2_苯基二氢苯并呋喃衍生物合成
苯并呋喃合成
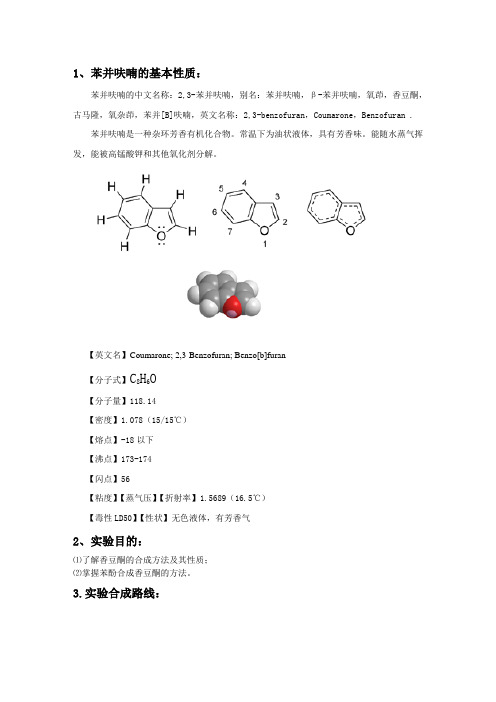
1、苯并呋喃的基本性质:苯并呋喃的中文名称:2,3-苯并呋喃,别名:苯并呋喃,β-苯并呋喃,氧茚,香豆酮,古马隆,氧杂茚,苯并[B]呋喃,英文名称:2,3-benzofuran,Coumarone,Benzofuran .苯并呋喃是一种杂环芳香有机化合物。
常温下为油状液体,具有芳香味。
能随水蒸气挥发,能被高锰酸钾和其他氧化剂分解。
【英文名】Coumarone; 2,3-Benzofuran; Benzo[b]furan【分子式】C8H6O【分子量】118.14【密度】1.078(15/15℃)【熔点】-18以下【沸点】173-174【闪点】56【粘度】【蒸气压】【折射率】1.5689(16.5℃)【毒性LD50】【性状】无色液体,有芳香气2、实验目的:⑴了解香豆酮的合成方法及其性质;⑵掌握苯酚合成香豆酮的方法。
3.实验合成路线:4.实验内容:1、实验试剂的基本性质(1)苯酚:为无色针状结晶或白色结晶熔块,可燃,腐蚀力强,有毒。
不纯品在光和空气作用下变为淡红或红色,遇碱变色更快。
与大约8水混合可液化。
可吸收空气中水分并液化。
有特殊臭味和燃烧味,极稀的溶液具有甜味。
相对密度1.0576,凝固点41℃,熔点43℃,沸点181.7℃(182℃),折射率1.54178,闪点79.44℃(闭杯),85℃(开杯),自燃点715℃,蒸气密度3.24,蒸气压0.13kPa(40.1℃),蒸气与空气混合物燃烧极限1.7-8.6。
1g苯酚溶于约15ml水(0.67,25℃加热后可以任何比例溶解)、12ml苯。
易溶于乙醇、乙醚、氯仿、甘油、二硫化碳、凡士林、挥发油、固定油、强碱水溶液。
几乎不溶于石油醚。
水溶液pH 值约为6.0。
(2)氯仿:无色透明、高折射率、易挥发的液体,有特殊香甜气味。
凝固点-63.5℃,沸点61.3℃,熔点-63.2℃,相对密度1.4984(15/4℃),1.4840(20/20℃),折光率1.4476,折射率1.4422,黏度(20℃)0.563mPa·s。
黄柏酮反应原理

黄柏酮反应原理一、引言黄柏酮是一种天然化合物,具有多种生物活性,如抗氧化、抗炎、抗菌等。
因此,黄柏酮及其衍生物被广泛应用于医药、保健品等领域。
本文将介绍黄柏酮的反应原理。
二、黄柏酮的结构黄柏酮(Baicalein)的分子式为C15H10O5,分子量为270.24g/mol。
它是一种黄色晶体,具有两个苯环和一个联苯环,并带有两个羟基和一个卡宾基(C=O)。
三、黄柏酮的反应类型1.氧化反应:在存在氧气或氧化剂的条件下,黄柏酮会发生氧化反应。
例如,在硫酸铜溶液中加热可得到2,3,4-三羟基苯甲醛。
2.还原反应:在还原剂的作用下,黄柏酮可以发生还原反应。
例如,在硫代乙醇钠(NaSCH3)和水的混合溶液中加热可得到2,3,4-三羟基苯甲醇。
3.烷基化反应:在强酸催化剂的作用下,黄柏酮可以发生烷基化反应。
例如,在硫酸催化下,黄柏酮和甲醇反应可得到3-羟基-4-甲氧基苯甲醛。
4.芳香性互变异构反应:在存在碱性条件下,黄柏酮可以发生芳香性互变异构反应。
例如,在氢氧化钾溶液中加热可得到5,6-二羟基-7-甲氧基联苯。
四、黄柏酮的光学活性黄柏酮是一种手性分子,具有光学活性。
它的两个立体异构体分别为(R)-Baicalein和(S)-Baicalein。
其中(R)-Baicalein的旋光度为+30°,而(S)-Baicalein的旋光度为-30°。
五、黄柏酮的合成方法1.从桦树皮中提取:桦树皮中含有丰富的黄柏素原,通过水提取、分离、纯化等步骤可制备出纯度较高的黄柏素。
2.化学合成:通过多步合成法可制备出黄柏酮。
例如,通过苯甲酸和苯乙烯的反应,得到2-苯基-2,3-二氢苯并[4,5]呋喃-4,7-二酮,再通过还原、羟基化、脱水等步骤得到黄柏酮。
六、黄柏酮的药理作用黄柏酮具有多种生物活性,如抗氧化、抗炎、抗菌等。
它可以通过多种途径发挥其药理作用。
例如:1.抗氧化:黄柏酮可以清除自由基,减轻氧化应激对细胞的损伤。
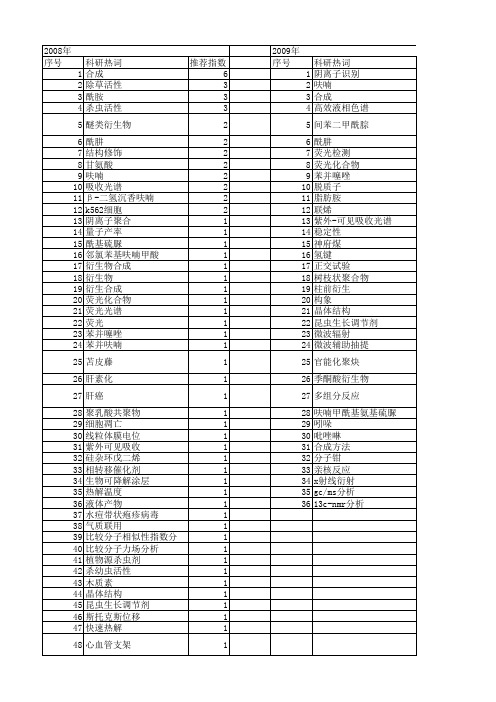
【国家自然科学基金】_呋喃衍生物_基金支持热词逐年推荐_【万方软件创新助手】_20140730
2012年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52
科研热词 推荐指数 合成 6 除草活性 3 酰胺 3 杀虫活性 3 醚类衍生物 2 酰肼 2 结构修饰 2 甘氨酸 2 呋喃 2 吸收光谱 2 β -二氢沉香呋喃 2 k562细胞 2 阴离子聚合 1 量子产率 1 酰基硫脲 1 邻氯苯基呋喃甲酸 1 衍生物合成 1 衍生物 1 衍生合成 1 荧光化合物 1 荧光光谱 1 荧光 1 苯并噻唑 1 苯并呋喃 1 苫皮藤 1 肝素化 1 肝癌 1 聚乳酸共聚物 1 细胞凋亡 1 线粒体膜电位 1 紫外可见吸收 1 硅杂环戊二烯 1 相转移催化剂 1 生物可降解涂层 1 热解温度 1 液体产物 1 水痘带状疱疹病毒 1 气质联用 1 比较分子相似性指数分析 1 比较分子力场分析 1 植物源杀虫剂 1 杀幼虫活性 1 木质素 1 晶体结构 1 昆虫生长调节剂 1 斯托克斯位移 1 快速热解 1 心血管支架 1 密度泛函理论 1 呋喃衍生物 1 呋喃[2,3-d]嘧啶核苷 1 吡唑啉 1
2008年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52
53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78
木脂素结构性质提取方法及其鉴别

● 氢核磁共振(1H-NMR)谱: 化学位移范围:在0~20 ppm 三大要素:化学位移(δH)、偶合常数(J)及峰面积。 灵敏度高,样品用量少(1-5 mg),测试时间短 ●碳核磁共振(13C-NMR)谱: 化学位移范围:在0~250 ppm 要素:化学位移(δC) 灵敏度较低,样品用量较多(5-20 mg),测试时间 长
O MeO
O O O
呋胡椒脂酮 futoenone
(十一)联苯类(biphenylenes)
OH
OH
厚朴酚 honokiol
(十二)倍半木脂素(sesquilignans)和二木脂素( dilignans)分别由3分子和4分子苯丙素聚合而成。
O O HO OMe
MeO O
OH
OMe
拉帕酚A lቤተ መጻሕፍቲ ባይዱppaol A
1、溶剂提取法
一般来说,两种基本母核相同的成分,其分子中功能基的 极性越大,或极性功能基数量越多,则整个分子的极性大 ,亲水性强,而亲脂性就越弱,其分子非极性部分越大, 或碳键越长,则极性小,亲脂性强,而亲水性就越弱。
1、溶剂提取法
各类溶剂的性质,同样也与其分子结构有关。 这样,我们就可以通过对天然产物成分结构分析,去估计 它们的此类性质和选用的溶剂。 总的说来,只要天然产物成分的亲水性和亲脂性与溶剂的 此项性质相当,就会在其中有较大的溶解度,即所谓“相 似相溶”的规律。这是选择适当溶剂自天然产物中提取所 需要成分的依据之一。
三、提取分离
一般宜先查阅有关资料,搜索比较该种或该类成分的各种 提取方案,尤其是工业生产方法,在根据具体条件加以选 用。从天然产物中寻找未知有效成分或有效部位时,情况 比较复杂。只能根据预先确定的目标,在适当的活性测试 体系指导下,进行提取、分离并以相应的动物模型筛选、 临床验证、反复实践,才能达到目的。 天然产物的有效成分往往需要从复杂的均相或非均相体系 中提取出来,然后通过分离和去除杂质以达到提纯和精制 的目的。
香豆素酯设计合成2-苯并呋喃甲酸

本科毕业论文香豆素酯设计合成2-苯并呋喃甲酸COUMARIN DESIGN AND SYNTHESIS OF COUMARILIC ACID学院(部):化学工程学院专业班级:应化06-2学生姓名:刘志彬指导教师:刘维新2010 年 6 月 8 日香豆素酯设计合成2-苯并呋喃甲酸摘要香豆素酯溶解于氯仿或其他有机溶剂后与液溴的氯仿溶液加成生成3,4-二溴-3,4-二氢香豆素中间体。
在一定温度下中间体经过水解、亲核取代、消去、酸化、水蒸气蒸馏、重结晶等一系列相关步骤最终获得产物2-苯并呋喃甲酸。
关键词:香豆素酯,氯仿,3,4-二溴-3,4-二氢香豆素,水解,酸化,水蒸气蒸馏,重结晶COUMARIN DESIGN AND SYNTHESIS OFCOUMARILIC ACIDABSTRACTCoumarilic acid preparation of first coumarin dissolved in chloroform or other organic solvents generate and the reaction of liquid bromine get 3,4 - dibromocoumarin,undera certain temperature through hydrolysis, acidification, distillation, recrystallization ultimately the product of Coumarilic acidKEYWORDS:Coumarin ,Chloroform, 3,4 - Dibromocoumarin, Hydrolysis, Acidification ,Distillation, Recrystallization目录摘要 (1)ABSTRACT (1)目录 (2)1绪论 (3)1.1有机合成发展前景 (3)1.2香豆素的研究现状 (3)1.3香豆素结构 (4)1.3.1简单香豆素类: (5)1.3.2呋喃香豆素类(furocoumarins) (线型和角型): (5)1.3.3吡喃香豆素类(pyranocoumarins) (线型和角型) (6)1.3.4其他香豆素类 (7)1.4香豆素物化性质 (7)1.4.1 香豆素的性状 (7)1.4.2 香豆素的溶解性 (7)1.4.3 香豆素的碱水解反应(内酯性质) (8)1.4.4 香豆素的酸的反应 (8)1.5 2-苯并呋喃甲酸的研究现状 (9)1.5.1 2-苯并呋喃甲酸的结构 (9)1.5.2 2-苯并呋喃甲酸的衍生物的合成及应用价值 (9)1.6课题的意义 (10)2 实验部分 (10)2.1实验试剂和实验设备 (10)2.1.1实验试剂 (10)2.1.2实验设备 (10)2.2香豆素酯制备2-苯并呋喃甲酸的反应设计过程 (11)2.3 实验的反应流程 (11)2.4 香豆素酯合成2-苯并呋喃甲酸的实验步骤 (14)2.4.1制备3,4-二溴-3,4-二氢香豆素 (14)2.4.2 由3,4-二溴-3,4-二氢香豆素制备2-苯并呋喃甲酸 (14)2.5产品的红外图谱表征 (15)3实验结果与讨论 (16)3.1实验讨论 (16)4. 结论 (17)参考文献 (18)谢辞 (20)1绪论1.1有机合成发展前景有机合成就是利用化学的方法,将有机物与单质、无机物合成出结构复杂的有机物的过程。
nickl苯并呋喃合成反应

nickl苯并呋喃合成反应
尼克尔-苯并呋喃合成反应是一种重要的有机合成方法,用于合成苯并呋喃类化合物。
该反应以芳香醛和乙炔为原料,在存在金属催化剂的条件下进行。
反应机理如下:
1. 氧化加成:芳香醛和乙炔首先在氧化剂(如铜盐)的存在下发生氧化加成反应,生成α,β-不饱和醛。
2. 减脱羧:α,β-不饱和醛经过减脱羧反应,失去一个CO2分子,生成羰基上的负离子中间体。
3. 芳香亲核取代:金属催化剂(如钯、铜等)促使负离子中间体与芳香化合物进行芳香亲核取代反应,生成苯环与呋喃环连接的产物。
尼克尔-苯并呋喃合成反应具有高效、选择性好、官能团容忍度广等优点,可以合成多样化的苯并呋喃化合物。
这些化合物在药物领域、材料科学等领域具有重要的应用价值。
类胡萝卜素和花青素研究方案

植物呈现出绚丽多姿的色彩,有的是为了光合同化作用,有的则是作为吸引信号或者植物防御信号,从而有利于它们的生存、授粉或种子传播,以便将自己的基因长久的遗传下去。
这些色彩是由植物体内卟啉类、类胡萝卜素类、类黄酮类和甜菜素类四种物质而引起的。
其中类胡萝卜素使高等植物呈现出黄色、橙红色或红色。
类黄酮可分为黄酮、黄酮醇和花青素3类,前两者大多呈浅黄色,花青素则会根据PH 值的不同,使高等植物呈现出红色到蓝色。
1.类胡萝卜素类胡萝卜素是一类呈黄色、橙红色或红色的多烯类物质, 一般由8个类异戊二烯单位组成,其具有抗氧化、防癌症、预防夜盲症等功能。
自然界中发现的类胡萝卜素种类繁多, 大约有700多种。
近10年来,每年大约有3000多篇类胡萝卜素相关的文献产出。
(1)类胡萝卜素在高等植物中的分布类胡萝卜素作为光合色素的辅助色素,广泛存在于高等植物中,主要是以光合色素-蛋白质复合体的形式存在于高等植物的叶绿体中,几乎所有有叶绿素的地方就有类胡萝卜素。
如许多黄色或橙色的高等植物花瓣和果实的颜色均源于存在于其组织细胞之中的类胡萝卜素化合物,如番茄、柑橘、辣椒、胡萝卜、玉米等。
(2)类胡萝卜素的分析检测方法类胡萝卜素的分离、定性、定量分析是从事类胡萝卜素研究工作的基本方法。
类胡萝卜素的共轭双键对热、光、氧和酸都是比较敏感的,所以在分析操作过程中要尽量减少氧的破坏、较大可能的避免光和热引起的变化。
分析试剂不要含过氧化物、酸和游离氧。
色谱质谱技术和光谱技术是类胡萝卜素研究工作中重要的分析方法,在定性、定量分析和分离、制备方面起着重要的作用。
(3)类胡萝卜素的功能类胡萝卜素在叶绿体光合作用中扮演着重要角色,一方面可帮助叶绿素接收光能,另一方面高温、强光下可通过叶黄素循环,耗散多余能量,此外类胡萝卜素还是ABA的前体。
高等植物的叶、花、果及根因富含类胡萝卜素,而呈现出黄色、橙红色或红色,因此胡萝卜素是决定园艺植物观赏价值的重要指标。
苯并呋喃与苯并二氧六环类新木脂素及其衍生物的合成与生物活性研究

苯并呋喃与苯并二氧六环类新木脂素及其衍生物的合成与生物活性研究汪秋安;徐雨;余玲敏;刘双艳【摘要】以3,4-二羟基苯丙烯酸(咖啡酸)为原料,经酯化和仿生氧化偶联反应得到苯并呋喃类化合物2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-羟基-2,3-二氢苯并呋喃(1)和苯并二氧六环类化合物2-(3′,4′-二羟基苯基)-3-甲氧羰基-6-甲氧羰基乙烯基-2,3-二氢-1,4-苯并二氧六环(2),然后经乙酰化、DDQ氧化脱氢、Pd/C 催化氢化、氢化铝锂还原、碱性条件下脱乙酰基等反应,合成了一系列苯并呋喃新木脂素类化合物3~7和苯并二氧六环新木脂素类化合物8~10.所合成化合物的结构已由核磁共振法(1 H NMR,13 C NMR)、质谱法(MS)进行了表征.其中5~7,9和10是未见文献报道的新化合物,8为天然产物异美商陆醇A.采用MTT法对所合成的苯并呋喃新木脂素类化合物1,3~5进行了生物活性测试.结果表明:化合物1,3,4和5对白血病细胞(HL-60)、肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、结肠癌细胞(SW-480)、肝癌细胞(SMMC-7721)有良好的体外生长抑制活性.%Benzofurans compound 2-(3',4'-dihydroxyphenyl)-3-methoxy carbonyl-5-methoxy carbonyl vinyl-7-hydroxy-2,3-dihydrobenzofuran (1)and benzodioxanes compound 2-(3',4'-dihydroxyphenyl)-3-me-thoxy carbonyl-6-methoxy carbonyl vinyl-2,3-dihydro-1,4-benzodioxane (2)were synthesized from caffeic acid through esterification and biomimetic oxidative coupling reactions.Moreover,a series of benzofuran-neolignan compounds 3~7 and benzodioxaneneolignan compounds 8~10 were synthesized from compounds 1 and 2 respectively throughacetylation,DDQ oxydehydrogenation,Pd/C catalytic hydrogenation,lithiumaluminium hydride reduction and deacetylation in alkaline condition.All of these synthesized compounds were confirmed with MS,IR,1 H NMR and 13 C NMR spectra.Among them,5~7,9 and 10 are new com-pounds.8 is the natural product isoamericanol A.The biological activities of benzofuranneolignan com-pounds 1 ,3~5 against five human cancer cell lines were evaluated in the standard MTT method,and the results have shown that compounds1,3,4 and 5 exhibit good inhibitory effect on leukemia cells (HL-60), lung carcinoma cell (A-549),breast cancer cell (MCF-7),colon cancer cell (SW-480),and hepatoma car-cinoma cell (SMMC-7 7 21 ).【期刊名称】《湖南大学学报(自然科学版)》【年(卷),期】2014(000)007【总页数】7页(P90-96)【关键词】合成(化学的);苯并呋喃类;苯并二氧六环类;新木脂素;生物活性【作者】汪秋安;徐雨;余玲敏;刘双艳【作者单位】湖南大学化学化工学院,湖南长沙 410082;湖南大学化学化工学院,湖南长沙 410082;湖南大学化学化工学院,湖南长沙 410082;湖南大学化学化工学院,湖南长沙 410082【正文语种】中文【中图分类】O626.11苯并呋喃和苯并二氢呋喃新木脂素类是存在于丹参、百部、龙血巴豆、西洋参、野花椒、水飞蓟、牛蒡子等药用植物中的天然有机化合物,它们具有良好的生物活性,如抗病毒、抗肿瘤、抗菌、抗氧化、免疫抑制剂、抗血小板聚集活性和神经营养作用等[1-5].例如:从南美洲大戟科龙血巴豆树茎中分离出来的苯并呋喃新木脂素3′,4-di-O-methylcedrusin具有良好的抗肿瘤活性[6].从羊角草中分离得到的苯并二氧六环新木脂素cleomiscosinde A也具有显著的抗肿瘤活性[7].从美洲商陆Phytolacca americana L种子中分离得到的苯并二氧六环新木脂素isoamericanol A,具有营养神经的活性,可提高胎鼠大脑半球胆碱乙酰转移酶的活性,改善神经条的形态[8].为了研究这类化合物的生理活性和构效关系,以及新药开发的需要,我们探索了简便高效的合成苯并呋喃类和苯并二氧六环类新木脂素的方法,并进一步研究这些化合物的生理活性.以3,4-二羟基苯丙烯酸(咖啡酸)为原料,以仿生氧化偶联和DDQ 脱氢反应为关键步骤,合成了一系列苯并呋喃新木脂素类化合物1,3~7和苯并二氧六环新木脂素类化合物2,8~10.其中5~7,9和10是未见文献报道的新化合物,8为天然产物美洲商陆醇A合成路线如图1所示.Reagent and conditions:(a) MeOH,concentrated sulfuric acid,reflux; (b)Ag2O,anhydrous toluene,anhydrous acetone,r.t,dark; (c) Ac2O,pyridine,r.t; (d) DDQ,1,4-dioxane,reflux,48h; (e) 10% Pd-C,H2,THF,r.t;(f)LiAlH4,anhydrous THF,-20 oC→r.t.图1 苯并呋喃和苯并二氧六环新木脂素类化合物的合成路线Fig.1 Synthesis route of benzofuran and benzoxioxane neolignans compounds1 实验部分1.1 仪器与试剂核磁共振仪:Bruker-AV400,400 MHz(各种氘代溶剂,TMS为内标);质谱(ESI)用VG Autospec-3000,SHIMADZ qp-500;红外光谱用Bruker Tensor-27(KBr压片法);熔点用XRC-1型显微熔点仪测定(温度未校正).所用试剂如无特殊说明均为市售化学纯或者分析纯;柱层析用硅胶300~400目(青岛海洋化工厂产品).3,4-二羟基苯基丙烯酸甲酯按文献[9]合成.1.2 2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-羟基-2,3-二氢苯并呋喃(1)和2-(3',4′-二羟基苯基)-3-甲氧羰基-7-甲氧羰基乙烯基-2,3-二氢-1,4-苯并二氧六环(2)的合成在100 mL的三颈圆底烧瓶中加入化合物3 2.5 g(12.88 mmol)和新制氧化银粉末1.99 g(8.59 mmol),再加入无水丙酮20 mL,无水甲苯40 mL.在N2保护下室温避光搅拌,TLC监测反应终点.约48 h后停止反应,过滤,用丙酮洗涤,减压旋除溶剂,得红褐色黏稠物.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)= 4∶1~3∶1],分别得2 0.9 g 和1 0.96 g,产率分别为36%和39%.化合物1:黄色黏稠物.1H NMR (400 MHz,CDCl3) δ(ppm):7.56 (1H,d,J=16.0 Hz,8-H),7.04 (1H,s,4-H),6.99 (1H,s,6-H),6.84 (1H,d,J=2.0 Hz,2′-H),6.79(1H,d,J=8.0 Hz,5′-H),6.76 (1H,dd,J=8.0,2.0 Hz 6′-H),6.26 (1H,d,J=16.0 Hz,9-H),6.01 (1H,d,J=7.6 Hz,2-H),4.26 (1H,d,J=7.6 Hz,3-H),3.80 (3H,s,10-OCH3),3.78 (3H,s,11-OCH3); MS (ESI) m/z:387 [M+H]+.化合物2:黄色油状物.1H NMR (400 MHz,CDCl3) δ(ppm):7.58 (1H,d,J=16.0 Hz,8-H),7.14 (1H,s,5-H),7.09 (1H,s,6-H),6.98 (1H,s,7-H),6.96 (1H,s,2′-H),6.85 (1H,s,5′-H),6.77 (1H,s,6′-H),6.59 (1H,s,3′-OH),6.46 (1H,s,4′-OH),6.29(1H,d,J=16.0 Hz,9-H),5.08 (1H,d,J=6.4 Hz,2-H),4.67 (1H,d,J=6.4 Hz,3-H),3.79 (3H,s,10-OCH3),3.64 (3H,s,11-OCH3); MS(ESI) m/z:387 [M+H]+.1.3 2-(3′,4′-乙酰氧基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-乙酰氧基-2,3-二氢苯并呋喃(3)的合成在100 mL的单口烧瓶中加入化合物1 238 mg(0.617 mmol),加入无水吡啶10 mL将其溶解.室温搅拌10 min后,加入乙酸酐5 mL,TLC监测反应,直至原料点消失.约3 h后停止反应,将反应液倾入装有30 mL冰水的烧杯中搅拌20 min,出现白色浑浊,用乙酸乙酯(3×20 mL)萃取,合并有机相依次用5%稀盐酸、饱和食盐水洗涤,最后用无水硫酸镁粉末干燥.蒸除溶剂得黄色固状物,用甲醇重结晶后得白色膨松固体285 mg,产率90%.m.p.127-128 oC; 1H NMR (400MHz,CDCl3) δ(ppm):7.63 (1H,d,J=15.6 Hz ,8-H),7.44 (1H,s,4-H),7.30(1H,dd,J=8.4,2.0 Hz 6′-H),7.24 (1H,d,J=2.0 Hz,2′-H),7.21 (1H,d,J=8.4 Hz,5'-H),7.19 (1H,s,6-H),6.33 (1H,d,J=16.0 Hz,9-H),6.22 (1H,d,J=7.6 Hz,2-H),4.32 (1H,d,J=7.6 Hz,3-H),3.85 (3H,s,10-OCH3),3.79 (3H,s,11-OCH3),2.32 (3H,s,7-OCH3),2.28 (6H,s,3′-COCH3,4'-COCH3); MS (ESI) m/z:513 [M+H]+.1.4 2-(3',4'-乙酰氧基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-乙酰氧基苯并呋喃(4)的合成在100 mL三颈烧瓶内加入化合物3 500 mg(0.976 mmol),DDQ(2,3-二氯-5,6-二氰基-1,4-苯醌)2 g(9.76 mmol),在N2氛围下加入无水1,4-二氧六环30 mL,继续通入N2 5 min,换无水氯化钙干燥管.加热回流,TLC监测反应,直至原料点基本消失.48 h后停止反应,将反应液冷却,过滤,滤液中倒入溶有NaHSO3 3.05 g(29.33 mmol)的水溶液100 mL,CH2Cl2 200 mL,充分搅拌后分液.水层用二氯甲烷萃取(3×20 mL),有机相合并用饱和食盐水洗涤,无水Na2SO4干燥.硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=3∶1],得白色固体3 15 mg,产率63%.m.p.157-158 oC; 1H NMR (400 MHz,DMSO-d6)δ(ppm):8.20(1H,s,4-H),7.93 (1H,s,6-H),7.91 (1H,dd,J=2.4,8.0 Hz,6'-H),7.82 (1H,d,J=8.0 Hz,5′-H),7.81 (1H,s,2′-H),7.50 (1H,d,J=16.4 Hz,8-H),6.74(1H,d,J=16.4 Hz,9-H ),3.91 (3H,s,10-OCH3),3.75 (3H,s,11-OCH3),2.43(3H,s,7-COCH3),2.33 (6H,s,3′-COCH3,4′-COCH3).MS (ESI) m/z:511 [M+H]+.1.5 2-(3′,4′-乙酰氧基苯基)-3-甲氧羰基-5-甲氧羰基乙基-7-乙酰氧基苯并呋喃(5)的合成在100 mL的单口圆底烧瓶内加入化合物4 100 mg(0.196 mmol),5% Pd-C 40 mg,加入无水THF 15 mL.氢气氛围下室温磁力搅拌12 h,TLC监测反应至原料点很浅,过滤,减压旋干溶剂.湿法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=3∶1],得白色固体92 mg,产率92%.m.p.152-153 oC; 1H NMR (400 MHz,CDCl3) δ(ppm):7.82 (1H,s,4-H),7.68 (1H,s,6-H),7.23(1H,dd,J=2.4,8.0 Hz,6′-H),7.19 (1H,s,2′-H),6.91 (1H,s,5′-H),3.86 (3H,s,10-OCH3),3.61 (3H,s,11-OCH3),3.00 (2H,t,J=8.0 Hz,8-CH2-),2.62 (2H,t,J=8.4 Hz,9-CH2-),2.33 (3H,s,7-COCH3),2.25 (6H,s,3′-COCH3,4′-COCH3).MS(ESI) m/z:513 [M+H]+.1.6 2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃(6)和2- (3',4'-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃(7)的合成称取四氢铝锂47.5 mg(1.25 mmol)于100 mL单口瓶中,加入10 mL无水乙醚.于-20 oC,N2氛围下将溶有化合物5 80 mg(0.156 mmol)的12 mL无水THF 缓慢滴加至其中.在-20 oC下磁力搅拌反应,反应2 h后,改室温反应,TLC监测直至原料点消失.缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×12 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)/V(甲醇)=15∶15∶1],得无色油状物6 32 mg和无色油状物7 25 mg,经表征,无色油状物6为2-(3′,4′-二羟基苯基)-3-羟基甲基-5-羟基甲基乙基-7-羟基苯并呋喃,产率57%.1H NMR(400 MHz,DMSO-d6)δ(ppm):9.07 (1H,s,7-OH),8.96 (1H,s,3′-OH),8.89 (1H,s,4′-OH),6.73 (1H,s,2′-H),6.67 (1H,d,J=8.0 Hz,5′-H),6.61 (1H,dd,J=2.0,8.0 Hz ,6′-H),6.52 (1H,s,6-H),6.47 (1H,s,4-H),5.00 (1H,s,10-OH),4.45 (1H,s,11-OH),3.30 (2H,t,J=8.0 Hz,10-CH2-),3.16 (2H,s,11-CH2-),2.44 (2H,t,J=8.0 Hz,8-CH2-),1.62-1.66 (2H,m,9-CH2-); MS(ESI) m/z:331 [M+H]+.无色油状物7为2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃,产率49%.1H NMR(400 MHz,DMSO-d6) δ(ppm):10.14 (1H,s,7-OH),9.62 (1H,s,3′-OH),9.33 (1H,s,4′-OH),7.44 (1H,s,2′-H),7.36 (1H,dd,J=2.0,8.0 Hz ,6′-H),7.17 (1H,s,6-H),6.87 (1H,d,J=8.4 Hz,5′-H),6.64 (1H,s,4-H), 4.50 (1H,s,10-OH),3.85 (3H,s,11-OCH3),3.43 (2H,t,J=7.6 Hz,10-CH2-),2.62 (2H,t,J=8.0 Hz,8-CH2-),1.70-1.74 (2H,m,9-CH2-); MS(ESI) m/z:359 [M+H]+.1.7 2-(3′,4′-二羟基苯基)-3-羟基甲基-7-羟基甲基乙烯基-2,3-二氢-1,4-苯并二氧六环(8)的合成在50 mL的单口圆底瓶中加入四氢铝锂12 mg(0.31 mmol),无水四氢呋喃10 mL 于-20 oC,N2氛围下将溶有化合物2 40 mg(0.11 mmol)的10 mL无水四氢呋喃缓慢滴加至其中.在-20 oC下磁力搅拌反应1 h,撤出低温,室温过夜反应,TLC监测至原料点消失,反应液呈灰绿色.缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×15 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.过滤旋干溶剂,干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=1∶1],得白色固体21 mg,产率59%.m.p.147-149 oC [文献值[10]:147-150 ℃]; 1H NMR (400 MHz,DMSO-d6) δ(ppm):8.90 (3H,s,3′-OH,4′-OH,11-OH),7.73(1H,d,J=15.8 Hz,8-H),6.92 (2H,s,5-H,6-H),6.71 (2H,s,5′-H,6′-H),6.68 (1H,s,2′-H),6.61 (1H,s,7-H),6.47 (1H,d,J=16.0 Hz,9-H),5.41 (1H,d,J=5.6 Hz,2-H),5.04 (1H,s,10-OH),4.77-4.79 (1H,m,3-H),4.08 (2H,d,J=4.4 Hz,11-CH2-),4.02(2H,d,J=6.4 Hz,10-CH2-).MS(ESI) m/z:331 [M+H]+.1.8 2-(3′,4′-二羟基苯基)-3-甲氧羰基-7-甲氧羰基乙基-2,3-二氢-1,4-苯并二氧六环(9)的合成在100 mL的圆底烧瓶中加入化合物2 530 mg(1.37 mmol),5% Pd/C 200 mg,然后加入无水THF 20 mL.氢气氛围下室温磁力搅拌12 h,TLC监测反应至原料点消失,过滤,减压蒸除溶剂.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=4∶1],得无色油状物489 mg,产率92%.1H NMR (400 MHz,CDCl3) δ(ppm):6.89 (1H,s,5-H),6.85 (1H,s,6-H),6.81 (1H,s,7-H),6.75 (1H,s,2′-H),6.70 (1H,s,3′-H),6.68 (1H,s,4′-H),5.10-5.80 (2H,s,3′-OH,4′-OH),5.00 (1H,d,J=6.8 Hz,2-H),4.58 (1H,d,J=6.0 Hz,3-H),3.66 (3H,s,10-OCH3),3.61 (3H,s,11-OCH3),2.84 (2H,t,J=7.6 Hz,8-CH2-),2.59 (2H,t,J=8.0 Hz,9-CH2-).MS(ESI)m/z:389 [M+H]+.1.9 2-(3′,4′-二羟基苯基)-3-羟基甲基-7-羟基丙基-2,3-二氢-1,4-苯并二氧六环(10)的合成在100 mL的单口瓶中加入四氢铝锂147 mg(3.86 mmol),加入无水乙醚10 mL 于-20 oC,N2氛围下将溶有化合物9 300 mg(0.77 mmol)的12 mL无水THF缓慢滴加至其中.在-20 oC下磁力搅拌反应,反应4 h后,TLC监测原料点消失.将反应撤至室温,缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×15 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=4∶1],得无色油状物132 mg,产率51%.1HNMR(400 MHz,DMSO-d6) δ(ppm):9.05 (1H,s,3′-OH),9.01 (1H,s,4′-OH),6.80 (1H,s,2′-H),6.78 (1H,d,7-H),6.73 (1H,s,5-H),6.70 (1H,d,6-H),6.67 (1H,d,5′-H),6.65 (1H,d,6′-H),4.90 (1H,s,11-OH),4.79 (1H,d,J=7.6 Hz,2-H),4.34(1H,s,10-OH),3.96 (1H,d,J=7.2 Hz,3-H),3.51 (2H,d,J=13.6 Hz,11-CH2-),3.37-3.40 (2H,m,10-CH2-),2.34 (2H,t,J=8.0 Hz,8-CH2-),1.62-1.69 (2H,m,9-CH2-);MS(ESI) m/z:333 [M+H]+.1.10 生物活性测试实验方法:1)接种细胞:用含10%胎牛血清的培养液(DMEM或者RMPI1640)配成单个细胞悬液,以每孔5 000~10 000个细胞接种到96孔板,每孔体积100μL,贴壁细胞需提前12 h接种培养.2)加入待测化合物溶液(固定浓度40 μM初筛,于该浓度对肿瘤细胞生长抑制在50%附近的化合物设5个浓度进入梯度复筛),每孔终体积200 μL,每种处理均设3个复孔.3)显色:37 oC培养48 h后,每孔加MTT溶液20 μL.继续孵育4 h,终止培养,吸弃孔内培养上清液,每孔加200 μL的SDS溶液(10%),过夜孵育(温度37 oC),使结晶物充分融解.4)比色:选择595 nm的波长,酶联免疫检测仪(Bio-Rad 680)读取各孔光吸收值,记录测定结果,以浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线,应用两点法(Reed and Muench法)计算化合物的IC50值.2 结果与讨论3,4-羟基苯基丙烯酸甲酯在氧化银催化下,以无水甲苯和无水丙酮作为溶剂,得到苯并二氢呋喃环结构化合物1和苯并二氧六环类化合物2.该步反应与天然苯并二氢呋喃和苯并二氧六环类的生物合成途径类似[11],属自由基仿生氧化偶联反应,其反应机理如图2所示.Ag2O催化仿生氧化偶联法同时合成了两种新木脂素化合物,1和2的产量接近1∶1.该反应条件温和,后处理简单,以1/1.5倍当量新制氧化银粉末作催化剂最宜.经多次实验发现,采用未重蒸的甲苯和丙酮溶剂对产率并无太大影响,因此简化了实验条件.此外还发现,反应时间约40 h可达到较好收率,反应时间延长对产率提高不大且有可能增加其它副产物的生成.化合物1用乙酸酐来保护酚羟基和DDQ脱氢反应,得到苯并呋喃类化合物4,化合物4的成功合成实现了苯并二氢呋喃向苯并呋喃环结构的转变,这为苯并呋喃新木脂素化合物的合成提供了一种有效的方法.在对酚羟基进行乙酰化保护的薄层色谱监测过程中,用稀盐酸先将反应液进行酸化,再点板,目的是消除溶剂吡啶在点板观察时的影响,此步反应可提高下一步氧化时的产率.实验发现,3.5倍当量的DDQ(2,3-二氯-5,6-二氰基-1,4-苯醌)可将原料全部脱氢氧化,后处理时以往采用的是硅胶柱过滤再经硅胶柱层析分离,虽然能达到尽可能除去DDQ的效果,但此过程需要使用大量的CH2Cl2且操作麻烦.因此,先用V丙酮/V甲醇=2∶1进行重结晶,再经硅胶柱层析分离得到纯品.化合物4在经催化氢化得5,5在氢化铝锂作用和无水无氧操作条件下,室温进行还原反应得到含有二个醇羟基的苯并呋喃新木脂素6,同时还生成了部分还原的产物7.在用氢化铝锂还原时,一定要采用重蒸THF作溶剂,将溶有原料的THF溶液在-20 oC的低温条件下缓慢滴加至氢化铝锂的THF溶液中,后处理用水猝灭反应时有大量氢气放出,所以加水过程一定要缓慢.化合物2在氢化铝锂作用和无水无氧操作条件下,室温进行还原反应则得到生物活性苯并二氧六环类天然产物isoamericanol A 化合物2经催化氢化和氢化铝锂还原分别得到未见文献报道的苯并二氧六环类化合物9和10.图2 苯并二氢呋喃和苯并二氧六环新木脂素类化合物的合成机理Fig.2 Synthetic mechanism of benzodihydrofuran and benzoxioxane neolignans compounds对所合成的苯并呋喃新木脂素类化合物1,3~5使用MTT法进行生物活性的测试.半数生长抑制浓度IC50值表明,化合物1,3,4对白血病细胞(HL-60)、肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、结肠癌细胞(SW-480)、肝癌细胞(SMMC-7721)有明显的体外肿瘤生长抑制活性;化合物5对白血病细胞(HL-60)、肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、肝癌细胞(SMMC-7721)有明显的体外肿瘤生长抑制活性,其中部分抗肿瘤细胞活性优于对照药物顺铂(MW300)(如表1).从1,3,4,5化合物的生物活性变化来看,羟基乙酰化的结构修饰明显提高了化合物对结肠癌细胞(SW-480)的抑制作用;由苯并二氢呋喃变为苯并呋喃的结构变化提高了化合物对白血病细胞(HL-60)和结肠癌细胞(SW-480)的生长抑制活性;8位、9位的乙烯基还原为乙基的结构变化使得化合物5对肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、结肠癌细胞(SW-480)和肝癌细胞(SMMC-7721)的生长抑制活性降低,但其对白血病细胞(HL-60)的抑制活性优于化合物3.表1 化合物对不同肿瘤细胞株的半数生长抑制浓度IC50Tab.1 IC50 values of synthetic benzofuran neolignans compounds on five hunman cancer cell lines μM样品编号不同肿瘤细胞株IC50值(μM)白血病细胞HL-60肺癌细胞A-549乳腺癌细胞MCF-7结肠癌细胞SW-480肝癌细胞SMMC-772110.070.950.1912.520.9532.380.970.221.601.1940.141.363.631.061.6650 .9313.5526.75>4023.09紫杉醇<0.008<0.008<0.008<0.008<0.008顺铂(MW300)1.6914.0920.8218.8512.49参考文献[1]CHAILIN KAO,JIWAN CHEM.A novel strategy for the synthesis of benzofuran skeleton neolignans,:application to ailanthoidol,XH-14 and obovaten [J].J Org Chem,2002,67:6772-6787.[2]蒲文臣,王飞,王淳.2-芳基苯并[b]呋喃衍生物的生物活性与合成策略[J].有机化学,2011,31:155-165.PU Wen-chen,WANG Fei,WANG Chun,Bioactivities and synthetic methodsof 2-arylbenzo[b]furans[J].Chin J Org Chem,2011,31:155-165.(In Chinese) [3]RAKOTONDRAMANANA D L,DELOMENEDE M,BALTAS M,et al.Synthesis of ferulic ester dimers,functionalisation and biological evaluation aspotential antiatherogenic and antiplasmodial agents[J].Bioorg Med Chem,2007,15(18):6018-6026.[4]FAN Hua-fang,REN Ying-mei,WU Xiu-ling,et al.Synthesis and cytotoxicity of novel benzofuran neolignan derivatives[J].J Chem Res,2010,34(4):233-235.[5]WU Zheng,LIANG Zhi-ying,LI We,et al.Synthesis of (+)-Demethylnitidanin,Herpetol and Salvinal as well as their glycosyl derivatives[J].Chem Res Chinese Univertsities,2011,27(6):949-954.[6]PIETERS L,VAN DYCK S,GAO M,et al.Synthesis and biological evaluation of dihydrobenzofuran lignans and related compounds as potential antitumor agents that inhibit tubulin polymerization[J].Journal of Medical Chemistry,1999,42(26):5475-5481.[7]HITOSHI T,ICHIRO K.Total synthesis of cleomiscosin A,a courmari-neolignoid[J].Chem Pharm Bull,1985,33:3218-3223.[8]FUKUYAMA Y,HASEGAWA T,TODA M,et al.Structure of americanin A and isoamericanol A having a neuotrophic property from the sead of phytolacca americana[J].Chem Pharm Bull,1992,40(1):252-254.[9]WANG E C,WEIN Y S,KUO Y H.A concise and effcient synthesis of salvinal from isoeugenol via a phenoxenium ion intermediate[J].Tetrahedron Lett,2006,47(52):9195-9197.[10]余竟光,李彤梅,孙兰,等.鹰爪种子化学成分的研究[J].药学学报,2001,36(4),281-286.YU Jing-guang,LI Tong-mei,SUN Lan,et al.Studies on the chemical constituents of the seeds from artabostrys hexapeta (Annonaceae)[J].ActaPharm Sin,2001,36(4):281-286.(In Chinese)[11]KUO Y H,WU C H.Synthesis of 5-(3-Hydroxypropyl)-7-methoxy- 2-(3-methoxy-4-hydroxyphenyl)-3-benzo[b]furancarbaldehyde,a novel adenosine A1 receptor ligand from the root of Salvia miltiorrhiza[J].J Nat Prod,1996,59(6):625-628.。
呋喃环结构

呋喃环结构呋喃结构式如下图:呋喃(oxole)是最简单的含氧五元杂环化合物,分子式为C4H4O,它存在于松木焦油中,为无色液体,具有类似氯仿的气味,难溶于水,易溶于有机溶剂。
结构由于其分子中氧原子的一对孤对电子在共轭轨道平面内形成大π键,使得共轭平面内共6个电子,符合4n+2结构,所以呋喃具有芳香性。
芳香性使得呋喃具有“易取代难加成”的性质。
氧的另外一对孤对电子向外伸展。
氧原子本身符合sp2杂化。
呋喃含有一个氧杂原子的五元杂环化合物,分子式C4H4O。
呋喃存在于少数的精油和松木焦油内。
有些糖类以分子内半缩醛的形式形成取代的呋喃环系,称为呋喃型糖。
呋喃的最重要衍生物糠醛是一种农副产品。
苯并呋喃和二苯并呋喃存在于某些天然产物内。
呋喃熔点-85.6°C,沸点31.4°C;微溶于水,溶于有机溶剂。
它与浸有盐酸的松木片反应,呈现绿色,这是鉴定呋喃的一种方法,叫做松片反应。
呋喃从结构上讲,是一个环状的烯醚,可以发生加成反应,例如,可与顺丁烯二酸酐反应,发生双烯合成。
也可以水解成1,4-二羰基化合物,即上述合成反应的逆反应。
它比苯活泼得多,硝化或卤化时,除得取代(见取代反应)产物外,还可得加成产物。
呋喃环上有其他吸电子基团时,活性降低,反应的进行较易控制。
例如,2-呋喃羧酸在5位氯化后再脱羧,即得2-氯代呋喃。
呋喃可由糠醛大量生产。
呋喃的同系物一般可以由1,4-二羰基化合物失水制取,例如:四氢呋喃THF的沸点为67°C,相对密度为0.8892(20/4°C);是重要的非质子极性溶剂,可与水以任何比例混溶;与乙醚类似,也容易形成易爆炸的过氧化物。
有些卤代烃在乙醚内不能生成格利雅试剂,而在THF中可以顺利地进行反应,这可能是由于THF是个环醚,分子中的氧更容易与金属镁络合(见配位化合物)。
THF也容易水解开环,利用这一反应可以制取1,4-二羟基化合物、二卤化合物等。
苯并呋喃,又称香豆酮,可用作工业原料。
二苯基吡唑类化合物及其制备方法和用途

二苯基丙烯酮就像化学界的酷孩子,用一个吸引药物,农用化学和材
料科学的滑翔机来搅拌它们的东西。
有了它们的真菌皮拉佐勒环系和两个苯基组挂在了所有正确的地方,这些pound就像化学的变色龙,能够转换它们的特性用于任何手头的任务。
这就像他们有一个衣柜
充满选择权从,使他们最终多功能玩家在化学游戏。
所以不管他们
是为了制药厂还是为了在农用化工领域下水和下水,这些二苯并呋喃
总是可以摇晃的!
有几种不同的方法来制造这些二苯并二恶英的东西。
一种方法是将羟基物质与乙酮或苯甲醛混合,然后将它们一起形成 p环。
另一种方法是利用金属来帮助将酰胺与烯烃混合,使二苯并二甲苯形成形状。
这
些方法让我们在组合中加入不同的东西,以获得我们想要的结果。
二苯并二甲苯的应用涉及广泛的行业,并具有相当大的前景。
在制药
部门,这些pound类表现出了一系列生物活动,包括抗癌、抗炎和抗病毒性质。
在农用化学品领域,二苯并二恶英的衍生物显示出强大的
杀虫和杀真菌活动,使它们成为开发新型作物保护剂的宝贵竞争者。
二苯并二甲苯特有的电子和光学特性在材料科学领域引起了人们的注意,有可能应用于有机电子、光伏和发光装置。
集体而言,二苯并二
甲苯的灵活性和潜力使它们成为积极研究和开发各种应用的协调中心。
苯并噻唑合成工艺

苯并噻唑合成工艺一、引言苯并噻唑是一种重要的有机化合物,具有广泛的应用价值。
它可以作为药物、染料和光敏材料等方面的原料。
因此,苯并噻唑的制备方法备受关注。
本文将介绍一种苯并噻唑合成工艺。
二、实验材料与仪器1. 2-氨基苯甲酸2. 硫酸3. 氢氧化钠4. 乙醇5. 乙酸乙酯6. 氨水7. 氢氧化钾8. 溶剂回收装置三、实验步骤1. 合成2-苯基-1,3-噻唑啉-4-酮中间体(1)将2-氨基苯甲酸加入硫酸中,搅拌至完全溶解。
(2)冷却至0℃以下,缓慢滴加浓氢氧化钠溶液,同时控制反应温度在0℃以下。
(3)继续搅拌30分钟后,在室温下静置过夜。
(4)过滤得到沉淀,用乙醇洗涤并干燥,得到2-苯基-1,3-噻唑啉-4-酮中间体。
2. 合成苯并噻唑(1)将2-苯基-1,3-噻唑啉-4-酮中间体加入乙酸乙酯中,加入氨水调节pH值至7.5左右。
(2)加入氢氧化钾固体,并在室温下搅拌反应24小时。
(3)反应结束后,用水洗涤产物,过滤得到固体。
(4)用乙醇重结晶并干燥,得到苯并噻唑产物。
四、实验结果分析经过实验验证,本工艺合成的苯并噻唑产率高达85%以上。
同时,该工艺操作简单、步骤少、易于扩大生产规模。
因此,在工业生产上具有广泛的应用前景。
五、安全注意事项1. 硫酸具有强腐蚀性,操作时需戴手套、护目镜等防护装置。
2. 氢氧化钠为强碱性物质,操作时需注意避免皮肤接触和吸入。
3. 氢氧化钾为强碱性物质,操作时需注意避免皮肤接触和吸入。
4. 实验室应配备溶剂回收装置,避免有机溶剂挥发对环境造成污染。
六、结论本文介绍了一种简单易行的苯并噻唑合成工艺,经过实验验证,该工艺产率高、操作简单、易于扩大生产规模。
因此,在工业生产上具有广泛的应用前景。
在实验过程中应注意安全事项,确保人身安全和环境保护。
苯并咪唑衍生物的合成_晶体结构及量子化学计算(精)

2004年第24卷第3期,300~305有机化学Chinese J ournal of Organic ChemistryVol.24,2004No.3,300~305研究论文苯并咪唑衍生物的合成、晶体结构及量子化学计算卑凤利陈海群杨绪杰陆路德汪信(南京理工大学化工学院材料化学实验室南京210094摘要合成了两种苯并咪唑衍生物:2 苄基-苯并咪唑(1和1 苄基 2 苯基-苯并咪唑(2.用元素分析、红外光谱和核磁共振氢谱对产物进行了表征,并且测定了它们的单晶结构.化合物1属正交晶系,空间群Pbca,a=0 93783(6nm,b=0 97155(6nm,c=2 49187(16nm,V=2 2705(2nm3,D c=1 218g/cm3,Z=8,F(000=880, =0 073m m-1,R=0 0816,wR=0 1698,GOF=0 948.化合物2属单斜晶系,空间群P21/c,a=060272(12nm,b=1 7059(3nm,c=1 5257(3nm, =91 45(3 ,V=1 5682(5nm3,D c=1 204g/cm3,Z=4,F(000=600, =0 071mm-1,R=0 0730,wR=0 1215,GOF=1 191.采用量子化学方法,在B3LYP/6 31G*水平上计算了标题化合物1和2的优化构型、电荷分布和热力学函数.关键词苯并咪唑,晶体结构,量子化学计算Synthesis,Crystal S tructure and Quantum Chemical Calculation ofBenzimidazole DerivativesB EI,Feng LiC HEN,Hai Qun YANG,Xu Jie LU,Lu De W ANG,Xin(Materials Chemistry Laboratory,N an jin g University o f Science and Technology,Nan j ing210094Abstract Two benzimidazole derivatives,2 benzyl 1H benzimidazole(1and1 benzyl 2 phenyl 1H benzimidazole(2,were synthesized and characterized by elemental analysis,IR and1H NMR spectra.Their crystal structures weredetermined by X ray single crystal diffraction.The compoud1belongs to orthorhombic system with space group Pbca, a=0 93783(6nm,b=0 97155(6nm,c=2 49187(16nm,V=2 2705(2nm3,D c=1 218g/cm3,Z= 8,F(000=880, =0 073mm-1,R=0 0816,wR=0 1698,GOF=0 948.The compoud2belongs to monoclinic system with space group P21/c,a=0 60272(12nm,b=1 7059(3nm,c=1 5257(3nm, =91 45(3 ,V=1 5682(5nm3,D c=1 204g/cm3,Z=4,F(000=600, =0 071mm-1,R=0 0730,wR=0 1215,GOF=1 191.The DF T B3LYP methods with6 31G*basis set have been applied to optimization of the title compounds1and2.Their charges distribution and thermodynamic function were investigated.Keywords benzimidazole derivative,crystal structure,quantum chemical calculation苯并咪唑衍生物及其金属配合物一直都是研究热点[1~6],之所以引起人们极大的研究兴趣,主要是由于这类化合物具有良好的生物活性[7,8].苯并咪唑是一个有趣的杂环化合物,许多药物是苯并咪唑衍生物或其配合物[9~12].其中最重要的两类是抗寄生虫药和质子泵抑制剂.1961年人们发现了噻并达唑具有抗肠道线虫作用,由此导致苯并咪唑类驱虫药的发展[13].苯并咪唑衍生物作为配体与过渡金属离子形成的配合物,可用于生物分子的模拟,例如维生素B12 (Vita mine B12就是苯并咪唑核苷酸与可啉环系(Corrin ring的内络盐[18].苯并咪唑衍生物还可用作铜铝等金属防腐剂、光敏剂、无银照相、表面活性剂、荧光增白剂及光敏染料[14~17]等.苯并咪唑可用作抗辐射剂[18],效果达90%.聚苯并咪唑[19]由于高热稳定性、化学稳定性、低温稳定性及粘结性好,又具不燃性及抗吸潮性,已广泛用于太空服装等空间E mail:wxin@;Tel:025 *******;Fax:025 *******.Received August12,2003;revised Oc tober13,2003;accepted November14,2003.国家自然科学基金(No.50372028资助项目.用材料.本文报道了两种苯并咪唑衍生物:2 苄基-苯并咪唑(1和1 苄基 2 苯基-苯并咪唑(2的晶体结构和量子化学计算结果.1 实验1.1 仪器和试剂Perkin El mer240C元素分析仪;Bruke r VECTOR 22型红外光谱仪(KBr压片;Bruke r DRX 300MHz核磁共振仪,D MF 为溶剂;Mo K ( =0 071073nmSieme ns CCD SMAR T1000型X射线衍射仪.所用试剂均为市售分析纯.1.2 标题化合物的合成1.2.1 2 苄基-苯并咪唑的合成室温(30 下,将5mmol邻苯二胺粉末(白色与5 mmol苯乙酸(无色晶体溶于乙二醇溶液中,加热回流6h,反应体系颜色为红色,停止反应并用冰水冷却,得到红色粗产物.用少量冷无水乙醇洗涤,室温干燥,重结晶得到标题化合物2 苄基-苯并咪唑.产率65%,1H N MR(DMF,300 MHz :4 40(s,C H2,7 67~8 23(m,9H,Ar H;IR(KBr :3622,3458,3162,3028,2927,2987,2854,1612,1567,1511,1444,1383,1348,1220,1021,907,876,835, 756,715c m-1.Anal.calcd forC14H12N2:C80 73,H5 81,N 13 46;found C80 56,H6 26,N13 78.1.2.2 1 苄基 2 苯基-苯并咪唑的合成室温(30 下,将5mmol邻苯二胺粉末(白色与5 mmol苯甲酸(无色晶体溶于乙二醇溶液中,加热回流6h,反应体系颜色为黄色,停止反应并用冰水冷却,得到黄色中间粗产物2 苯基-苯并咪唑,产率82%.将中间产物2 苯基-苯并咪唑与适量的苄氯和K2CO3,溶于甲醇/水溶液中,加热回流3h,得到棕色沉淀,过滤、室温干燥,重结晶得到标题化合物1 苄基 2 苯基-苯并咪唑,产率30%.1H NMR(D MF, 300MHz :485(s,CH2,7 13~8 67(m,14H,Ar H;IR (KBr :3107,3048,2915,2948,2847,1609,1509,1438, 1387,1351,1212,1019,910,878,837,757,713c m-1. Anal.calcd for C20H16N2:C84 47,H5 68,N9 86;found C 84 21,H5 80,N9 96.1.3 晶体结构测定将适量产物1和2分别溶于乙醇溶液中,在室温下缓慢挥发,约三周后得到红色1和黑色2单晶.选取大小适合的化合物1的单晶(0 38mm 0 16mm 0 12mm和化合物2的单晶(0 42mm 0 18mm 0 12mm分别置于Sie mens CCD SMAR T1000型X射线衍射仪上,用石墨单色化的Mo K ( =0 071073nm射线进行数据收集.其中化合物1在1 63 28 36 范围内,以 /2 扫描方式,于293(2K下共收集到14953个衍射点,独立衍射点2802个(R i nt=0 1635,其中1171个可观测点[I>2 (I]用于晶体结构解析,最终差值残余电子密度的最高峰为563e/nm3,最低峰为-487e/ nm3.其晶体属正交晶系,空间群Pbca,a=0 93783(6nm,b=0 97155(6nm,c=2 49187(16nm,V=2 2705(2nm3, D c=1 218g/c m3,Z=8,F(000=880, =0 073mm-1, R=0 0816,wR=0 1698,GOF=0 948;化合物2在1 7927 46 范围内,以 /2 扫描方式,于293(2K下共收集到4242个衍射点,独立衍射点2547个(R i nt=0 0334,其中2056个可观测点[I>2 (I]用于晶体结构解析,最终差值残余电子密度的最高峰为126e/nm3,最低峰为-135e/nm3,其晶体属单斜晶系,空间群P21/c,a=0 60272(12nm, b=1 7059(3nm,c=1 5257(3nm, =91 45(3 ,V= 1 5682(5nm3,D c=1 204g/c m3,Z=4,F(000=600, = 0 071mm-1,R=0 0730,wR=0 1215,GO F=1 191.全部强度数据经经验吸收校正.晶体结构采用S HELEXS97软件[20]由直接法解出,全部氢原子由理论加氢得到.全部非氢原子的坐标和各向异性热参数经全矩阵最小二乘法修正收敛.1.4 计算方法以Hype rc he m程序搭建模型,以AM1方法[21]优化构型为初始值,借助Gaussia n98[22]程序Be rny能量梯度法[23]对标题物的分子几何实施DF T B3LYP/6 31G*水平的全优化计算.收敛精度取程序内定值.振动分析无虚频,证明所得优化构型对应势能面上极小点.基于统计热力学[24]方法,求得了200~1200K温度范围的标准热力学函数.所有计算在PI V 微机和Compaq ALP HA DS20E服务器上进行.2 结果与讨论2.1 晶体结构描述标题化合物1和2的晶体结构测定过程及相关数据列于表1;晶体结构实验键长、键角及理论计算键长、键角列于表2;图1和图2分别为化合物1的分子结构透视图和晶胞堆积图;图3和图4分别为化合物2的分子结构透视图和晶胞堆积图.化合物1和2的计算几何的原子标号与晶体结构相一致,见图1和图3.由表2可知,两个化合物的苯并咪唑环骨架上的各原子之间的键长均介于单、双键之间,因而在分子中电子具有高度的离域作用,且与已报道的苯并咪唑衍生物的键长基本相符[25].咪唑环上的内环键角基本上接近109 ,苯环上内环键角基本上接近120 ,与所报道的相关化合物的键角基本相符[26].由于化合物2的N(2原子上连接了苄基,导致其N(2 C(7键长(0 13876nm比相应的化合物1中的N(1 C(7键(0 1345nm略长一些.苯并咪唑环骨架上9个原子共面,构成的最小二乘面(A方程:化合物1为4 343x-1 720y+21 640z=2 921[最大偏离原子为N(2,偏差为0 0031(2nm];化合物2为2 5224x+152492y+ 2 6106z=1 0275[最大偏离原子为C(7,偏差为0 0005 nm].从标题化合物1的晶胞堆积图看,分子间氢键是以N(1 H N(2#[#所表示的等效位置为:1/2-x,1/2+ y,z]形式存在,N(1 H N(2#之间的平均距离为0 2871(3nm,N(1 H之间的距离为0 08593nm,H N(2#之间的距离为0 20237nm,键角N(1 H N(2为301No.3卑凤利等:苯并咪唑衍生物的合成、晶体结构及量子化学计算168 46 .标题化合物1在晶胞中沿b轴方向形成一维链状分子间氢键结构.另外,从表2可知,晶体结构实测键长比采用密度泛函理论,在B3LYP/6 31G*水平上计算所得键长略短一些,这是由于晶体中分子受晶体场作用所致;而计算键角则与实验键角基本相符.表1 标题化合物1和2的晶体数据测定和结构修正Table1 Crystal data and structure refinement for1and2Identification code12Empirical formula C14H12N2C20H16N2Formula weigh t208.26284.35T emperature/K293(2293(2Wavelength/nm0.0710730.071073Crystal system,space group Orthorhombic,Pbca Monoclinic,P21/cUni t cell dimensi ons a=0.93783(6nm a=0.60272(12n mb=0.97155(6nm b=1.7059(3nmc=2.49187(16nm c=1.5257(3n m=90 =90=90 =91.45(3=90 =90Volume/n m3 2.2705(2 1.5682(5Z,Calculated density/(Mg m-38, 1.2184, 1.204Absorpti on coefficient/mm-10.0730.071F(000880600Crystal size/mm30.38 0.16 0.120.42 0.18 0.12range for data collection/( 1.63~28.36 1.79~27.46Li miting indices-12 h 10-12 k 12-32 l 290 h 7-21 k 22-19 l 1914953/28024242/2547Reflections collected/unique[R(int=0.1635][R(int=0.0334]Absorpti on correction Empirical EmpiricalRefi nement method Full matrix least squares on F2Full matrix least squares on F2 Data/restraints/parameters2802/0/1462547/0/200Goodness of fi t on F20.948 1.191Final R indices[I>2 (I]R1=0.0816R1=0.0730wR2=0.1698wR2=0.1215Extinction coefficient0.062(60.0114(13Largest diff.peak and hole/(e nm-3563and-487126and-135302 有机化学Vol.24,2004表2 标题化合物的实验和计算键长(nm、键角(Table 2 Bond lengths (nmand angles ( of the title compounds by experiment and calculation12实验值计算值实验值计算值N(1 C(70.1345(30.1381N(1 C(70.1323(20.1366N(1C(10.1383(30.1384N(1 C(200.1392(20.1445N(2 C(70.1315(30.1312N(2 C(150.1385(20.1423N(2 C(60.1391(30.1390N(2C(70.13876(180.1439C(1 C(60.1384(30.1417N(2 C(80.1459(20.1497C(1C(20.1391(40.1396C(1 C(20.1389(30.1405C(2 C(30.1376(40.1393C(1C(60.1395(20.1421C(3 C(40.1379(50.1409C(2 C(30.1383(30.1411C(7 N(1 C(1107.0(2107.25C(7 N(1C(20105.35(12103.31C(7 N(2 C(6104.5(2105.09C(15 N(2 C(7106.25(12105.96N(1 C(7 C(8121.9(3121.80N(2 C(7 C(6125.33(14124.22C(7 C(8 C(9112.7(2113.71N(2 C(8C(9113.53(13114.15C(10 C(9 C(14119.0(4118.61C(12 C(13 C(14120.61(17120.12C(10 C(9 C(8120.1(3120.42C(13 C(14 C(9120.91(17120.31图1 标题化合物1的分子结构图Figure 1 Molecular structure of the title compound 1图2 标题化合物1的晶胞堆积图Figure 2 Crystal packing of the ti tle compound 1图3 标题化合物2的分子结构图F igure 3 Molecular structure of the title compound 2图4 标题化合物2的晶胞堆积图Figure 4 Crystal packing of the title compound 2303No.3卑凤利等:苯并咪唑衍生物的合成、晶体结构及量子化学计算2.2 TG/DTA在等速升温、氮气保护下,对标题化合物1和2进行了TG/D TA分析.TG表明,化合物1在200~300 范围内已分解完全;而2在250 才开始分解,直到800 分解仍未完全,这说明2比1的热稳定性好.化合物2在250~500 的温度区间内出现一个持续的热分解过程,失重74%;在500~ 700 区间内TG曲线上出现平台(无重量损失,但对应的DTA曲线上却出现了很强的放热峰,这可能是由于稠环化作用所致,稠环化是一个放热过程,但无重量损失;700 以后再次出现失重,直到800 仍未分解完全,这可能对应稠环的分解,但由于稠环较为稳定,所以800 时分解仍未完全.2.3 标题化合物分子的电荷分布及热力学函数计算运用Gaussian98程序,在B3LYP/6 31G*水平上计算了两个化合物分子中各原子的净电荷分布和热力学性质,结果见表3和表4.从表3可知,N(1和N(2原子的净电荷最大,在-0 55787~-0 72939e之间.另外,对比化合物1和2可知:1 取代作用使得1位N原子所带负电荷有大幅下降(由-0 72939降至-0 56109.整个体系其它原子电荷分布趋于均匀化,这主要是由于电子的离域作用使体系的自由能达到最小.表3 原子净电荷数(eTable3 Atomic net charges(e21N(1-0.5530N(1-0.7294N(2-0.5611N(2-0.5479C(150.3314C(10.3482C(200.2390C(60.2271C(60.0964C(70.5174C(8-0.3381C(8-0.4341表4 化合物1和2的热力学性质Table4 Thermodynamic properties of1and2at different temperatures aCompd.T/KC0p,m/(J mol-1 K-1S0m/(J mol-1 K-1H0m/(kJ mol-1298.2222.48481.5635.51 400297.4557.6662.07 1600407.3700.69133.37 800477.73828.21222.36 1000525.4940.25322.96 1200559.191039.19431.6298.2294.74578.2847.27 400394.38679.0982.46 2600544.14869.49177.39800640.81040.24296.56 1000706.411190.71431.67 1200753.351323.87577.89 在全优化构型和振动分析基础上,按统计热力学公式求得标题物在298~1200K 温度范围的标准恒压热容C0P,m,标准熵S0m和标准焓H0m,择少数列于表4.由表4可见,随温度升高各热力学函数值均增大,这是由于分子振动的贡献随温度升高而大幅度增加.通过拟合求得H0m与温度之间具有良好的线性关系,相关系数为1为0 996;2为0 987.References1Ewa,S.;Jerzy,S.Pol.J.Chem.1979,53,2339.2M alatesh, A.P.;Timmanagoud, D. B.Transition Met.Chem.1998,13,423.3M atthews, C.J.;William, C.;Heath,S.L.;Martin,N. C.;Strart Hill,M.N.Inorg.Chem.1998,37,199.4Gayathri,V. E.;Leelamani,G.N.;Gowda,M.N.;Reddy,G.K.N.Polyhedron1999,18,2351.5Rajnikanr, F.;David,J.W.A cta Cry sta llogr.1995,C51, 2388.6Neil,J. C.;Eberhard,.Chem.1997,42, 3485.7Bei, F.L.;Jian, F. F.;Yang,X.J.;Lu,L.D.;Wang, X.;Raj,S.S.S.;Fun,H.K.Acta Crystallogr.2000,C56, 718.8Ozbey,S.;Ide,S.;Kendi, E.J.Mol.Struct.1998,442,23.9David,S.S.;Abhijit,M.;David,M.P.Chem.Rev.1993, 93,2295.10Raban,M.;Chang,H.;Craine,L.;Hortelano, .Chem.1985,50,2005.11Cole, E.R.;Crank,G.;Sheikh, A.S.J.Chromatogr.1973,78,323.12Chaudhury,S.;Debroy, A.;Mahajan,M.P.Can.J.Chem.1982,60,1122.13Bai,X.Flotation Rea gent,Metallurgy Industry Publi shing House, Beiji ng,1981(in Chinese.(百熙,浮选药剂,冶金工业出版社,北京,1981.14Chauhan,P.M.S.;B hakuni, D.S.Indian J.Chem.1986, 25B,1146.15Matthews, C.J.;Clegg,W.;Elsegood,M.R.J.;Leese,T.A.;Thorp, D.;Thornton,P.;Lockhart,J. C.J.Chem.Soc.,Dalton Trans.1996,1531.16Albada,G.V.;Lakin,M.T.;Velaman,N.;Spek, A.L.;Reedijk,J.I norg.Chem.1995,34,4910.17Raban,M.;Chang,H.;Craine,L.;Hortelano, .Chem.1985,50,2205.18Han, C. L.;Song,X. P.T he Me dicine Produce Technique, Science and Technology Literature Publishi ng House,Beijing, 2000(in Chinese.(韩长日,宋小平,药物制造技术,科学技术文献出版社,北京,2000.304 有机化学Vol.24,2004No. 3 19 20 21 22 卑凤利等: 苯并咪唑衍生物的合成、晶体结构及量子化学计算 305 Ueda, M. ; Sato , M. ; Mochizuki, A . Macromolecules 1985, 18 , 2723. Sheldrick, G. M . SH ELXTL 97, Program for Crystal Structure Refinement, University of Gottingen, Germany, 1997. Dewar, M . J. S. ; Zoebisch, E. G. ; Healy, E. F. J . Am . Chem. Soc. 1985, 107, 3902. Frisch, J. ; Trucks, G. W. ; Schlegel, H. B. ; Scuseria, G. E. ; Robb, M. A. ; Cheeseman, J. R. ; Zakrzewski, V. G. ; Montgomery, J. A. , Jr . ; Stratmann, R. E. ; Burant, J. C. ; Dapprich, S. ; Millam, J. M. ; Daniels, A. D. ; Kudin, K. N. ; Strain, M . C. ; Farkas, O. ; Tomasi, J. ; Barone, V. ; Cossi, M. ; Cammi, R. ; M ennucci, B. ; Pomelli, C. ; Adamo, C. ; Clifford, S. ; Ochterski, J. ; Petersson, G. A . ; Ayala, P. Y. ; Cui, Q. ; Morokuma, K. ; Malick, D. K. ; Rabuck, A. D. ; Raghavachari, K. ; Foresman, J. B. ; Cioslowski, J. ; Ortiz, J. V. ; Baboul, A . G. ; Stefanov, B. B. ; Liu, G. ; Liashenko , A. ; Piskorz, P. ; Komaromi, I. ; Gomperts, R. ; Martin, R. L. ; Fox, D. J. ; Keith, T. ; Al Laham, M. A. ; Peng, C. Y. ; Nanayakkara, A. ; Gonzalez, C. ; Challacombe, M. ; Gill, P. M. W. ; Johnson, B. ; Chen, W. ; Wong , M . W. ; Andres, J. L. ; Gonzalez, C. ; Head Gordon, M . ; Replogle, E. S. ; Pople, 28 27 26 25 24 23 J. A . Gaussian 98, Revision A. 7, Gaussian, Inc. , Pittiburgh PA, 1998. Peng, C. ; Ayala, P. Y. ; Schlegel, H. B. ; Frisch, M. J. J . Comput . Chem. 1996, 17 , 49. Xiao, H. M . ; Li, Y. F. The Band and Electronic Structrue f or Metal Az ides , Science Press, Beijing, 1996 ( inChinese . ( 肖鹤鸣, 李永富, 金属叠氮化物的能带和电子结构, 科学出版社, 北京, 1996. Ozbey, S. ; Ide, S. ; Kendi, E. J . Mol . Struct. 1998, 442, 23. Rajnikant, W. D. J. ; Tranter, G. A cta Crystallogr . 1995, C51, 2388. Pople, J. A. ; Schlegel, H. B. ; Krishnan, R. ; Defrees, D. J. ; Binkley, J. S. ; Frisch, M. J. ; Whiteside, R. A. ; Hout, R. F. ; Hehre, W. J. Int. J . Quantum Chem. , Quantum Chem. Symp . 1981, 15 , 269. Xiao, J. J. ; Zhang, J. ; Yang, D. ; Xiao, H. M. Acta Chim. Sinica 2002, 60 , 2110 ( in Chinese . ( 肖继军,张骥, 杨栋, 肖鹤鸣, 化学学报, 2002, 60, 2110. ( Y0308123 QIN, X. Q. ; DONG, H. Z.。
苯并呋喃酮工艺技术概况及选择

苯并呋喃酮工艺技术概况及选择:
A工艺路线:以邻硝基甲苯为原料在乙醇钠催化下与草酸二乙酯缩合后,经水解、双氧水氧化、酸化后制得邻硝基苯乙酸;再经还原、重氮化、水解反应得到苯并呋喃酮。
原料:邻硝基甲苯、金属钠、草酸二乙酯、乙醇、氢氧化钠、30%双氧水、8%硫化铵溶液、亚硝酸钠、浓硫酸等九种。
反应原理:
B工艺路线:以邻氯苯乙腈为原料,经过皂化、水解、酯化、环化合成苯并呋喃酮。
原料:邻氯苯乙腈、氢氧化钠、催化剂A、盐酸、催化剂B、甲苯等六种。
反应原理:
选用B工艺路线,只有六种原材料,两步合成步聚,具有原料少,反应工艺步聚少的优点,也具有更加节能降排的优点。