、平安性和生活质量(Quality of Life, QoL)变动.研究到达了主要终点.哌柏西利 + 来曲唑组与抚慰剂 + 来曲唑组患者的中位 PFS 分别为 24.8 个月(95% CI:22.1, NE)和14.5 个月(95% CI:12.9, 17.1).风险比(Hazard Ratio, HR)为 0.576(95% CI:0.46, 0.72), 单侧分层对数秩检验 p 值 <0.000001.PALOMA-2 研究的疗效数据总结于表6中, PFS 的 Kaplan-Meier 曲线见图 1.主要终点结果基于根据RECIST 1.1证实和未经证实的缓解.图 1.PALOMA-2 研究—无进展生存期(研究者评估, 意向治疗人群)的 Kaplan-Meier 曲线03691215182124273033Time (Month)P r o g r e s s i o n -F r e e S u r v i v a l P r o b a b i l i t y (%)4443953603282952632381546929102PAL+LET 222171148131116988154221242PCB+LETNumber of patients at riskPAL = 哌柏西利;LET = 来曲唑;PCB = 抚慰剂. 基于预后因素和基线特征进行一系列预先界说的亚组 PFS 分析以考察治疗效果的内部一致性.在所有个体患者亚组(按分层因素以时间(月) 有风险的患者人数 PAL + LET PCB +LET无进展生存概率(%)及基线特征界说)中观察到了哌柏西利 + 来曲唑组可降低疾病进展风险或死亡风险.该结果在以下患者中较为显著:内脏转移患者(HR = 0.67 [95% CI:0.50, 0.89], mPFS为 19.2 个月与 12.9 个月), 或不伴内脏转移的患者(HR = 0.48 [95% CI:0.34, 0.67], mPFS 为未到达 [NR] 与 16.8 个月), 或仅发生骨转移的患者 (HR = 0.36 [95% CI:0.22, 0.59], mPFS 为 NR 与 11.2 个月), 或没有仅骨转移的患者 (HR = 0.65 [95% CI:0.51, 0.84], mPFS 为 22.2 个月与 14.5 个月).与之相似, 在 512 例通过免疫组化(Immunohistochemistry, IHC)检测肿瘤 Rb 卵白质表达结果呈阳性的患者中, 观察到哌柏西利 + 来曲唑的疾病进展或死亡风险下降(HR = 0.531 [95% CI:0.42, 0.68], mPFS 为24.2 个月与 13.7 个月).在 51 例通过 IHC 检测肿瘤 Rb 卵白质表达结果呈阴性的患者中, 虽不具有统计学显著性, 但哌柏西利联合来曲唑有利于疾病进展或死亡风险的下降 (HR = 0.675 [95% CI:0.31, 1.48], mPFS 为 NR 与 18.5 个月).在伴或不伴内脏转移的患者亚组中评估的其它疗效指标(ORR 和TTR)见表7.【药理毒理】药理作用哌柏西利是细胞周期卵白依赖性激酶(CDK)4和6的抑制剂.周期卵白 D1 和 CDK4/6 位于细胞增殖信号通路的下游.在体外, 通过阻滞细胞从 G1 期进入 S 期, 而减少雌激素受体(ER)阳性乳腺癌细胞系的细胞增殖.哌柏西利和雌激素拮抗剂联合作用于乳腺癌细胞系时, 可降低视网膜母细胞瘤(Rb)卵白磷酸化, 从而招致 E2F 表达, 及其信号传导下降, 与药物各自单用相比具有更强的生长抑制作用.哌柏西利和雌激素拮抗剂联合作用于ER 阳性的乳腺癌细胞系时, 与药物各自单用相比, 可使细胞老化增加, 这一效应在哌柏西利停药后最多维持 6 天, 但抗雌激素治疗继续进行时, 可招致更年夜水平的细胞老化.人源性ER 阳性乳腺癌异种移植模型体内研究显示, 与药物各自单用相比, 哌柏西利与来曲唑联用可对Rb 磷酸化、下游信号传导以及肿瘤生长发生更强的抑制作用.人骨髓单核细胞体外给予哌柏西利, 无论有无抗雌激素处置, 未见细胞发生老化, 去除哌柏西利后细胞可恢复增殖.毒理研究一般毒性:在犬遥测试验中, 给药剂量在人体临床流露量(Cmax)4倍以上时, 可见心血管影响(QTc 延长、心率下降、RR 间期延长和收缩压升高).在一项年夜鼠27周重复给药毒性试验中, 年夜鼠在试验早期尚未。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Home·Pfizer’s Potential Mega-Blockbuster Breast Cancer Drug 辉瑞口服乳腺癌药物帕博西尼(palbociclib)Pfizer’s PotentialMega-Blockbuster Breast Cancer Drug 辉瑞口服乳腺癌药物帕博西尼(palbociclib)2015年9月5日辉瑞公司的口服乳腺癌药物帕博西尼(palbociclib, Ibrance, 帕博西林)能否成为重磅炸弹产品?帕博西尼(Palbociclib)是近来备受关注的也是辉瑞最重要试验药物之一,2015年2月3日FDA提前批准辉瑞CDK4/6双抑制剂Palbociclib (商品名Ibrance)作为一线药物治疗ER阳性、HER2阴性乳腺癌,比原定4月13日的PDUFA日期提前两个多月。
分析人士预计,palbociclib每年销售额最高可达到30亿-50亿美元之多。
在一项2期临床研究中,使晚期乳腺癌患者的无进展生存期(PFS)平均增加一倍,但对患者总生存期(OS)未显示有统计学意义上的明显改善。
据研究人员2014年4月在圣地亚哥举行的美国癌症研究协会会议上发布的最新数据显示,雌激素受体阳性转移性乳腺癌患者在合并使用Palbociclib与抗雌激素药物来曲唑时,其PFS平均为20.2个月,相比之下,单独使用来曲唑的患者其PFS平均为10.2个月。
辉瑞公司决定在今年第三季度根据乳腺癌试验药物Palbociclib的2期临床试验结果即向美国食品药监局(FDA)申请上市批准,Palbociclib(又名PD-0332991)最早进入人们视野的是在2012年圣安东尼奥乳腺癌会议上(SABCS),一经发布就引起行业广泛关注。
Palbociclib是一种口服的细胞周期素依赖性激酶4、6的抑制药物,主要通过调节细胞周期发挥作用。
Palbociclib主要通过抑制CDK4/6活性来阻止细胞由G1期到S期进而抑制DNA的合成。
辉瑞的Palbociclib到底何时上市还不得而知。
不过,辉瑞公司于决定在今年底三季度根据2期临床试验结果即向美国食品药监局申请上市批准,而不会等待3期临床试验结果。
预计,如果这款药物能够获得批准,其年销售额可能会达到50亿美元(约合人民币309.98亿元)。
礼来公司表示,目前研究人员正在进行这种药物的三期临床研究实验设计,今年7月份有望启动3期研究,预计在2017年完成。
分析师预测ABemaciclib(LY2835219)到2026年的销售峰值会达到6.5亿美元。
辉瑞在乳腺癌患者中正进行这款药物的3期临床试验,以及治疗其它类型癌症的早期试验。
Palbociclib是一种CDK-4/6抑制剂,以在癌症中起作用的一对周期素依赖性激酶为靶点。
目前正在开发同类药物的公司有诺华的LEE011与礼来药物Bemaciclib(LY2835219)。
一项小型试验发现,礼来试验药物LY2835219在单独使用时,能使转移性雌激素阳性乳腺癌妇女的肿瘤缩小25%,同组中55%妇女的肿瘤得到稳定。
Palbociclib在这一竞赛中仍是一款有待观察的药物,但礼来在这一竞赛中正逐步缩小与辉瑞的差距。
商品名:Ibrance通用名:Palbociclib别名:GPD-0332991中文名:帕博西尼, 帕博西林, 帕波克利, 帕布昔利布英文化学名:6-acetyl-8-cyclopentyl-5-methyl-2-((5-(piperazin-1-yl)pyridin-2-yl) amino) pyrido[2,3-d] pyrimidin-7(8H)-oneCAS 登录号:571190-30-2((palbociclib, 帕博西尼), 827022-33-3 (palbociclib isethionate, 帕博西尼羟乙基磺酸盐)适应症:乳腺癌作用机制: 全球首个CDK4/6激酶抑制剂批准时间:2015年2月3日(美国)费用: 每月$9850销售额(2020年):50亿美元美国专利号码: US6936612(从下载US6936612专利),US7208489, US7456168知识产权情况: 专利到期: 2023年1月22日(US6936612), 2023年1月16日(US7208489, US7456168)药物公司:辉瑞Sources:Dhillon, Sohita. Palbociclib: First Global Approval.Drugs (2015),75(5), 543-551.Xu, Xuenong.Method for preparing palbociclib. Faming Zhuanli Shenqing (2015), CN104496983 A 20150408[发明公布]一种帕博西尼的制备方法. 申请公布号:CN104496983A. 申请公布日:2015.04.08. 申请号:2014106930911 申请日:2014.11.26. 申请人:苏州明锐医药科技有限公司. 发明人:许学农地址:江苏省苏州市工业园区联丰商业广场1幢1305室215000摘要:本发明揭示了一种帕博西尼(Palbociclib,I)的制备方法,其制备步骤包括:通过易得原料1-(4-氨基-2-取代基-5-嘧啶)乙酮(II)与乙酰乙酸酯(III)发生环合反应生成6-乙酰基-5-甲基-2-取代基-吡啶并[2,3-d]嘧啶-7(8H)-酮(IV);该中间体(IV)与卤代环戊烷(V)发生取代反应生成6-乙酰基-8-环戊基-5-甲基-2-取代基-吡啶并[2,3-d]嘧啶-7(8H)-酮(VI);中间体(VI)与4-(6-氨基-吡啶-3-基)-哌嗪-1-甲酸叔丁基酯(VII)发生缩合和水解反应制得帕博西尼(I)。
该制备方法原料易得,工艺简洁,经济环保,适合工业化生产Xu, Xuenong.Method for preparation of palbociclib. Faming Zhuanli Shenqing (2015), CN104447743 A 20150325.[发明公布]帕博西尼的制备方法. 申请公布号:CN104447743A. 申请公布日:2015.03.25. 申请号:2014106912330 申请日:2014.11.26. 申请人:苏州明锐医药科技有限公司. 发明人:许学农地址:江苏省苏州市工业园区联丰商业广场1幢1305室215000摘要:本发明揭示了一种帕博西尼(PalbOciclib,I)的制备方法,其制备步骤包括:2-乙酰基-2-丁烯酸甲酯与丙二睛在碱性条件下发生环合反应生成1,4,5,6-四氢-2-甲氧基-4-甲基-5-乙酰基-6-氧-3-吡啶甲腈(II);中间体(II)与卤代环戊烷(III)在缚酸剂作用下发生取代反应生成N-环戊基-1,4,5,6-四氢-2-甲氧基-4-甲基-5-乙酰基-6-氧-3-吡啶甲腈(IV);中间体(IV)与N-[5-(1-哌嗪基)-2-哌啶基]胍(V)发生缩合反应生成6-乙酰基-8-环戊基-5-甲基-2-[[5-(1-哌嗪基)-2-吡啶基]氨基]-5,6-二氢吡啶并[2,3-d]嘧啶-7(8H)-酮(VI);中间体(VI)与硒酸钠发生脱氢反应制得帕博西尼(I)。
该制备方法原料易得,工艺简洁,经济环保,适合工业化生产。
Wu, Yusheng; Niu, Chengshan; Zou, Dapeng; Zhang, Sen; Guo, Ruiyun; Li, Jingya. Deuterated palbociclib derivative, its preparation and application. Faming Zhuanli Shenqing (2015), CN104447739 A 20150325.[发明公布] 一种氘代Palbociclib衍生物、制备方法及应用.申请公布号:CN104447739A. 申请公布日:2015.03.25.申请号:201410623485X. 申请日:2014.11.07.申请人:郑州泰基鸿诺药物科技有限公司. 发明人:吴豫生;牛成山; 邹大鹏; 张森; 郭瑞云; 李敬亚. 地址:450052 河南省郑州市大学路75号摘要:本发明公开了一种氘代Palbociclib衍生物、制备方法及应用,该氘代Palbociclib衍生物的结构式如式(I)、式(II)、式(III)或式(IV)所示。
本发明的氘代Palbociclib衍生物,通过对Palbociclib的选择性氘代,改善了药物的药代性质,进而提高了药物的疗效、安全性和耐受性;本发明的氘代Palbociclib盐,提高了药物的溶解度和溶出速率;氘代Palbociclib衍生物的合成,为合成新型抗肿瘤药物提供了新的化合物,其与Palbociclib具有类似的生物活性,具有良好的药物应用前景。
|Ibrance Palbociclib Synthesis Patent|帕博西尼的制备专利| Mark Barvian, Richard John Booth, John Quin, III, Joseph Thomas Repine, Derek J. Sheehan, Peter Laurence Toogood, Scott Norman Vanderwel, Hairong Zhou,2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones. US patent number: US6936612 (download pdf file from ), Also published as: CA2473026A1, CA2473026C, CN101001857A,CN101001857B, CN101906104A, CN101906104B, CN102295643A, CN102295643B, DE60303009D1, DE60303009T2, EP1470124A1,EP1470124B1, US7208489, US7456168, US20030149001,US20050137214, US20070179118, WO2003062236A1,WO2003062236A8. Publication date: Aug 30, 2005.Original Assignee: Warner-Lambert CompanyMark Barvian, Richard John Booth, John Quin, III, Joseph Thomas Repine, Derek J. Sheehan, Peter Laurence Toogood, Scott Norman Vanderwel, Hairong Zhou, 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones . US patent number: US7208489 (download pdf file from ), Also published as: CA2473026A1,CA2473026C, CN101001857A, CN101001857B, CN101906104A, CN101906104B, CN102295643A, CN102295643B, DE60303009D1, DE60303009T2, EP1470124A1, EP1470124B1, US6936612,US7456168, US20030149001, US20050137214, US20070179118, WO2003062236A1, WO2003062236A8. Publication date: Apr 24, 2007. Original Assignee: Warner-Lambert CompanyFinn, Richard S.; Crown, John P.; Lang, Istvan; Boer, Katalin; Bondarenko, Igor M.; Kulyk, Sergey O.; Ettl, Johannes; Patel, Ravindranath; Pinter, Tamas; Schmidt, Marcus; et al. The cyclin- dependent kinase 4 /6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first- line treatment of oestrogen receptor- positive, HER2- negative, advanced breast cancer (PALOMA- 1 /TRIO- 18) : a randomised phase 2 study. Lancet Oncology (2015), 16(1), 25-35.Morgan, Adam J. Deuterated palbociclib as therapeutic agents. PCT Int. Appl. (2014), WO 2014150925 A2 20140925Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors ofCyclin-Dependent Kinase 4; Journal of MedicinalChemistry,2005,48(7),2371-2387;Erdman, David Thomas et al;Preparation of2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157 (download PDF file from ) Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997 (download PDF file here)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381 (download PDF file here)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.Phase III Study Evaluating Palbociclib (PD-0332991), aCyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”; number:NCT01864746;currently recruiting participants(as of January 2, 2013)A Randomized, Multicenter, Double-Blind Phase 3 Study OfPD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For AdvancedDisease; number:NCT01740427;currently recruiting participants(as of January 2, 2013)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991(Palbociclib) +/- Goserelin In Women With HormoneReceptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy; number:NCT01942135;currently recruiting participants(as of January 2, 2013)A Randomized, Multicenter, Double-Blind Phase 3 Study OfPD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For AdvancedDisease; number:NCT01740427;currently recruiting participants(as of Feb 3, 2014)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With HormoneReceptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy(PALOMA-3); number:NCT01942135;currently recruiting participants(as of Feb 3, 2014)Update (April 23, 2015): In April, 2015, Pfizer announced that the phase III PALOMA-3 trial of its cyclin-dependent kinase 4/6 (CDK 4/6) inhibitor palbociclib was stopped early after meeting its primary endpoint of improved progression-free survival when combined with fulvestrant in women with hormone receptor (HR)-positive, HER2-negative metastatic breast cancer who had previously progressed on endocrine therapy.close .blog-post-excerpt#post-35Author: hwansunPosted in: 未分类险些被扼杀在摇篮里的抗癌新药2015年8月27日在数年的药物研发进程中,一种可以干扰细胞分裂的化合物的出现,给癌症研究领域面临的困境带来了新的生机。