关于NDA 505 (b)(2)的理解
2015-2021年FDA批准的505b2药物

APPLICATIONNUMBERPROPRIETARY NAME ESTABLISHED NAME通用名APPLICANTNDA208746(1)PEMETREXED培美曲塞HOSPIRA INC NDA 206610ACETAMINOPHEN对乙酰氨基酚RISINGNDA 214313NOREPINEPHRINEBITARTRATE IN 5%DEXTROSENOREPINEPHRINEBITARTRATE酒石酸去甲肾上腺素BAXTER HEALTHCARECORPNDA 204803POSIMIR BUPIVACAINE布比卡因DURECT CORPNDA 204957ACETAMINOPHEN对乙酰氨基酚B BRAUN MEDICAL INCNDA 212994AZSTARYS SERDEXMETHYLPHENIDATE ANDDEXMETHYLPHENIDATE对甲苯磺酸盐和对甲苯磺酸盐COMMAVETHERAPEUTICS SANDA 211844MIDAZOLAM咪达唑仑INFORLIFE SANDA 213072ROSZET ROSUVASTATIN ANDEZETIMIBE瑞舒伐他汀和依折麦布ALTHERAPHARMACEUTICALSLLCNDA 214154NEXTSTELLIS DROSPIRENONE ANDESTETROL TABLETS屈螺酮和雌醇片MAYNE PHARMA LLCNDA 212045KLOXXADO NALOXONE HCL盐酸纳洛酮HIKMA PHARMACEUTICALS USA INCNDA214657(1)PEMETREXED SANDOZ INCNDA 211988ZYNRELEF BUPIVACAINE ANDMELOXICAM布比卡因和美洛昔康HERON THERAPEUTICSINCNDA 214253LEVOTHYROXINESODIUMCUSTOPHARM INCNDA 211488CAMCEVI LEUPROLIDE亮丙瑞林FORESEE PHARMACEUTICALS CO LTDNDA 214846MYFEMBREE RELUGOLIX 40 MG,ESTRADIOL 1 MG,AND NORETHINDRONEACETATE 0.5MGRELUGOLIX 40 mg、雌二醇 1 mg 和醋酸炔诺酮0.5 mgMYOVANT SCIENCESGMBHNDA 213378LYBALVI OLANZAPINE ANDSAMIDORPHAN奥氮平和沙美芬ALKERMES INCNDA 215025SODIUMPHENYLACETATE ANDSODIUM BENZOATE苯乙酸钠和苯甲酸钠MAIAPHARMACEUTICALSINCNDA 213218SOAANZ TORSEMIDE托拉塞米SARFEZ PHARMACEUTICALS INCNDA 213536REZIPRES EPHEDRINEHYDROCHLORIDE盐酸麻黄碱ETONPHARMACEUTICALSINCNDA 212156MICAFUNGIN米卡芬净PAR STERILE PRODUCTS LLCNDA 210282DAPTOMYCIN达托霉素HOSPIRA INC NDA 214965VERKAZIA CYCLOSPORINE环孢素SANTEN INCNDA 212303(1)DULUTEGRAVIR,LAMIVUDINE, ANDTENOFOVIRDISOPROXIL度替拉韦、拉米夫定和富马酸替诺福韦酯LUPIN LTDNDA 214902TWYNEO TRETINOIN ANDBENZOYL PEROXIDE维 A 酸和过氧化苯甲酰SOL-GELTECHNOLOGIES LTDNDA 215143SUCCINYLCHOLINECHLORIDE琥珀胆碱氯化物HIKMAPHARMACEUTICALSUSA INCNDA 210735CYCLOPHOSPHAMIDE环磷酰胺EUGIA PHARMA SPECIALITIES LTDNDA 213895VANCOMYCIN万古霉素XELLIA PHARMACEUTICALS APSNDA 214826LOREEV XR LORAZEPAM劳拉西泮ALMATICA PHARMA LLCNDA 211566(1)SITAGLIPTIN西他列汀ZYDUS WORLDWIDEDMCCNDA 213436TRUDHESA DIHYDROERGOTAMINEMESYLATE甲磺酸二氢麦角胺IMPEL NEUROPHARMANDA 215133SERTRALINEHYDROCHLORIDE盐酸舍曲林ALMATICA PHARMALLCNDA 212854ZIMHI NALOXONEHYDROCHLORIDE盐酸纳洛酮ADAMISPHARMACEUTICALSCORPNDA 213426SEGLENTIS CELECOXIB ANDTRAMADOLHYDROCHLORIDE塞来昔布和盐酸曲马多KOWAPHARMACEUTICALSAMERICA INCNDA 213978TYRVAYA VARENICLINESOLUTION伐尼克兰溶液OYSTER POINTPHARMA INCNDA 211950XIPERE TRIAMCINOLONEACETONIDE曲安奈德BAUSCH AND LOMBINCNDA 214028VUITY PILOCARPINEHYDROCHLORIDE盐酸匹洛卡品ABBVIE INCNDA 210526DYANAVEL XR AMPHETAMINE安非他明TRIS PHARMA INCNDA 213005(1)YUTREPIA TREPROSTINIL曲前列尼尔LIQUIDIATECHNOLOGIESNDA 214679EPRONTIA TOPIRAMATE托吡酯AZURITY PHARMACEUTICALS INCNDA 214869DHIVY CARBIDOPA ANDLEVODOPACARBIDOPA 和左旋多巴AVIONPHARMACEUTICALSLLCNDA 215668(1)BENDAMUSTINEHYDROCHLORIDE盐酸苯达莫司汀DR REDDYSLABORATORIES LTDNDA 213312FYARRO SIROLIMUS PROTEIN-BOUND PARTICLESSIROLIMUS 蛋白结合颗粒AADI BIOSCIENCEINCNDA 215422LYVISPAH BACLOFEN巴氯芬SAOL THERAPEUTICS RESEARCH LTDNDA 215650XACIATO CLINDAMYCINPHOSPHATE磷酸克林霉素DARE BIOSCIENCEINCNDA 215423ENTADFI TADALAFIL ANDFINASTERIDE他达拉非和非那雄胺VERU INCNDA 215935TARPEYO BUDESONIDE布地奈德CALLIDITAS THERAPEUTICS ABNDA 215019DARTISLA ODT GLYCOPYRROLATE甘氨酰吡咯酸EDENBRIDGE PHARMACEUTICALS LLCNDA 2I4032ILLUCCIX KIT FOR THEPREPARATION OF GA-68 PSMA-11GA-68 PSMA-11 制备试剂盒TELIXPHARMACEUTICALS USINCNDA 215395LANREOTIDE ACETATE醋酸来那度胺INVAGEN PHARMACEUTICALS INCNDA 214133RECORLEV LEVOKETOCONAZOLE左酮康唑STRONGBRIDGE DUBLIN LTDREVIEW CLASSIFI CATION 505(B)(2)APPROVALAPPROVAL DATE批准类型S Y1/8/2021Type 3 - New Dosage FormS Y1/15/2021Type 5 - New Formulation or New ManufacturerS Y1/15/2021Type 5 - New Formulation or New ManufacturerS Y2/1/2021Type 3 - New Dosage FormS Y2/18/2021Type 5 - New Formulation or New ManufacturerS Y3/2/2021Type 1 - New Molecular Entity and Type 4 - New CombinationS Y3/22/2021Type 5 - New Formulation or New ManufacturerS Y3/23/2021Type 4 - New CombinationS Y4/15/2021Type 1 - New Molecular EntityS Y4/29/2021Type 5 - New Formulation or New ManufacturerS Y5/6/2021Type 5 - New Formulation or New ManufacturerP Y5/12/2021Type 4 - New CombinationS Y5/17/2021Type 5 - New Formulation or New ManufacturerS Y5/25/2021Type 2 - New Active IngredientS Y5/26/2021Type 4 - New CombinationS Y5/28/2021Type 1 - New Molecular Entity and Type 4 - New CombinationS Y6/10/2021Type 5 - New Formulation or New ManufacturerS Y6/14/2021Type 5 - New Formulation or New ManufacturerS Y6/14/2021Type 5 - New Formulation or New ManufacturerNew ManufacturerS Y6/21/2021Type 5 - New Formulation or New ManufacturerS,O Y6/23/2021Type 5 - New Formulation or New ManufacturerS Y6/25/2021Type 4 - New Combination S Y7/26/2021Type 4 - New CombinationS Y8/20/2021Type 5 - New Formulation or New ManufacturerS Y8/25/2021Type 5 - New Formulation or New ManufacturerP Y8/26/2021Type 5 - New Formulation or New ManufacturerS Y8/27/2021Type 3 - New Dosage FormS Y9/2/2021Type 2 - New Active IngredientS Y9/2/2021Type 5 - New Formulation or New ManufacturerS Y10/4/2021Type 3 - New Dosage FormS Y10/15/2021Type 5 - New Formulation or New ManufacturerS Y10/15/2021Type 4 - New Combination S Y10/15/2021Type 3 - New Dosage Form S Y10/22/2021Type 3 - New Dosage FormS Y10/28/2021Type 5 - New Formulation or New ManufacturerS Y11/4/2021Type 3 - New Dosage Form S Y11/4/2021Type 3 - New Dosage FormS Y11/5/2021Type 3 - New Dosage FormS Y11/12/2021Type 5 - New Formulation or New ManufacturerNew ManufacturerP,O Y11/22/2021Type 5 - New Formulation or New ManufacturerS Y11/22/2021Type 3 - New Dosage FormP Y12/7/2021Type 5 - New Formulation or New ManufacturerS Y12/9/2021Type 4 - New CombinationP,O Y12/15/2021Type 5 - New Formulation or New ManufacturerS Y12/16/2021Type 3 - New Dosage FormS Y12/17/2021Type 3 - New Dosage Form and Type 4 - New CombinationS Y12/17/2021Type 5 - New Formulation or New ManufacturerS,O Y12/30/2021Type 2 - New Active Ingredient适应症NSCLC、Mesothelioma间皮瘤(注射剂)止痛、退烧(注射剂)升高低血压(注射剂)局部麻醉(注射剂)止痛、退烧CNS兴奋剂注射麻醉剂降低LDL-C预防妊娠阿片类拮抗剂,用于阿片类过量NSCLC、Mesothelioma间皮瘤术后止痛(注射剂)粘液性水肿昏迷myxedema coma晚期前列腺癌子宫平滑肌瘤月经量过多成人精神分裂症、成人双相 i 型障碍急性高血氨症成人心力衰竭、肾病相关水肿麻醉状态的低血压念珠菌血症、急性播散性念珠菌病、念珠菌腹膜炎和脓肿cSSSI、Bacteremia菌血症春季角结膜炎(Tentative Approval)痤疮局部治疗作为全身麻醉的辅助治疗、便于气管插管、在手术或机械通气期间提供骨骼肌松弛label not available败血症、感染性心内膜炎、皮肤和皮肤结构感染、骨感染、下呼吸道感染、艰难梭菌相关性腹泻、金黄色葡萄球菌引起的小肠结肠炎label not availablelabel not available偏头痛急性治疗MDD、OCD紧急治疗阿片类药物过量治疗需要阿片类镇痛剂且替代治疗效果不佳的成人急性疼痛。
药品试验数据保护制度及其实施要点的思考

“8+2+1”模式
5
TRIPS 39.3 规定的数据保护制度
试验数据保护的对象及条件- “……未披露试验数据或其它数据”
当成员要求以提交未披
必须是应政府部门要求所提交的数据;
露过的试验数据或其它数据, 作为批准采用新化学成分的
必须是为了获取产品上市许可而依法收集、提交的数据;
保护范围主要是针对包含NCE的新药,但各国也可以根据国 情对已上市药品的新剂型及新的适应证等具有临床比较优势 的创新产品提供时间略短一些的保护。
保护方法主要是通过对第二个申请人设定条件,采用“不受 理”或“不批准”的方式来履行“不依赖”的保护义务。
“不依赖”事实 上是试验数据保护 制度独有的核心义 务,而“不披露” 则是关于“保密” 的一般义务,通常 是由各国的《反不 正当竞争法》或 《刑法》规定调整。
10
《药品管理法实施条例》第53条 2002.8.15
国家对获得生产或者销售含有新型化学成份药品许可的生产者或者销售者提交 的自行取得且未披露的试验数据和其他数据实施保护,任何人不得对该未披露的试 验数据和其他数据进行不正当的商业利用。
自药品生产者或者销售者获得生产、销售新型化学成份药品的许可证明文件之 日起6年内,对其他申请人未经已获得许可的申请人同意,使用前款数据申请生产、 销售新型化学成份药品许可的,药品监督管理部门不予许可;但是,其他申请人提 交自行取得数据的除外。
8
药品试验数据保护制度的基本认识--法律渊源
TRIPS39.3,1991年12月20日 《中国加入工作组报告书》284段,2001年11月23日 《药品管理法实施条例》A53,A72,2002年8月15日
《药品注册管理办法》A14,2002年10月30日
2020年FDA批准上市的505b2药物清单
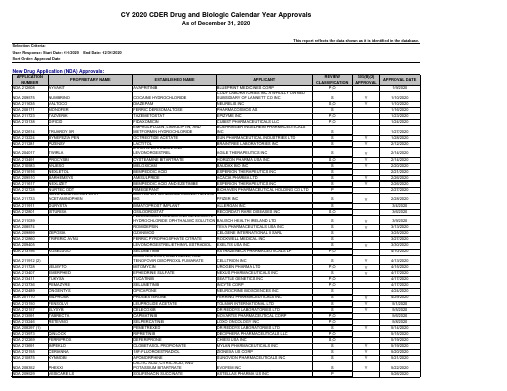
CAPMATINIB
NOVARTIS PHARMACEUTICAL CORP
SELPERCATINIB
LOXO ONCOLOGY INC
PEMETREXED
DR REDDYS LABORATORIES LTD
RIPRETINIB
DECIPHERA PHARMACEUTICALS LLC
DEFERIPRONE
S P,O
S P,O P,O
S S S S P,O P,O S P,O S,O S S S
S P
505(B)(2) APPROVAL
APPROVAL DATE 1/9/2020
Y
1/10/2020
Y
1/10/2020
1/16/2020
1/23/2020
1/24/2020
1/27/2020
Y
1/28/2020
INCYTE CORP
OPICAPONE
NEUROCRINE BIOSCIENCES INC
PROGESTERONE
FERRING PHARMACEUTICALS INC
LEUPROLIDE ACETATE
TOLMAR INTERNATIONAL LTD
CELECOXIB
DR REDDYS LABORATORIES LTD
OCTREOTIDE ACETATE
SUN PHARMACEUTICAL INDUSTRIES LTD
LACTITOL ETHINYL ESTRADIOL AND LEVONORGESTREL
BRAINTREE LABORATORIES INC AGILE THERAPEUTICS INC
CYSTEAMINE BITARTRATE
2018年FDA批准上市的505b2药物清单
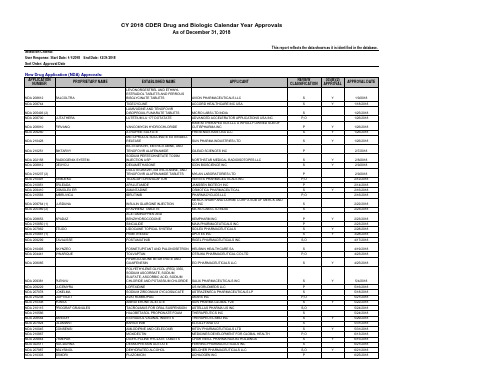
AKARX INC SUN PHARMA GLOBAL FZE ASTELLAS PHARMA US INC THERAPEUTICS INC THERAPEUTICSMD INC ELI LILLY AND CO KITOV PHARMACEUTICALS LTD
MOXIDECTIN DOXYCYCLINE HYCLATE TABLETS DESMOPRESSIN ACETATE
S
PIMAVANSERIN
ACADIA PHARMACEUTICALS INC
S
ARIPIPRAZOLE LAUROXIL
ALKERMES INC
S
FULVESTRANT
FRESENIUS KABI USA LLC
S
ATROPINE
RAFA LABORATORIES LTD
P
VANCOMYCIN HYDROCHLORIDE
S,O
Y
6/21/2018
P
6/25/2018
NDA 210365 NDA 210496 NDA 210498 NDA 210361 NDA 210793 NDA 209830 NDA 210326 (1) NDA 212319 NDA 209481 NDA 210997 NDA 208627
NDA 208313
NDA 207356 NDA 209863 NDA 209521 NDA 209816
ANNOVERA ONPATTRO
CEQUA
DIACOMIT DIACOMIT
INVELTYS ALTRENO XERAVA
LUTRATE DEPOT
PIFELTRO
DELSTRIGO TIGLUTIK CASSIPA XELPROS SEIZALAM COPIKTRA VIZIMPRO
【综述】使用FDA505(b)(2)新药申请监管途径简化非临床药物开发

【综述】使用FDA505(b)(2)新药申请监管途径简化非临床药物开发原创翻译者:康苗研如玉 4月24日编者语:我们连续推送了关于505(b)(2)文章如:【大力推荐】505(b)(2)药物批准途径【综述】505(b)(2)申请的审评时间分析【案例揭秘】505(b)(2)新药申请批准案例分析:愈创甘油醚缓释片在健康受试者的药代动力学研究今天我们继续为大家推送使用FDA 505(b)(2)新药申请监管途径简化非临床药物开发,同时潜在地保持市场独占权的方法与同行共享。
本文来源见以下截图使用FDA 505(b)(2)新药申请监管途径简化非临床药物开发作者:William F.Salminen, Marc E. Wiles, Ruth E. Stevens提示:新药上市申请(NDA)使用FDA 505(b)(2)监管途径可以简化和减少非临床药物开发要求,同时潜在地保持市场独占权。
强调:l 可以简化和减少505(b)(2)NDA非临床计划;l 可以依赖创新药物的非临床数据;l 505(b)(2)NDA反映了一种简化药物审批的途径和方法;l 可以降低药物开发成本和时间压力,同时保持市场独占权;在美国,药物通过三种主要的监管途径获得FDA的批准:(i)505(b)(1)新药上市申请(NDA);(ii)505(b)(2)新药上市申请;以及(iii)505(j)仿制药注册申请(ANDA)。
根据活性成分,已获批的药品,药物配方,临床适应症,暴露途径等其他因素选择合适的申请途径。
505(b)(2)NDA途径就是一种监管审批的途径,允许申请人使用现有的公开数据代替所要开展的研究;因此,有可能会提供重要的药物开发和市场优势。
根据505(b)(2)申报,非临床试验项目通常会被减少,有些情况下甚至不需要。
本文概述了505(b)(2)监管途径,重点关注如何简化和加速非临床计划。
缩写:代谢/内分泌,代谢/内分泌学。
关键词:505(b)(2)NDA非临床研究;非临床药物开发;新药申请;NDA.1.引言在美国,新药通过三种主要的监管途径获得FDA的批准:505(b)(1)和505(b)(2)新药上市申请,以及(iii)505(j)仿制药注册申请(ANDA)。
2017年FDA批准上市的505b2药物清单
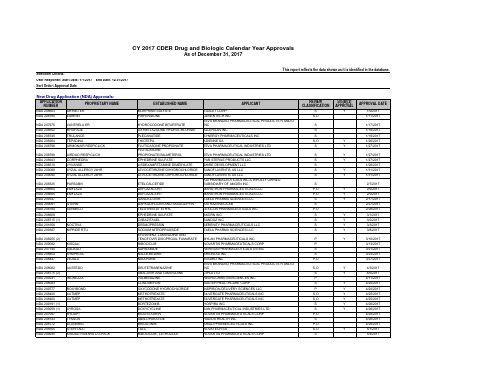
VANTRELA ER RHOFADE TRULANCE TEPADINA ARMONAIR RESPICLICK
NDA 208799 NDA 208943 NDA 208510 NDA 209089 NDA 209090
AIRDUO RESPICLICK CORPHEDRA VYVANSE XYZAL ALLERGY 24HR XYZAL ALLERGY 24HR
NOCTIVA NIPRIDE RTU
NDA 208255 (2) NDA 209092 NDA 207145 NDA 208854 NDA 208447
KISQALI XADAGO SYMPROIC ZEJULA
NDA 208082 NDA 208775 (2) NDA 209241 NDA 208083 NDA 209777 NDA 208400 NDA 208400 NDA 209191 (1) NDA 209259 (1) NDA 207997 NDA 208743 NDA 208772 NDA 205555 NDA 209935
S
1/28/2017
S
Y
1/31/2017
S
Y
1/31/2017
S
2/7/2017
P,O
Y
2/9/2017
P,O
Y
2/9/2017
S
Y
2/17/2017
S
2/27/2017
P,O
2/28/2017
S
Y
3/1/2017
S
Y
3/2/2017
S
Y
3/3/2017
S
Y
3/8/2017
P
Y
3/10/2017
505b2 Shanghai

YES
NO
505(b)(1)
6
505(b)(2)
505(j)
In the last decade, the U.S. healthcare system has saved more than $824 billion by using generic medicines
• According to a study released by the Generic Pharmaceutical Association: Key findings from the report include: 1. In 2009, use of FDA-approved generics saved $139.6 billion, a 15% growth over the prior year's savings or about $382 million every day; 2. Savings generated by new generics will continue to increase as $89 billion in branded drug sales will lose patent protection over the next five years.
2
4
New Drug Application (NDA)
• (1) an application that contains full reports of investigations of safety and effectiveness (section 505(b)(1));
• (2) an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference (section 505(b)(2)); and • (3) an application that contains information to show that the proposed product is identical in active ingredient, dosage form, strength, route of administration, labeling, quality, performance characteristics, and intended use, among other things, to a previously approved product (section 505(j)).
Applications Covered by Section 505(b)(2)

Applications Covered by Section505(b)(2)DRAFT GUIDANCEThis guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should be submitted within 60 days of publication of the Federal Register notice announcing the availability of the draft guidance. Submit comments to Dockets Management Branch (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20857. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.For questions on the content of the draft document contact Virginia Beakes, (301) 594-2041.U. S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)October 1999Applications Covered by Section505(b)(2)DRAFT GUIDANCEFor additional copies, contact:Drug Information BranchDivision of Communications Management, HFD-210Center for Drug Evaluation and Research (CDER)5600 Fishers LaneRockville, MD 20857(Tel) 301-827-4573/cder/guidance/index.htm.U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)October 1999Table of ContentsI.WHAT IS THE PURPOSE OF THIS GUIDANCE? (1)II.WHAT IS A 505(B)(2) APPLICATION? (2)A.W HAT TYPE OF INFORMATION CAN AN APPLICANT RELY ON? (2)B.W HAT KIND OF APPLICATION CAN BE SUBMITTED AS A 505(B)(2) APPLICATION? (3)III.WHAT ARE SOME EXAMPLES OF 505(B)(2) APPLICATIONS? (4)IV.WHAT CAN'T BE SUBMITTED AS 505(B)(2) APPLICATIONS? (6)V.WHY DOES IT MATTER IF AN NDA IS A 505(B)(2) APPLICATION? (6)VI.PATENT AND EXCLUSIVITY PROTECTIONS THAT COULD AFFECT A 505(B)(2) APPLICATION (7)A.W HAT TYPE OF PATENT AND/OR EXCLUSIVITY PROTECTION IS A 505(B)(2) APPLICATION ELIGIBLE FOR? (7)B.W HAT COULD DELAY THE APPROVAL OR FILING OF A 505(B)(2) APPLICATION? (7)VII.WHAT SHOULD BE INCLUDED IN 505(B)(2) APPLICATIONS? (7)REFERENCES (10)GLOSSARY (11)GUIDANCE FOR INDUSTRY1Applications Covered by Section 505(b)(2)I.WHAT IS THE PURPOSE OF THIS GUIDANCE?This guidance identifies the types of applications that are covered by section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act (the Act). A 505(b)(2) application is a new drug application (NDA) described in section 505(b)(2) of the Act. It is submitted under section 505(b)(1) of the Act and approved under section 505(c) of the Act. This guidance also provides further information and amplification regarding FDA's regulations at 21 CFR 314.54.Section 505 of the Act describes three types of new drug applications: (1) an application that contains full reports of investigations of safety and effectiveness (section 505(b)(1)); (2) an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference (section 505(b)(2)); and (3) an application that contains information to show that the proposed product is identical in active ingredient, dosage form, strength, route of administration, labeling, quality, performance characteristics, and intended use, among other things, to a previously approved product (section 505(j)). Note that a supplement to an application is a new drug application.Section 505(b)(2) was added to the Act by the Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Amendments). This provision expressly permits FDA to rely, for approval of an NDA, on data not developed by the applicant. Sections 505(b)(2) and (j) together replaced FDA's paper NDA policy, which had permitted an applicant to rely on studies published in the scientific literature to demonstrate the safety and effectiveness of duplicates of certain post-1962 pioneer drug products (see 46 FR 27396, May 19, 1981). Enactment of the generic drug approval provision of the Hatch-Waxman Amendments ended the need for approvals of duplicate drugs through the paper NDA process by permitting approval under 505(j) of duplicates of approved drugs (listed 1This guidance has been prepared by the Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration. This guidance document represents the Agency's current thinking on the types of applications that may be submitted pursuant to section 505(b)(2) of the Act. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute, regulations, or both.drugs) on the basis of chemistry and bioequivalence data, without the need for evidence from literature of effectiveness and safety. Section 505(b)(2) permits approval of applications other than those for duplicate products and permits reliance for such approvals on literature or on an Agency finding of safety and/or effectiveness for an approved drug product.Definitions for specific terms used throughout this guidance are given in the Glossary.II.WHAT IS A 505(B)(2) APPLICATION?A 505(b)(2) application is one for which one or more of the investigations relied upon by the applicant for approval "were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted" (21 U.S.C. 355(b)(2)).A.What type of information can an applicant rely on?What type of information can an applicant rely on in an application that is based upon studies“not conducted by or for the applicant and for which the applicant has not obtained a right ofreference?”1.Published literatureAn applicant should submit a 505(b)(2) application if approval of an application will relyto any extent on published literature (a literature-based 505(b)(2)). If the applicanthas not obtained a right of reference to the raw data underlying the published study orstudies, the application is a 505(b)(2) application; if the applicant obtains a right ofreference to the raw data, the application may be a full NDA (i.e., one submitted undersection 505(b)(1)). An NDA will be a 505(b)(2) application if any of the specificinformation necessary for approval is obtained from literature or from another source towhich the applicant does not have a right of reference, even if the applicant alsoconducted clinical studies to support approval. Note, however, that this does not meanany reference to published general information (e.g., about disease etiology, support forparticular endpoints, methods of analysis) or to general knowledge causes theapplication to be a 505(b)(2) application. Rather, reference should be to specificinformation (clinical trials, animal studies) necessary to the approval of the application.2.The Agency’s finding of safety and effectiveness for an approved drugAn applicant should submit a 505(b)(2) application for a change in a drug whenapproval of the application relies on the Agency's previous finding of safety and/oreffectiveness for a drug. This mechanism, which is embodied in a regulation at 21 CFR314.54, essentially makes the Agency's conclusions that would support the approval ofa 505(j) application available to an applicant who develops a modification of a drug.Section 314.54 permits a 505(b)(2) applicant to rely on the Agency's finding of safetyand effectiveness for an approved drug to the extent such reliance would be permittedunder the generic drug approval provisions at section 505(j). This approach is intended to encourage innovation in drug development without requiring duplicative studies todemonstrate what is already known about a drug while protecting the patent andexclusivity rights for the approved drug.It is possible that an applicant could submit a 505(b)(2) application that relies both on literature and upon the Agency’s finding of safety and effectiveness for a previously approved drug product (e.g., to support a new claim).B.What kind of application can be submitted as a 505(b)(2) application?1.New chemical entity (NCE)/new molecular entity (NME)A 505(b)(2) application may be submitted for an NCE when some part of the datanecessary for approval is derived from studies not conducted by or for the applicantand to which the applicant has not obtained a right of reference. For an NCE, this data is likely to be derived from published studies, rather than FDA's previous finding ofsafety and effectiveness of a drug. If the applicant had a right of reference to all of theinformation necessary for approval, even if the applicant had not conducted the studies, the application would be a considered a 505(b)(1) application.2.Changes to previously approved drugsFor changes to a previously approved drug product, an application may rely on theAgency's finding of safety and effectiveness of the previously approved product,coupled with the information needed to support the change from the approved product.The additional information could be new studies conducted by the applicant orpublished data. This use of section 505(b)(2), described in the regulations at 21 CFR314.54, was intended to encourage innovation without creating duplicate work andreflects the same principle as the 505(j) application: it is wasteful and unnecessary tocarry out studies to demonstrate what is already known about a drug. The approachwas described in a letter to industry dated April 10, 1987, from Dr. Paul D. Parkman,then Acting Director of the Center for Drugs and Biologics. This guidance helps toclarify and amplify the approaches stated in the April 10, 1987, letter and in theregulations.An applicant should file a 505(b)(2) application if it is seeking approval of a change toan approved drug that would not be permitted under section 505(j), because approval will require the review of clinical data. However, section 505(b)(2) applications shouldnot be submitted for duplicates of approved products that are eligible for approvalunder 505(j) (see 21 CFR 314.101(d)(9)).In addition, an applicant may submit a 505(b)(2) application for a change in a drugproduct that is eligible for consideration pursuant to a suitability petition under Section505(j)(2)(C) of the Act. In the preamble to the implementing regulations for the Hatch-Waxman amendments to the Act, the Agency noted that an application submittedpursuant to section 505(b)(2) of the Act is appropriate even when it could also besubmitted in accordance with a suitability petition as defined at section 505(j)(2)(C) ofthe Act (see 57 FR 17950; April 28, 1992).III.WHAT ARE SOME EXAMPLES OF 505(B)(2) APPLICATIONS?Following are examples of changes to approved drugs for which 505(b)(2) applications should be submitted. Please note that in particular cases, changes of the type described immediately below may not require review of information other than BA or BE studies or data from limited confirmatory testing.2 In those particular cases, approval of the drug may also be sought in a 505(j) application based on an approved suitability petition as described in section 505(j)(2)(C) of the Act. The descriptions below address the situation in which the application should be filed as a 505(b)(2) application because approval of the application will require review of studies beyond those that can be considered under section 505(j). Some or all of the additional information could be provided by literature or reference to past FDA findings of safety and effectiveness for approved drugs, or it could be based upon studies conducted by or for the applicant or to which it has obtained a right of reference.•Dosage form. An application for a change of dosage form, such as a change from a solid oral dosage form to a transdermal patch, that relies to some extent upon the Agency's finding ofsafety and/or effectiveness for an approved drug.•Strength. An application for a change to a lower or higher strength.•Route of administration. An application for a change in the route of administration, such as a change from an intravenous to intrathecal route.•Substitution of an active ingredient in a combination product. An application for a change in one of the active ingredients of an approved combination product for another active ingredient that has or has not been previously approved.Following are additional examples of applications that may be accepted pursuant to section 505(b)(2) of the Act. Some or all of the additional information could be provided by the literature or reference to2 Limited confirmatory testing is explained in further detail in 54 FR 288872, 28880 (July 10, 1989) and 57 FR 17950, 17957-58 (April 28, 1992).past FDA findings of safety and effectiveness for approved drugs, or it could be based on studies conducted by or for the applicant or to which it has obtained a right of reference.•Formulation. An application for a proposed drug product that contains a different quality or quantity of an excipient(s) than the listed drug where the studies required for approval arebeyond those considered limited confirmatory studies appropriate to a 505(j) application.•Dosing regimen. An application for a new dosing regimen, such as a change from twice daily to once daily.•Active ingredient. An application for a change in an active ingredient such as a different salt, ester, complex, chelate, clathrate, racemate, or enantiomer of an active ingredient in a listeddrug containing the same active moiety.•New molecular entity. In some cases a new molecular entity may have been studied by parties other than the applicant and published information may be pertinent to the new application. This is particularly likely if the NME is the prodrug of an approved drug or the active metabolite ofan approved drug. In some cases, data on a drug with similar pharmacologic effects could beconsidered critical to approval.•Combination product. An application for a new combination product in which the active ingredients have been previously approved individually.•Indication. An application for a not previously approved indication for a listed drug.•Rx/OTC switch. An application to change a prescription (Rx) indication to an over-the-counter (OTC) indication.•OTC monograph. An application for a drug product that differs from a product described in an OTC monograph (21 CFR 330.11), such as a nonmonograph indication or a new dosageform.•Naturally derived or recombinant active ingredient. An application for a drug product containing an active ingredient(s) derived from animal or botanical sources or recombinanttechnology where clinical investigations are necessary to show that the active ingredient is thesame as an active ingredient in a listed drug.•Bioinequivalence. Generally, an application for a pharmaceutically equivalent drug product must be submitted under section 505(j) of the Act and the proposed product must be shown to be bioequivalent to the reference listed drug (21 CFR 314.101(d)(9)). Applications forproposed drug products where the rate (21 CFR 314.54(b)(2)) and/or extent (21 CFR314.54(b)(1)) of absorption exceed, or are otherwise different from, the 505(j) standards forbioequivalence compared to a listed drug may be submitted pursuant to section 505(b)(2) of theAct. Such a proposed product may require additional clinical studies to document safety andefficacy at the different rate and extent of delivery. Generally, the differences in rate and extent of absorption should be reflected in the labeling of the 505(b)(2) product. The proposedproduct does not need to be shown to be clinically better than the previously approvedproduct; however, a 505(b)(2) application should not be used as a route of approval for poorly bioavailable generic drug products unable to meet the 505(j) standards for bioequivalence. If the proposed product is a duplicate of an already approved product, it should not be submitted as a 505(b)(2) application (21 CFR 314.101(d)(9)).For example, a 505(b)(2) application would be appropriate for a controlled release productthat is bioinequivalent to a reference listed drug where:1.The proposed product is at least as bioavailable as the approvedpharmaceutically equivalent product (unless it has some other advantage, suchas smaller peak/trough ratio); or2.The pattern of release of the proposed product, although different, is at least asfavorable as the approved pharmaceutically equivalent product.IV.WHAT CAN'T BE SUBMITTED AS 505(B)(2) APPLICATIONS?•An application that is a duplicate of a listed drug and eligible for approval under section 505(j) (see 21 CFR 314.101(d)(9)); or,•An application in which the only difference from the reference listed drug is that the extent to which the active ingredient(s) is absorbed or otherwise made available to the site of action isless than the listed drug (21 CFR 314.54(b)(1)); or,•An application in which the only difference from the reference listed drug is that the rate at which its active ingredient(s) is absorbed or otherwise made available to the site of action isunintentionally less than that of the listed drug (21 CFR 314.54(b)(2)).V.WHY DOES IT MATTER IF AN NDA IS A 505(B)(2) APPLICATION?Unlike a full NDA for which the sponsor has conducted or obtained a right of reference to all the data essential to approval, the filing or approval of a 505(b)(2) application may be delayed due to patent or exclusivity protections covering an approved product. Section 505(b)(2) applications must include patent certifications described at 21 CFR 314.50(i) and must provide notice of certain patent certifications to the NDA holder and patent owner under 21 CFR 314.52.VI.PATENT AND EXCLUSIVITY PROTECTIONS THAT COULD AFFECT A 505(B)(2) APPLICATIONA.What type of patent and/or exclusivity protection is a 505(b)(2) applicationeligible for?A 505(b)(2) application may itself be granted 3 years of Waxman-Hatch exclusivity if one ormore of the clinical investigations, other than BA/BE studies, was essential to approval of theapplication and was conducted or sponsored by the applicant (21 CFR 314.50(j);314.108(b)(4) and (5)). A 505(b)(2) application may also be granted 5 years of exclusivity if it is for a new chemical entity (21 CFR 314.50(j); 314.108(b)(2)). A 505(b)(2) application may also be eligible for orphan drug exclusivity (21 CFR 314.20-316.36) or pediatric exclusivity(section 505A of the Act).A 505(b)(2) application must contain information on patents claiming the drug or its method ofuse (21 CFR 314.54(a)(1)(v)).B.What could delay the approval or filing of a 505(b)(2) application?Approval or filing of a 505(b)(2) application, like a 505(j) application, may be delayed because of patent and exclusivity rights that apply to the listed drug (21 CFR 314.50(i), 314.107, and314.108 and section 505A of the Act). This is the case even if the application also includesclinical investigations supporting approval of the application.VII.WHAT SHOULD BE INCLUDED IN 505(B)(2) APPLICATIONS?The Act (sections 505(b)(1) and (b)(2)) and FDA regulations (21 CFR 314.54) distinguish between 505(b)(1) and (b)(2) applications. Although the two types of applications must meet the same standards for approval (see section 505(b) and (c) of the Act), they differ in source of information to support safety and effectiveness, the patent certification requirements, BA/BE evidence, exclusivity bars, and processing within the FDA. The requirements for 505(b)(1) and 505(b)(2) applications are described at 21 CFR 314.50. Additional requirements for certain 505(b)(2) applications are described at 21 CFR 314.54.A 505(b)(2) application should include the following:•Identification of those portions of the application that rely on information the applicant does not own or to which the applicant does not have a right of reference (for example, for reproductive toxicity studies).•If the 505(b)(2) seeks to rely on the Agency's previous finding of safety or efficacy for a listed drug or drugs, identification of any and all listed drugs by established name, proprietary name (ifany), dosage form, strength, route of administration, name of the listed drug's sponsor, and the application number (21 CFR 314.54(a)(1)(iii)).Even if the 505(b)(2) application is based solely upon literature and does not rely expressly on an Agency finding of safety andeffectiveness for a listed drug, the applicant must identify the listed drug(s) on which the studies were conducted, if there are any. If the 505(b)(2) application is for an NCE and the 505(b)(2) applicant is not relying on literature derived from studies of an approved drug, there may not bea listed drug. If there is a listed drug that is the pharmaceutical equivalent to the drug proposedin the 505(b)(2) application, that drug should be identified as the listed drug.•Information with respect to any patents that claim the drug or the use of the drug for which approval is sought (21 CFR 314.50(h)). This patent information will be published in the Orange Book when the application is approved.•Information required under 314.50(j) if the applicant believes it is entitled to marketing exclusivity (21 CFR 314.54(a)(1)(vii)).• A patent certification or statement as required under section 505(b)(2) of the Act with respect to any relevant patents that claim the listed drug and that claim any other drugs on which the investigations relied on by the applicant for approval of the application were conducted, or that claim a use for the listed or other drug (21 CFR 314.54(a)(1)(vi)).If there is a listed drug that is the pharmaceutical equivalent of the drug proposed in the505(b)(2) application, the 505(b)(2) applicant should provide patent certifications for thepatents listed for the pharmaceutically equivalent drug. Patent certifications should specify the exact patent number(s), and the exact name of the listed drug or other drug even if all relevant patents have expired.•If an application is for approval of a new indication, and not for the indications approved for the listed drug, a certification so stating (21 CFR 314.54(a)(1)(iv).• A statement as to whether the listed drug(s) identified above have received a period of marketing exclusivity (21 CFR 314.108(b)). If a listed drug is protected by exclusivity, filing or approval of the 505(b)(2) application may be delayed.• A Bioavailability/Bioequivalence (BA/BE) study comparing the proposed product to the listed drug (if any).•Studies necessary to support the change or modification from the listed drug or drugs (if any).Complete studies of safety and effectiveness may not be necessary if appropriate bridgingstudies are found to provide an adequate basis for reliance upon FDA’s finding of safety and effectiveness of the listed drug(s).Before submitting the application, the applicant should submit a plan to the appropriate new drug evaluation division identifying the types of bridging studies that should be conducted. The applicant should also identify those components of its application for which it expects to rely on FDA’s finding of safety and effectiveness of a previously approved drug product. The division will critique the plan and provide guidance.REFERENCESApril 10, 1987, letter from then Acting Director of the Center for Drugs and Biologics to all NDA and ANDA holders and applicants."Abbreviated New Drug Application Regulations; Proposed Rule," Federal Register. Vol. 54, No. 130, Monday, July 10, 1989, page 28872."Abbreviated New Drug Regulations; Final Rule," Federal Register. Vol. 57, No. 82, Tuesday, April 28, 1992, page 17950."Abbreviated New Drug Application Regulations; Patent and Exclusivity Provisions; Final Rule," Federal Register. Vol. 59, No. 190, Monday, October 3, 1994, page 50338.GLOSSARY505(b)(2) application: an application submitted under section 505(b)(1) of the Act for a drug for which one or more of the investigations relied on by the applicant for approval of the "application were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted" (21 U.S.C. 355(b)(2)).Active ingredient: "any component that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease, or to affect the structure or any function of the body of man or of animals. The term includes those components that may undergo chemical change in the manufacture of the drug product and be present in the drug product in a modified form intended to furnish the specified activity or effect" (21 CFR 60.3(b)(2)).Active moiety: "the molecule or ion, excluding those appended portions of the molecule that cause the drug to be an ester, salt (including a salt with hydrogen or coordination bonds), or other noncovalent derivative (such as a complex, chelate, or clathrate) of the molecule, responsible for the physiological or pharmacological action of the drug substance" (21 CFR 314.108(a)).Investigations relied on for approval: those without which the application cannot be approved (i.e., animal and human safety tests as well as clinical investigations of effectiveness).Listed drug: "a new drug product that has an effective approval under section 505(c) of the act for safety and effectiveness or under section 505(j) of the act, which has not been withdrawn or suspended under section 505(e)(1) through (e)(5) or (j)(5) of the act, and which has not been withdrawn from sale for what FDA has determined are reasons of safety or effectiveness. Listed drug status is evidenced by the drug product's identification as a drug with an effective approval in the current edition of FDA's “Approved Drug Products with Therapeutic Equivalence Evaluations” (the list) or any current supplement thereto, as a drug with an effective approval. A drug product is deemed to be a listed drug on the date of effective approval of the application or abbreviated application for that drug product" (21 CFR 314.3(b)).Literature: published reports of well-controlled studies that support safety or effectiveness; proposed and final monographs published in the Federal Register; the data supporting a Federal Register notice announcing a product’s safety and/or effectiveness.Orange Book:Approved Drug Products with Therapeutic Equivalence Evaluations and any current supplement to the publication.Pharmaceutical equivalent or duplicate:"drug products that contain identical amounts of the identical active drug ingredient, i.e., the same salt or ester of the same therapeutic moiety, in identical dosage forms, but not necessarily containing the same inactive ingredients, and that meet the identical compendial or other applicable standard of identity, strength, quality, and purity, including potency and,where applicable, content uniformity disintegration times and/or dissolution rates" (21 CFR 320.1(c)). Products with different mechanisms of release can be considered to be pharmaceutical equivalents or duplicates.Referenced listed drug: "the listed drug identified by FDA as the drug product upon which an applicant relies in seeking approval of its abbreviated application" (21 CFR 314.3(b)).Right of reference or use: "the authority to rely upon, and otherwise use, an investigation for the purpose of obtaining approval of an application, including the ability to make available the underlying raw data from the investigation for FDA audit, if necessary" (21 CFR 314.3(b)).Sponsors have the right of reference to any studies: (1) they conduct, (2) that are conducted for them, or (3) for which they formally obtain a documented right of reference.An applicant is not considered to have a right of reference to published studies, because the applicant does not have access to the raw data. However, if the raw data are in the public domain, a right of reference is unnecessary.Suitability petition: A citizen petition submitted to the Agency seeking permission to file an abbreviated new drug application for a change from a listed drug in dosage form, strength, route of administration, or active ingredient in a combination product. (See section 505(j)(2)(C) of the Act)。
The 505(b)(2)

The 505(b)(2) Drug Development Pathway:When and How toTake Advantage of aUnique AmericanRegulatory Pathway By Mukesh Kumar, PhD, RAC andHemant Jethwani, MSThe 505(b)(2) regulation offers a less expensive and faster new drug development pathway that may be particularly attractive to a manufacturer with experience in developing generic products. It involves making significant changes to an existing product approved by the US Food and Drug Administration (FDA), called the reference product, to create a new drug with its own indica-tion, formulation, target population and/or other differences that need to be supported with clini-cal studies. A major advantage of this pathway is that it allows a sponsor to rely, at least in part, on FDA’s findings of safety and/or effectiveness for the previously approved drug, thereby reducing the number of clinical trials required for approval. Another incentive is three to five years of market exclusivity for 505(b)(2) products, depending upon the extent of changes to the reference prod-uct and the type of clinical data included in the approved New Drug Application (NDA).However, like all drug development strat-egies, the 505(b)(2) pathway requires careful consideration and planning. Important issues to consider include intellectual property concerns, the amount and quality of supporting informa-tion available from reference products or the literature, the logistics of conducting clinical trials with generic-like products, market compe-tition for approved products and requirements for an international product launch. Here we discuss practical strategies for drug development via the 505(b)(2) regulatory pathway.Drugs Can be Approved via One of Three Regulatory PathwaysNew drug products can belong to one of two broad categories: brand new drugs and identical or close copies of previously approved drugs, also called generics. Globally, separate regulatory pathways for innovator products and generic drugs are well established. US regulations, how-ever, divide these drugs into three categories: (1) new drugs, covered under Section 505(b)(1) of the Food, Drug, and Cosmetics Act (FD&C Act); (2) generic drugs, covered under Section 505(j) of the FD&C Act; and (3) “similar” drugs, covered under Section 505 (b)(2). It is the third category that is discussed here.The generic and 505(b)(2) categories were added by the Drug Price Competition and Patent Term Restoration Act of 1984, usually referred to as the Hatch-Waxman Act. The Hatch-Waxman Act aimed to promote generics while leaving intact a financial incentive for new product research and development. It was an attempt to balance the need for innovation with the desire for lower-cost alternatives within a reasonable length of time. Drug companies were given the opportu-nity to create not only exact copies of previously approved drugs, provided there was no infringe-ment of patents, but also improved versions of previously approved drugs by updating formu-lations or finding new uses. Table 1 describes the three pathways under the FD&C Act.Despite existing for more than 25 years, along with generic drugs, the 505(b)(2) prod-ucts have only recently become popular with drug companies due to increased challenges to discover and develop new chemical entities. As with innovator drugs, products following the 505(b)(2) pathway are subject to the full userfee under the Prescription Drug User Fee Act (PDUFA). They also may require several clinical and nonclinical studies that could involve signifi-cant resources, albeit less than for an innovator product but much higher than for a generic drug. Some key parameters for the three product cat-egories are listed in Table 2.The 505(b)(2) Pathway is Unique to the USThe 505(b)(2) application is intended to encour-age sponsors to develop improved generics, i.e., drugs similar to an approved product with some significant changes that are not permitted under Abbreviated New Drug Application (ANDA) rules. The 505(b)(2) pathway replaced the “Paper NDA” pathway used prior to the Hatch-WaxmanTable 1. Regulatory Pathways for New Drug ProductsAct, whereby FDA could approve NDAs that relied on published studies and lacked any reference to innovator safety and effectiveness data. Paper NDAs were frequently challengedby innovator product manufacturers, citing lack of sufficient safety and efficacy data. Under the 505(b)(2) regulation, FDA has the authority to approve new products based on fewer new stud-ies to demonstrate their safety and efficacy and relying extensively on the agency’s previous findings of safety and efficacy for the reference product. The sponsor of a 505(b)(2) product is not required to obtain a right of reference from the innovator product manufacturer. However, the sponsor needs to include data from bridging stud-ies to support changes from the reference drug.As mentioned, the 505(b)(2) application applies when certain changes are made to the innovator drug to either create a new formu-lation or include new uses/indications. The following are examples of changes to approved drugs that would fall under the 505(b)(2) mechanism:changes in dosage form, strength, for-•mulation, dosing regimen or route ofadministrationnew combination product, including •substitution of an active ingredientmodified active ingredient (e.g., salt, •chelate, ester, complex, etc.)new indications for previously•approved drugsover-the-counter switch of an approved •prescription drugBecause 505(b)(2) products are considered new products, they are subject to the PDUFA user fee requirement. Review by FDA is similar in duration to that of traditional NDAs, and the approved product is eligible for a minimum of three years of market protection from generics if the bridging studies were other than bioavailabil-ity (BA) and bioequivalence (BE) studies. This regulatory process is unique to the US. Products approved under the 505(b)(2) pathway typically are approved either as generics or new products in other countries. The 505(b)(2) Pathway Offers Many Advantages to Manufacturers and PatientsThere are advantages for all stakeholders from developing 505(b)(2) products. This pathway eliminates duplication of experiments and encourages developers to conduct new stud-ies that add value to the final product, suchas a better understanding of mechanisms of action, improved formulation and utilization of the same product for multiple diseases. Also, development of such products creates new intel-lectual property while protecting the rights of the original product, and providing a fair incentive for the investment. Since 505(b)(2) products are derived from reference products for which exten-sive safety and efficacy information is available, they generally carry less risk, cost less and can achieve FDA approval in a much shorter time. Some 505(b)(2) products have been created with less than $30 million in additional investment (in terms of new clinical and nonclinical studies conducted) and in about three years, which is remarkable compared to the cost and timeline for a traditional new drug.Perhaps the biggest incentive to develop 505(b)(2) products is the three to five years of market exclusivity in the US, depending upon the extent of changes to the previously approved drug and the amount of data submitted to FDA. This is an apparent advantage when compared to ANDA approval, where exclusivity can be held for only 180 days and applies only to the first generic product. Table 3 lists the different terms of market exclusivity available by product cat-egory and target indication.Market exclusivity enables manufacturersto take advantage of greater pricing flexibil-ity. During the market exclusivity period they can promote their product over the innovator drug and build their own brand with an attrac-tive price without fear of price erosion due to generic competition. Most importantly, 505(b)(2) products may receive an “AB” substitutability rating in the Orange Book. Thus, from a thera-peutic substitution perspective and under state formulary laws, the 505(b)(2) product is not at a disadvantage relative to a generic drug.Table 2. Comparison Between Conventional NDA, ANDA and 505(b)(2) Drug SubmissionsChallenges for Developing 505(b)(2) Drug ProductThere are some unique challenges facing 505(b) (2) applications. They often require substantial additional innovative work to bring the prod-uct to market. Since similars involve significant changes to the reference product formulation, either by including additional components or making changes to the active pharmaceuti-cal ingredient, the impact of these changeson safety and efficacy must be evaluated via clinical and/or nonclinical studies. Such stud-ies could uncover new issues, leading to further investigations and associated costs. Also, unlike generic drugs, such products involve extensive interactions with FDA to proactively understand regulatory, scientific and technical requirements. Such products are considered new and unique by FDA; hence, the review process is analogous to that for traditional NDAs.Also, since portions of the 505(b)(2) appli-cation could utilize self-generated proprietary data, this information needs to be protected via a patent or trade secret agreement, as applicable. Still, significant portions of the information could be in the public domain in existing patents for the innovator product. Unlike the traditional NDA, wherein the sponsor owns all the data necessary for approval (or has obtained the right to reference), the filing or approval of a 505(b) (2) application may be delayed due to reference drug patent or exclusivity protection. Sponsors filing 505(b)(2) applications must include patent certifications in their applications and must also provide notice of certain patent certifications to reference drug NDA and patent holders.Determining what additional information may be required for approval is a critical strategic requirement. Information requirements usually are subject to case-by-case determination by FDA. FDA guidance documents and discussions with regulatory professionals experienced in the 505(b) (2) approval route, as well as with the relevant FDA review division, are critical in understand-ing what data are necessary and adequate. The biggest risk: if the required studies are only BA/ BE studies, the product will receive a 505(b)(2) designation and be subject to associated user fees (which are about $1.4 million (US) in 2010) without being eligible for any market exclusivity and thus subject to generic competition from the beginning of market approval.There are few additional challenges associated with the use of the 505(b)(2) pathway. These products face fierce competition from generics with similar biological properties and since they are more expensive than generics, a robust marketing campaign may be required to attract customer attention. On the other hand, 505(b)(2) products offer certain advantages over innovator and/or generic drugs, enabling the manufacturer to promote these benefits directly to patients. These could be marketing advantages such as a formulation that is easier for patients to take, extended dosage, different strength, etc.Strategies for Developing 505(b)(2) ProductsFor small drug companies, the 505(b)(2) pathway for a new product could prove an attractive busi-ness model for the simple reason that it takes much less time, cost and risk to get the product onto the market compared to innovator drugs, and could yield significantly higher returns on investment compared to generic drugs.A good strategy could mean the difference between a successful, i.e., profitable, and unsuc-cessful product. The following are key strategic considerations for a 505(b)(2) product:extent of innovation/modification made •to the innovator product:these modifi-cations decide whether the product isapplicable for a 505(b)(2) review or not,and help determine the number of yearsof market exclusivity grantedthorough analysis of available data:•before embarking on manufacturing a505(b)(2) product, a company shouldthoroughly analyze the data available,including the scientific basis of approvalof the reference drug, published litera-ture, particularly since the innovatordrug was approved, market competi-tion, etc. (The amount of available datapreviously submitted to FDA deter-mines whether this is a viable project.)development strategy:• careful analysis of data should lead to a list of the addi-tional studies that may be required for agiven 505(b)(2) product; bridging stud-ies are required to show that changesto the innovator product lead to thedesired impact on safety, efficacy andtolerance of the proposed drug productTable 3. Market Exclusivity Available to FDA-Approved ProductsFDA discussions: there is no substitute •for robust discussions with the relevantFDA review division regarding theproposed and executed developmentstrategy; FDA offers significant adviceregarding final requirements for anapproval, and it has been statisticallydemonstrated that companies thatinvolve FDA in discussions early intheir product development plans andimplement the agency’s suggestionsincrease their chances for first cycleapproval almost three-fold, leading toenormous time and cost savings and,hence, higher returns on investmentimplementation of strategy: exhaustive •implementation planning is the path tosuccess; timelines should be diligentlyobserved and any deviations aggres-sively addressedcost control:• cost incurred depends upon the preclinical and clinical stud-ies required, amount of informationavailable regarding the reference drug,advancements in analytical technologyand various other such factors; bridgingstudies should be scientifically justifiedand strategically executed to control costmarketing and branding strategy:• as 505(b)(2) products are generally moreexpensive than generic versions ofthe innovator drug, the manufacturershould have a robust marketing plan ConclusionOver the years, the 505(b)(2) regulatory pathway has become very attractive to companies of all sizes. It is the proverbial “low-hanging fruit” for manufacturers due to the short time of marketing with attractive returns on investment. Every year FDA approves about twice as many 505(b)(2) applications as traditional 505(b)(1) applications. It is projected that due to increased challenges in creating new products, 505(b)(2) products might comprise more than 70% of all FDA approvals within 10 years. This pathway is particularly attractive to manufacturers transitioning from generic drugs to innovator products. Due to the similarities to traditional drug development, these products offer a low-risk market entry point by training the work force in the traditional development processes. However, there are unique scientific, regulatory, logistical and finan-cial challenges to developing such products––all of which could convert a potentially attractive project into a constant headache.AuthorsMukesh Kumar, PhD, RAC is a senior director, Regulatory Affairs, at Amarex Clinical Research, LLC, located in Germantown, MD, which is a full-service CRO offering regula-tory consultancy, strategic planning, trial management, data management and statistical analysis services for global clinical trials. Kumar is a member of the RAPS Board of Editors for Regulatory Focus and can be reached at mukeshk@amarexcro. com. Hemant Jethwani, MS is a regulatory affairs associate at Amarex Clinical Research LLC. He can be reached athemantj@.Do you want to be a part of an organization whose mission it is to protectthe health of the public by ensuring the safety and effectiveness ofmedical devices? Do you have a BA/BS degree in the sciences, and areyou knowledgeable in the fields of Engineering, Interdisciplinary Scienceand/or Nursing? Then, the Office of Compliance, Center for Devicesand Radiological Health, FDA wants you!The Office of Compliance mission is to promote and protect the healthof the public by ensuring the safety and effectiveness of medical devices.We enforce regulations and laws to which regulated industry is subject,without hindering innovation or access to medical devices. We are anorganization that hires scientists interested in protecting the public health,performing regulatory activities and working in a state of thescientific environment.To apply, email/send resume to:Collin Figueroa, Program Management Officer, OC,CDRH10903 New Hampshire Ave., Bldg. 66 Room 3438Silver Spring, Maryland 20993-0002e-mail: collin.figueroa@。
【技术篇】FDA药品申请介绍—505(b)(1),505(b)(2)和505(j)的区别

【技术篇】FDA药品申请介绍—505(b)(1),505(b)(2)和505(j)的区别本文由北京睿知而行科技有限公司首发,转载请注明出处。
欢迎关注北京睿知而行科技有限公司微信公众平台:bjruizhierxing。
一.总结505(b)(1),NDA,即新药申请,需要提供完整的临床前和临床安全性和有效性数据;505(b)(2),“文献”NDA,即“文献”新药申请,可依赖之前发表的安全性和有效性数据。
可能包括临床数据但是有时可仅仅基于生物利用度数据。
505(j),ANDA,简要新药申请,与现有产品具有生物等效性的药品。
注意ANDA可以不用包含临床前或临床安全性或有效性数据。
注意,ANDA可能会被要求提供“临床等效”数据,并且在理论上,如果杂质在其它方面不受限制,需提供毒理学数据。
二.背景介绍在FDA药品申请中经常提到的505(b)(1),505(b)(2)和505(j),实际上来源于美国用于管理药品的联邦法律,它分为两个部分,1.成文法-《联邦食品、药品和化妆品法》;2.行政法—《美国联邦法规》。
成文法由美国国会通过,而行政法由公职人员是贯彻执行法令的规定,由公职人员制定。
其中《联邦食品、药品和换妆品法》中的505章节,描述了新药的划分和要求。
以下是505章节的相关规定:505(a):除非依据小节(b)或小节(j)提交的与药品有关的有效申请获得批准,否则任何人不应当将任何新药引入或送交洲际贸易。
505(b)(1)新药申请(NDA),“任何人可依据小结(a)条款的要求向部长”递交与任何药品对象有关的申请。
作为申请的一部分,此人应当提交给部长:(A)完整的证明该药品使用是否安全和使用是否有效的研究报告;(B)完整的用作该药组分的成分清单;(C)完整的该药组分说明;(D)用于该药生产、加工和包装的方法及设施及控制的完整叙述;(E)部长可能要求的该药样品及用作组分的成分样品;(F)拟用于该药的标签样本;(G)依据505B条款要求的任何评价;505(b)(2)“文献”NDA新药申请,“依据段(1)提交的该段条款(A)所描述的药品研究的一种申请【即505b(1)】,申请者未开展过或没有为申请者开展过研究,在这种情况下,申请者不能从开展方或研究针对方获得引述或使用权……”允许参考已发表的科学文献建立药物临床前和临床的安全性和有效性。
干货|FDA的505(b)(2)新药申请途径

干货|FDA的505(b)(2)新药申请途径一、法律基础自1938年《联邦食品、药品和化妆品法案》(FD&C Act)颁布以后,所有新药在美国上市之前都必须向FDA提出正式的新药申请。
FDA需根据申请者提供的信息对三个方面做出评判:▪对于拟申报适应症,药物是否安全有效?药物的获益是否大于风险?▪药物的标签内容是否合适?需要包含哪些内容?▪药物的制造方法和质量控制是否足以保证其特性、规格、质量和纯度?1984年国会通过了《药品价格竞争和专利期修正案》(Hatch-Waxman Amendment),修订后的《联邦食品、药品和化妆品法案》505部分为新药申请提供了三条路径:▪505(b)(1):申请包含完整安全性和有效性研究报告▪505(b)(2):申请包含完整安全性和有效性研究报告,但至少有部分信息来源于非申请者开展或申请者无权引用的研究。
▪505(j) :申请包含信息证明拟申报药物与参比制剂有着完全相同的活性成分、剂型、规格、给药途径、标签信息、质量、特性和适应症等。
505(b)(1)和505(b)(2)被称为新药申请(NDA),而505(j)则被称为简略新药申请(ANDA)。
505(b)(2)和(j)部分共同替代了FDA先前的“文献NDA”政策,即可以用发表的文献作为药物安全性和有效性的证据。
图1:FDA新药申请的三条途径二、适用范围505(b)(2)路径可以适用于两种类型的申报,一种是新化学分子(NCE),另一种是已批准药物的改变。
一般情况下,NCE通过505(b)(1)途径申报,但是当部分申报所用数据不是由申请者开展的试验得来,同时申请者又没有权限引用这些试验原始数据时,就必须通过505(b)(2)路径进行申请。
大多数情况下NCE申请引用数据的来源是发表的文献。
505(b)(2)路径用的最多的是对已批准药物的改变,包括适应症、配方、剂型、给药途径、用药方案等。
法规的初衷是鼓励创新,同时避免重复不必要的已经明了的试验。
生物等效性原理及原则

生物等效性试验原理和原则1.背景美国对药品质量监管的三项制度安排,使得它在制定和颁布行业法规方面领先于世界。
首先,美国国会授予美国药典(US P)和国家处方集(NF)修订委员会制定药品及其制剂的规格、质量和纯度标准的权利。
尽管USP和NF是私人机构,对美国食品药品监督管理局(FDA)没有管理权。
其次,FDA也由美国国会授权,为开发和制造安全有效的药物制定法规。
最后,主要由美国食品及药物管理局制定,药品生产商实施的药品生产质量管理规范,确保了药品的质量。
FDA还颁布了药品的生物利用度(BA)和生物等效性(BE)的规范。
所有新药申请(NDAs)和新药补充申请必须通过体外的测试阐明药品在体内的生物利用度,以确保各个批次的质量,通常用溶出度测试的方法。
表5.1展示了各种监管法规对不同注册类型的要求。
根据联邦食品、药品和化妆品(FD&C)法案第505(b)节的规定,提交NDA或新动物药品申请(NADA)必须记录BA(21CFR 320.21(a))。
如果药品获得批准,NDA药品可能随后成为参比制剂(RLD)。
根据505(j)章节的规定,申请人提交简化新药申请(ANDA)或简化动物新药申请(ANADA)时必须达到药学等效,再达到生物等效,才能被视为和RLD药品治疗等效。
BE是利用相对生物利用度的方法,比较仿制药和参比制剂的体内行为。
(药学等效是指药品含有相同的活性成分、相同的BE在21 CFR 320.1中被定义为“在相似的试验条件下给以相同摩尔剂量的药物后,受试制剂的活性成分、活性分子的吸收速度和程度与参比制剂相比,无显著性差异。
”FDA通常考虑用血浆中的药物浓度作为药物作用位点的浓度的替代指标。
21 CFR 320.24给出了实现BE的途径。
证明生物等效需要综合多项研究证据,如PK、PD、临床试验、体外实验,以及其他能证明等效的研究资料。
2.获得上市许可的等效性文件药学等效的不同厂家的医药产品必须证明治疗等效,才可以相互替换。
你了解RLD与RS吗?
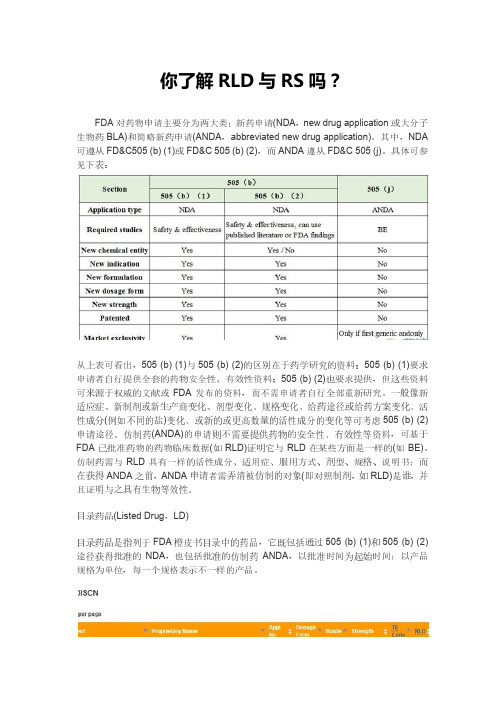
你了解RLD与RS吗?FDA对药物申请主要分为两大类:新药申请(NDA,new drug application或大分子生物药BLA)和简略新药申请(ANDA,abbreviated new drug application)。
其中,NDA 可遵从FD&C505 (b) (1)或FD&C 505 (b) (2),而ANDA遵从FD&C 505 (j)。
具体可参见下表:从上表可看出,505 (b) (1)与505 (b) (2)的区别在于药学研究的资料:505 (b) (1)要求申请者自行提供全套的药物安全性、有效性资料;505 (b) (2)也要求提供,但这些资料可来源于权威的文献或FDA发布的资料,而不需申请者自行全部重新研究。
一般像新适应症、新制剂或新生产商变化、剂型变化、规格变化、给药途径或给药方案变化、活性成分(例如不同的盐)变化、或新的或更高数量的活性成分的变化等可考虑505 (b) (2)申请途径。
仿制药(ANDA)的申请则不需要提供药物的安全性、有效性等资料,可基于FDA已批准药物的药物临床数据(如RLD)证明它与RLD在某些方面是一样的(如BE)。
仿制药需与RLD具有一样的活性成分、适用症、服用方式、剂型、规格、说明书;而在获得ANDA之前,ANDA申请者需弄清被仿制的对象(即对照制剂,如RLD)是谁,并且证明与之具有生物等效性。
目录药品(Listed Drug,LD)目录药品是指列于FDA橙皮书目录中的药品,它既包括通过505 (b) (1)和505 (b) (2)途径获得批准的NDA,也包括批准的仿制药ANDA,以批准时间为起始时间;以产品规格为单位,每一个规格表示不一样的产品。
FDA批准的每一个产品都会列于橙皮书中,或于“处方药Rx”目录中,或于“非处方药OTC”目录中,或于“撤市产品DISCN”目录中。
其中,撤市产品是指已被FDA批准但不再上市的产品,一般是因产品销量问题而撤市;而对于因产品自身的安全性、有效性问题而撤市的产品,会直接剔除橙皮书中。
2016年FDA批准上市的505b2药物清单

As of December 31, 2016
This report reflects the data shown as it is identified in the database. Selection Criteria: User Response: Start Date: 1/1/2016 Sort Order: Approval Date End Date: 12/31/2016
DEXILANT SOLUTAB ADZENYS XR-ODT ONZETRA XSAIL ZENAVOD ZEMBRACE SYMTOUCH ZEPATIER CETYLEV
TAKEDA PHARMACEUTICALS USA INC NEOS THERAPEUTICS AVANIR PHARMACEUTICALS DR REDDYS LABORATORIES INC DR REDDYS LABORATORIES LTD MERCK SHARP AND DOHME CORP ARBOR PHARMACEUTICALS LLC ACCORD HEALTHCARE INC PROMIUS PHARMA LLC UCB INC UBC INC UCB INC PFIZER INC ALLERGAN INC ALCON RESEARCH LTD GILEAD SCIENCES INC SPECTRUM PHARMACEUTICALS INC SPECTRUM PHARMACEUTICALS INC GENTIUM SPA GILEAD SCIENCES INC PROVEPHARM SAS INSITE VISION INC ABBVIE INC
2019年FDA批准上市的505b2药物清单
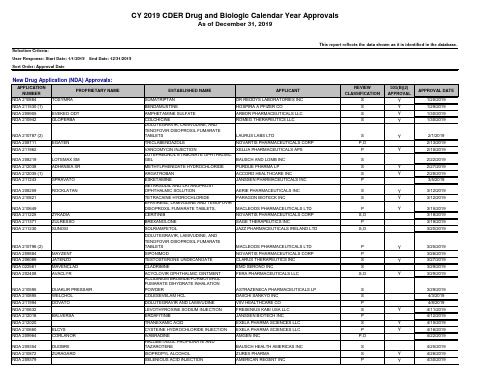
EXELA PHARMA SCIENCES LLC AMGEN INC
BAUSCH HEALTH AMERICAS INC ZUREX PHARMA AMERICAN REGENT INC
REVIEW CLASSIFICATION
S S S S
505(B)(2) APPROVAL
Y Y Y Y
APPROVAL DATE
P
ZINC SULFATE
AMERICAN REGENT INC
S
DULOXETINE DELAYED-RELEASE
CAPSULES
SUN PHARMA GLOBAL FZE
S
GLUCAGON
ELI LILLY AND CO
S
BIVALIRUDIN
MAIA PHARMACEUTICALS INC
S
NDA 208219 NDA 212038 NDA 212035 (1) NDA 211243
NDA 208259 NDA 210821
NDA 210649 NDA 211225 NDA 211371 NDA 211230
EGATEN
LOTEMAX SM ADHANSIA XR SPRAVATO ROCKLATAN
FERA PHARMACEUTICALS LLC ASTRAZENECA PHARMACEUTICALS LP
COLESEVELAM HCL DOLUTEGRAVIR AND LAMIVUDINE LEVOTHYROXINE SODIUM INJECTION
DAIICHI SANKYO INC VIIV HEALTHCARE CO FRESENIUS KABI USA LLC
FERRIC MALTOL
生物等效性原理及原则

生物等效性试验原理和原则1.背景美国对药品质量监管的三项制度安排,使得它在制定和颁布行业法规方面领先于世界。
首先,美国国会授予美国药典(US P)和国家处方集(NF)修订委员会制定药品及其制剂的规格、质量和纯度标准的权利。
尽管USP和NF是私人机构,对美国食品药品监督管理局(FDA)没有管理权。
其次,FDA也由美国国会授权,为开发和制造安全有效的药物制定法规。
最后,主要由美国食品及药物管理局制定,药品生产商实施的药品生产质量管理规范,确保了药品的质量。
FDA还颁布了药品的生物利用度(BA)和生物等效性(BE)的规范。
所有新药申请(NDAs)和新药补充申请必须通过体外的测试阐明药品在体内的生物利用度,以确保各个批次的质量,通常用溶出度测试的方法。
表5.1展示了各种监管法规对不同注册类型的要求。
根据联邦食品、药品和化妆品(FD&C)法案第505(b)节的规定,提交NDA或新动物药品申请(NADA)必须记录BA(21CFR 320.21(a))。
如果药品获得批准,NDA药品可能随后成为参比制剂(RLD)。
根据505(j)章节的规定,申请人提交简化新药申请(ANDA)或简化动物新药申请(ANADA)时必须达到药学等效,再达到生物等效,才能被视为和RLD药品治疗等效。
BE是利用相对生物利用度的方法,比较仿制药和参比制剂的体内行为。
(药学等效是指药品含有相同的活性成分、相同的说明书需要需要需要需要需要BE在21 CFR 320.1中被定义为“在相似的试验条件下给以相同摩尔剂量的药物后,受试制剂的活性成分、活性分子的吸收速度和程度与参比制剂相比,无显著性差异。
”FDA通常考虑用血浆中的药物浓度作为药物作用位点的浓度的替代指标。
21 CFR 320.24给出了实现BE的途径。
证明生物等效需要综合多项研究证据,如PK、PD、临床试验、体外实验,以及其他能证明等效的研究资料。
2.获得上市许可的等效性文件药学等效的不同厂家的医药产品必须证明治疗等效,才可以相互替换。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
This lesson is supported by an educational grant fromDrug Store News is approved by theAccreditation Council for Pharmacy Education(ACPE) as a provider oflogical advances necessary to create bio-pharmaceuticals, these medications car-ry a higher price tag. These medications also target a smaller patient population, such as those with rare cancers or im-munologic disorders. In order to recov-er the cost of creating and marketing these technological miracles, manufac-turers are forced to charge huge prices. Unfortunately, no generic formulations of these medications are available cur-rently, predominantly because of patent coverage. However, the patents on nu-merous biotech agents have expired or will expire soon, including human in-sulin and human growth hormones. THE HATCH-WAXMAN ACTThe Hatch-Waxman Act, formally known as the Drug Price Competition and Patent Term Restoration Act of 1984, was an attempt to balance the need for innovation with the desire for lower-cost alternatives within a reasonablelength of time. The two categories of ap-plications for Food and Drug Adminis-tration (FDA) medication approval with which health care professionals are most familiar are NDAs, used for innovator drugs, and ANDAs, used for generic products. The NDA process for innova-tor products is described in section 505(b)(1) of Federal Food, Drug and Cosmetic Act. The approval process for these products requires full reports of investigations of safety and effective-ness conducted by or for the applicant or for which the applicant has the right of reference for the information submitted. These medications are innovator prod-ucts protected by patent laws.The right of reference means the authority to rely upon an investigation for the purpose of obtaining approval of an application, including the abili-ty to make available the underlying raw data from the investigation for FDA audit. Data used in the 505(b)(1) process typically are based on research for which the applicant has the right of reference because the applicant has conducted the research or paid to have it done.The ANDA process for generic prod-ucts is described in section 505(j). In the ANDA process, the generic manufac-turer only needs to complete studies that demonstrate the generic product to be bioequivalent to the innovator product.All drug products do not fall neat-ly into one of these two categories. For example, a product that has the same active ingredient as a product ap-proved through the 505(b)(1) process, but has a different formulation, cannot be considered for approval as a gener-ic product. Yet, if treated strictly as a “new” drug, it would require all the clinical testing for safety and efficacy that is required of innovator drugs. Keeping in mind that part of the intent of the Hatch-Waxman Act was to in-crease efficiencies in the drug approval process and to avoid unnecessary de-lay and expense in bringing a drug product to market, it is not surprising that the FDA created some special pro-visions for the approval of such prod-ucts. These provisions are embodiedin section 505(b)(2) of the act. Funda-mentally, they enable a drug manu-facturer to submit a drug for approvalbased on existing data about the drugor a similar drug, thus avoiding theneed to conduct research that wouldduplicate that data. Instead, what maybe submitted are full reports of inves-tigations of safety and effectivenesswhere at least some portion of the in-formation submitted for approvalcomes from studies not conducted byor for the applicant and for which theapplicant has not obtained the right ofreference for the information undersection 505(b)(2) of the Act. The typesof information an applicant can rely oninclude published literature describ-ing study results and the FDA’s find-ings of safety and effectiveness from apreviously approved medication.Products approved through the505(b)(2) process are considered to bepharmaceutical alternatives to an ap-proved medication.APPROVAL UNDER SECTION 505(B)(1)When a pharmaceutical manufacturerdevelops a new medication, the FDArequires extensive testing to provethe safety and efficacy of the productbefore it can be marketed. The approvalprocess is regulated by numerous lawsoutlining strict requirements. The firstattempt to regulate medications manu-factured in the United States was theFederal Food and Drug Act of 1906, re-quiring that medications marketed ininterstate commerce meet minimal stan-dards of strength, purity and quality.This law made no attempts to regulatetherapeutic claims. This act soon wasfollowed by the Sherley Amendment in1912, allowing for the regulation offraudulent claims of therapeutic effectsthrough the regulation of labeling. TheFDA finally gained the authority toevaluate and approve medicationsbased on their safety profile until theFood and Drug Act was updated by theFederal Food, Drug and Cosmetic Actof 1938. It was not until 1962 whenthe Kefauver-Harris Amendment waspassed that the FDA gained authorityto require proof of efficacy in order togain FDA approval.In order to prove safety and efficacy,pharmaceutical manufacturers are re-quired to complete extensive clinicaltrials, consisting of three phases. PhaseI clinical trials are intended to assess thepharmacologic and pharmacokinetic ac-tivity of the drug in humans. In thesestudies, the medication is administeredto a small number of healthy volunteers.Phase II clinical studies are intended toobtain preliminary data related to theeffectiveness of the medication in pa-tients suffering from the disease orcondition being evaluated. Along withassessing effectiveness, information isgathered pertaining to side effects andrisks associated with the medication. Iffavorable results are obtained in PhaseII trials, Phase III trials are performed.The size of trial is determined by theprevalence of the condition being treat-ed, the power necessary to discern clin-ical outcomes and the medication’ssafety profile. Typically, several hun-dred to several thousand participantsare required in order to yield sufficientinformation pertaining to safety, effica-cy and the overall risk-benefit ratio foruse of this product in humans. When allthree phases are complete, the manu-facturer submits an NDA containingresults from these studies to the FDA.FDA scientists review the NDA to as-sess whether the trials demonstrate theproduct’s benefit, compared with itsrisks. During the evaluation process, theFDA reviews the study outcomes, chem-istry, manufacturing, controls, labeling,bioavailability, stability and compliancewith good manufacturing standards.By the time an NDA is approved, themanufacturer has invested numerousyears and many millions of dollars fordevelopment, clinical trials and regula-tory approval. As compensation, theFDA grants the company the exclusiveright to manufacture the product underpatent protection. Prior to 1995, patentprotection typically lasted 17 years fromthe date the patent was issued. Follow-ing the Uruguay Rounds AgreementsAct (Public Law 103-465) in 1995, man-ufacturers are granted patent protectionfor 20 years from the date of the firstfiling of the patent application. Becauseproducts usually are patented beforethey reach clinical trials, many years ofpatent life have passed before the NDAis approved. The Patent Term Restora-tion, another component of the Hatch-Waxman Act, does allow the manufac-turer to reclaim some of the time lostduring development and approval. TheFDA can extend the patent by the lengthof time required for the regulatoryprocess plus one-half of the time re-quired for the clinical trials, the total ofwhich is not to exceed five years. Oncethe patent has expired, other manufac-turers can petition the FDA for approvalof a generic version.GENERIC APPROVAL PROCESSSECTION 505(J)Before 1984, generics manufacturers hadto complete the same time-intensive andcostly clinical trials that the innovatingmanufacturer submitted with its NDA.This act created an abbreviated proc-ess for generic product approval, theANDA. Before submitting an ANDA,the generic manufacturer only needs tocomplete studies that demonstrated thegeneric product to be bioequivalent tothe innovator product. No longer re-quired to repeat clinical trials, genericsmanufacturers are able to obtain ap-proval in less time and with less expen-diture of resources. This enables themto offer their product at a lower price.In order to manufacture a genericproduct, the FDA mandates that themanufacturer prove its medication:•contains the same active ingredient.•is the same strength, in the samedosage form and is administeredvia the same route.•has the same indications and dosing.•is bioequivalent.•meets the same batch-to-batchrequirements for strength, purityUnderstanding the 505(b)(2) approval processand quality.•is manufactured using good manu-facturing practice regulations simi-lar to those of the brand product.Bioequivalent medications show no significant difference in the rate and extent to which the active ingredient becomes available at the site of action. This is assessed by looking for differ-ences in the plasma concentration-time profile. The first component is the peak concentration of the drug in the blood, known as C max. The second component is the time required to reach the peak concentration after the medication is ad-ministered, referred to as t max. The final component is the area under the curve (AUC), representing the total area un-der the concentration-time curve. The C max, t max and AUC of the generic product cannot be statistically different from those of the innovator product. The results of the concentration-time profile must fall within a 90 percent confidence interval for the ratio of the mean re-sponse and fall within the limits of 80 percent to 125 percent, using log trans-formed data.Based on FDA-established bioequiv-alence of products, substitution of one product for another can be recom-mended without any adjustment in dose or additional therapeutic monitoring. To identify bioequivalent medications, the pharmacist can refer to the “Ap-proved Drug Products with Therapeutic Equivalence Evaluations,” commonly referred to as the “Orange Book.” The Orange Book is so named for the color of the book’s cover and also is available on the FDA’s Web site.UTILIZING THE ORANGE BOOKFOR SUBSTITUTIONSWhen considering medication for sub-stitution, the pharmacist should beaware of the coding system the FDAuses in the Orange Book (Table 3). TheFDA assigns each multisource producta two-letter code, providing informa-tion about equivalency to a referencelisted drug.The first letter, representing theequivalency of the product, is eitheran “A” or “B.” The FDA considers med-ications rated “A” to be therapeuticallyequivalent to its pharmaceuticallyequivalent products. To be consideredtherapeutically equivalent, all actual orpotential bioequivalence problems mustbe resolved with adequate in vivo or invitro bioequivalence evidence.The FDA does not consider medica-tions rated “B” as therapeutically equiv-alent to other pharmaceutically equiva-lent products. “B”-rated products fallinto three main categories:1) products containing an active in-gredient or in a dosage form with doc-umented or potential bioequivalenceproblems for which adequate data tosupport bioequivalence has not beensubmitted to the FDA, or2) products with inadequate qualitystandards or insufficient basis to deter-mine therapeutic equivalence, or3) products currently undergoingFDA review.The second letter represents anyadditional information the FDA dis-covered during its evaluation. Theseletters are usually “A,” “B,” “N,” “O,”“P” or “T.” The information providedby this letter often is related to thedosage form of the product. AB-ratedproducts are those for which “actual orpotential bioequivalence problemshave been resolved with adequate invivo and/or in vitro evidence support-ing bioequivalence.”12Although all“A”-rated products are consideredbioequivalent, this bioequivalence hasbeen proven scientifically with AB-rat-ed products. AA, AN, AO, AP, AT-rat-ed products do not have any suspectedor proven bioequivalence problems,but the necessary bioequivalence in-formation is presumed. Despite thislack of information, these products arestill considered bioequivalent.In the late 1970s, states beganencouraging substitution with genericproducts. As of 2004, 11 states mandat-ed generic substitution when anappropriate generic is available, whilethe other 39 permit the pharmacist tosubstitute equivalent products. States’regulations vary on which medicationsare substitutable. Many states allow sub-stitution only with AB-rated medica-tions. Some states, such as Massachu-setts, utilize positive formularies for sub-stitution. Within positive formularies,only medications listed in the formula-ry are eligible for substitution.APPROVAL UNDER SECTION 505(B)(2)Section 505(b)(2) of the Hatch-WaxmanAct is intended to promote innovation,while saving time and resources andavoiding unnecessary duplicate hu-man testing. Medications approved un-der this section are not completely newproducts, yet they do not qualify forapproval as generics. These medica-tions contain some differences from areference listed drug, while offering atherapeutic alternative. NDA approvalunder section 505(b)(2) is not intend-ed to infringe on patent and exclusiv-ity protections of other medications.Since 1987, the FDA has approvedmore than 100 applications submittedunder section 505(b)(2).In the 505(b)(2) application, the ap-plicant relies on information from stud-ies it did not conduct and does not havethe right of reference.Although the manufacturer may nothave performed the studies, it must sub-mit clinical and non-clinical data todemonstrate the medication is safe andeffective. It also must be able to providedata and information, including bio-availability or comparative bioavail-ability studies, to establish sufficientlythe appropriateness of relying on mate-rial without the right of reference.Not all applications are eligible forsubmission under section 505(b)(2). A505(b)(2) application is not appropriatefor medications eligible for ANDA ap-proval as a generic or medications inwhich the only difference from the ap-proved product is the extent or rate inwhich the active ingredient is absorbedor otherwise made available to the siteof action. Submissions that do qualifyfor 505(b)(2) approval include productsthat have the following characteristics:•dosage form change,such as a solidoral preparation to a transdermalmatch, that relies to some extenton FDA safety and effectivenessfindings.•change in strength.•change in the route of administration,such as a change from subcutane-ous to intravenous route.•substitution of an activ e ingredientin a combination product for an-other active ingredient (both ofwhich previously have been ap-proved individually).•formulation change,including pro-posed drug products that containa different quality or quantity ofan excipient.•dosing regimen change.•different activ e ingredient,such as adifferent salt, ester, complex, chelateor enantiomer.•new molecular entity if the entity wasstudied by parties other than the ap-plicant, particularly if the proposedmedication is the prodrug or metabo-lite of an approved medication.•new combination product in whichthe active ingredients previouslyhave been approved individually.•new indications.•change a prescription to an ov er-the-counter indication.•naturally derived or recombinant activeingredient for which clinical investi-gations are necessary to show theactive ingredient is the same as anapproved medication.Important medication approvalshave occurred through this approvalpathway. In particular, section 505(b)(2) is crucial in areas of drug development where limited or uncertain markets war-rant maximum utilization of available knowledge. For example, pharmaceuti-cal manufacturers often formulate med-ications in dosage forms and regimens intended for adults. After approval of the adult formulation, manufacturers may seek approval for a different for-mulation for children. Along with drain-ing resources and delaying approval, repeating extensive clinical trials to demonstrate effectiveness could put study subjects at unnecessary risk.Even the United States Army has utilized section 505(b)(2) to expedite ap-proval of agents necessary to protect military personnel during times of war. In this era of concern about biological weapons, military personnel require pre-treatment against nerve agents. In the event of exposure, military person-nel require easy and rapid administra-tion of treatments. In order to obtainapproval for use of pyridostigmine bro-mide as pretreatment to increase sur-vival after exposure to the nerve agentSoman, the Army was required to sub-mit data demonstrating safety and effi-cacy. The exposure of subjects to Somanin order to prove safety and efficacywould have been unethical. Therefore,the Army utilized efficacy data from an-imal studies and safety informationfrom pyridostigmine bromide use forthe treatment of myasthenia gravis. Toprovide treatment in the event of nerveagent exposure, military personnelcarry atropine and pralidoxime autoin-jectors. For quicker administration ofthese medications, the Army submittedan NDA to combine these products inone autoinjector.LEGAL ISSUESApproving medications under section505(b)(2) has been challenged by someparties. The application process allowsapplicants to utilize published litera-ture and the FDA’s prior findings ofsafety and effectiveness of an approvedmedication. Some innovator manufac-turers have filed citizens’ petitionsagainst the use of FDA’s prior findings.These FDA findings are often based onfindings from studies submitted as partof an approved NDA. While publishedliterature is available in the public do-main, data from NDA submissions re-mains proprietary. Although the statu-tory language clearly allows for fullNDA applications that rely on data towhich the applicant does not haveright of reference, language does notclearly specify if that information canextend past published literature. Somepharmaceutical manufacturers arguethe intent of section 505(b)(2) was toallow referencing only portions of anNDA application available in the pub-lished literature, not proprietary por-tions of data. The FDA has upheld itsposition that section 505(b)(2) permitsreliance on previous FDA findings ofsafety and efficacy.Some innovator manufacturers alsocontend that 505(b)(2) approvals infringeon their patents, while the FDA contendsthat 505(b)(2) medications are designedaround the patent without infringingupon it. Pfizer and Dr. Reddy’s Labora-tories (DRL) waged a highly publicizedlegal battle over amlodipine patents. In1986, Pfizer was granted a patent on theamlodipine molecule. The followingyear, Pfizer filed a subsequent patent ap-plication specifically for the amlodipinebesylate salt, later marketed as Norvasc,®due to superior blood pressure-lower-ing activity than amlodipine maleate. Asthe patent neared expiration, Pfizer filedfor a Patent Term Extension in order torecoup some of the patent life lost dur-ing the approval process. After review-ing Pfizer’s patents, lawyers for DRLconcluded that the Patent Term Exten-sion only covered the besylate salt, ratherthan the amlodipine molecule. DRL sub-mitted an NDA under section 505(b)(2)for the besylate form. In 2002, the FDAgranted approval for amlodipine besy-late (AmVaz®). Meanwhile, Pfizer suedDRL for patent infringement, arguingthat the patent extension covered the am-lodipine molecule. In this case, the U.S.Appeals Court sided with Pfizer’s inter-pretation of its patent.Innovator manufacturers that do notpatent an entire molecule, rather opting topatent only the salt, isomer or activemetabolite that will be FDA approved,often have found that another manufac-turer is waiting to produce other formu-lations. While traditional albuterol prod-ucts contained a mixture of two enan-tiomers, Xopenex®(levalbuterol) containsonly the isomer that exhibits fewer ad-verse effects. Although albuterol is off-patent and available from numerous man-ufacturers, Xopenex®maintains patentprotection and market exclusivity, cost-ing more than twice as much. Sepracoralso co-markets fexofenadine (Allegra®)and desloratadine (Clarinex®), based onprevious patents for terfenadine(Seldane®) and loratadine (Claritin®).Even some generic manufacturershave challenged 505(b)(2) approvals. In2000, Synthon filed a 505(b)(2) applica-tion for paroxetine mesylate tablets (Pex-eva®). The application proved safety andefficacy from published literature, clin-ical trials Synthon performed and pre-vious FDA findings of safety and effi-cacy of a related chemical compound,paroxetine hydrochloride hemihydrate(Paxil®). The FDA approved Pexeva®in 2003. Pexeva®was not AB-ratedto Paxil,®but was a less expensivepharmaceutical alternative offeringequivalent dosing regimens. Within aCASE STUDYUnderstanding the 505(b)(2) approval processmonth after approval of Pexeva,®the FDA approved Torpharm’s generic ver-sion of paroxetine hydrochloride tablets, providing the product with an AB rating to Paxil.®Torpharm submitted a citizens petition to the FDA on the grounds that Pexeva®infringed on its 180-day mar-keting exclusivity as the first generic equivalent to Paxil.®According to the FDA, disallowing 505(b)(2) submissions in their current state would divert industry resources, increase drug costs, strain FDA review resources and slow the approval process without providing an improved health benefit. In addition, the need to dupli-cate trials utilized in previous NDAs has the potential to raise ethical questions pertaining to unjustified testing of ani-mals and humans.WILL 505(B)(2) PROVIDE A BIOGENERIC APPROVAL PATHWAY?Most biotech medications are regulated by laws different from those controlling traditional medications. Because of the complex cellular processing necessary to manufacture biopharmaceuticals, in-novator biopharmaceuticals undergo a different approval process, known as biologics licensing applications.Some concern exists pertaining to the difficulty of replicating all aspects of these molecules, leading to questions about approving biogenerics through the standard ANDA process. Being as the Hatch-Waxman Act was instituted prior to the development of most biopharma-ceutical products, it does not specifical-ly address the approval process for bio-generics. A few simpler biologic med-ications, including recombinant insulin and human growth hormone, do fall un-der the jurisdiction of Hatch-Waxman because the innovator was approved under section 505(b)(1), rather than a biologics licensing application. As of yet, no regulations and guidelines are available in the United States to address approval of biogenerics.The European Medicine Evaluation Agency (EMEA) has issued guidelines concerning biogenerics. The EMEA Committee for Proprietary of Medicinal Product’s guidelines, entitled “Guide-line on comparability of medicinal prod-ucts containing biotechnology-derived proteins as active substance: quality issues,” are primarily devoted to com-parability issues related to a manufac-turer’s alteration in its own production process, but briefly addresses produc-tion of multisource products from oth-er manufacturers. The EMEA recog-nized that generic manufacturers may not have complete access to the innova-tor company’s production information, making them unable to compare all aspects of the products. The guideline recognized that simple biochemical characteristics would be insufficient to establish equivalency, but do not clearly define manufacturing practices and sub-mission requirements for such products.In the United States, one potential but not universally supported approach for dealing with biogenerics is approval under section 505(b)(2). After Sandoz submitted a 505(b)(2) application for Omnitrope®(somatropin [rDNA origin]for injection) to the FDA, citizens’ peti-tions were submitted to the FDA on behalf of Pfizer, the patent holder for Genotropin,®a somatropin product. Ac-cording to the petition, Pfizer requested the FDA deny Sandoz’s application on the grounds that the company deemed it “scientifically and legally improper for the FDA to rely on, reference or other-wise use the clinical and manufacturing information that establishes the safety and effectiveness of Genotropin®… to approve Omnitrop.®” The petition states that the “data submitted to support ap-proval of Omnitrope ‘do not adequately address … the specific product differ-ences between Genotropin®and Omni-trop.®’” Pfizer’s petition was based on Sandoz’s use of FDA’s finding of safety and effectiveness for Genotropin,®which is approved for the same indications as those sought by Sandoz, in conjunction with studies Sandoz performed.Despite Pfizer’s petition, the FDA approved Omnitrope®under section 505(b)(2) in May 2006, following simi-lar approvals by the European Com-mission and Australia. The FDA con-sidered Omnitrope®different from more complex or less well-understood biotech products, being as the FDA has ap-proved eight different recombinant human growth hormone product lines for various indications. Although San-doz was able to demonstrate the pro-teins in Genotropin®and Omnitrope®are highly similar, it did not need to prove they are the same. Sandoz was re-quired only to prove they were similar enough that utilizing FDA findings on safety and efficacy for Genotropin®is appropriate. Submission of an NDA un-der section 505(b)(2) does not intend to prove “sameness.” Being as Sandoz was not seeking an “A” therapeutic rating for Omnitrope,®it will be designated with a “BX” rating in the Orange Book, therefore, making Genotropin®and Omnitrope®not bioequivalent.HOW SHOULD PHARMACISTS DEAL WITH 505(B)(2) MEDICATIONS?Most pharmacists are aware, at least to some extent, of the approval process in-volving traditional NDAs and ANDAs. Pharmacists also must be aware of the 505(b)(2) process in order to make appropriate decisions pertaining to product selection.Section 505(b)(2) allows the FDA to determine what studies are necessary to support approval of a medication, re-ducing the unneeded studies. Therefore, manufacturers of these medications spend less time and money doing re-search and development. These savings are typically passed on to the consumer through lower pricing. For example, the average wholesale price (AWP) for 30 tablets of Paxil®30 mg (paroxetine hydrochloride) is $20 higher than an equivalent amount of Pexeva®(paroxe-tine mesylate), which was approved under section 505(b)(2). The AWP for Pexeva®is similar to that of generic paroxetine hydrochloride products.Although these medications often are less expensive, the pharmacist must remember that these medications are not bioequivalent to a reference-listed。