药物临床试验立项申请表
医院与卫生院新药申请表及承诺书

医院临床科室新药申请
廉洁承诺书
为了严把新药准入关,坚决抵制医药购销中的不正之风,医院药
事管理委员会和临床科室工作人员将严格遵守廉洁自律承诺,承诺内容如下:
1、加强廉政建设,增强自身的法制观念和反腐能力,廉洁行医。
2、坚持原则,秉公办事,自觉抵制医药购销中的不正之风,做到:
不接受药品经销商送的礼金礼品、有价证券、购物券、回扣、佣金;
不私下接触、参与宴请和娱乐活动;不索要好处费、赞助费和宣传费:不代支付旅游费用、报销各种消费凭证。
3、拒绝药品经销商给予的礼金、有价证券等。
4、作为新药引进的申请人,我保证所申请的新药____ ___ 等个品种(见申请表)是我科室专业治疗必须的,并对所在科室所申请新药的合理使用进行监督管理,若发生所申请新药临床不合理
应用及其它违规、违纪行为,本人愿承担监督管理等相关责任。
5、本承诺书一式二份,将留存医院存档。
承诺人:
年月日
医院新药引进申请表
药事管理委员会意见:
参会委员:共()名
评审结果:共收回选票()份,通过()票,不通过()票。
评审决议:□通过□不通过
签名:
年月日主管院长意见:
签名:
年月日院长意见:
签名:
年月日。
药物临床试验申请报告

药物临床试验申请报告
药物临床试验机构:
专业申请参加:
本专业确定作为该试验项目的负责人,作为
该试验项目的质控员,为该试验项目的药品专管员。
本项目的负责人及质控员已阅读并熟悉该项目的相关资料,同意执行试验方案中所有细节,确保参加项目的其他研究人员在试验过程遵守临床试验方案。
本专业具备完成该临床试验的医疗条件,有足够的病例资源,可以在规定的时间内、在符合GCP等法律法规的前提下按临床试验方案要求完成该项工作。
专业负责人签字:
日期:。
药物临床试验申请审批表

药物临床试验申请审批表申办者声明我保证以上信息真实准确,并负责该临床试验全过程的质量保证,承诺该临床试验数据真实可靠,操作规范,符合NMPA 《药物临床试验质量管理规范》(GCP )要求。
如有失实,愿意承担相关责任。
申办者代表签名(盖章):年月日CRO 公司声明我保证以上信息真实准确,并负责该临床试验全过程的质量保证,承诺该临床试验数据真实可靠,操作规范,符合NMPA 《药物临床试验质量管理规范》(GCP )要求。
如有失实,愿意承担相关责任。
CRo 代表签名(盖章):年月日主要研究者声明我保证以上信息真实准确,并负责该临床试验全过程中的质量保证,承诺该临床试验数据真实可靠,操作规范,符合NMPA 《药物临床试验质量管理规范》(GCP )要求。
本人承诺本研究团队人员与该项目无利益冲突。
如有失实,愿意承担相关责任。
主要研究者签字:_________________________________ 年月日审批意见(手签)主要研究者对本试验的评估及意见:L 试验的入排标准是否合理:2 .病源病种是否能够满足方案要求:3 .研究人员是否有足够的试验时间:4 .科室的场地和设施是否能保障:5 .是否能对试验质量进行保证:6 .是否保证能在约定时间内完成试验:7 .科室在研项目情况:是否有竞争入组临床试验在研:在研药物临床试验数量:项,其中处于筛选期和治疗期的项 8 .主要研究者决定:同意承接口主要研究者(签字):年月日科室意见:同意承接口不同意承接口专业负责人(签字):机构办公室意见:同意承接口项目编号: 不同意承接口机构办主任(签字):机构意见]同意承接口不同意承接口机构主任(签字):注:(1)表格内的选择框内勾选均采用“国)”:(2)请使用A4纸双面打印。
□ □□□□□ □ 否否否否否否 否 □□□□□□ □ 是是是是是是 是。
临床试验Timeline模板
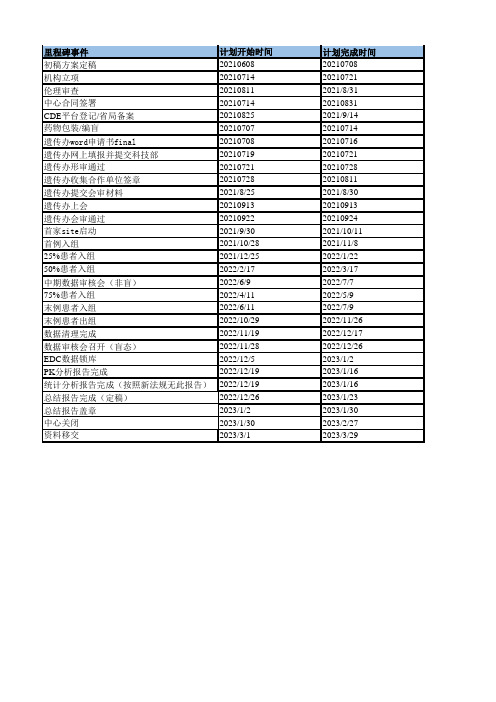
实际完成时间
备注 PI审核后定稿 立项约需1周时间 伦理上会到出批件约需2周时间 立项资料递交时,同步进行主协议签署流程
与申办方沟通为此时间段内完成
撰写,审阅,final
填写,QC,提交
预审查审批5个工作日,预计一次性通过
预计2周
主要受限于EC批件,最晚30号要拿到
预计第18批会议(09.13)
网上公示通过
计划开始时间 20210608 20210714 20210811 20210714 20210825 20210707 20210708 20210719 20210721 20210728 2021/8/25 20210913 20210922 2021/9/30 2021/10/28 2021/12/25 2022/2/17 2022/6/9 2022/4/11 2022/6/11 2022/10/29 2022/11/19 2022/11/28 2022/12/5 2022/12/19 2022/12/19 2022/12/26 2023/1/2 2023/1/30 2023/3/1
获得遗传办纸版批件
126
启动后立即筛选,筛选期最长28天
50
104
100例受试者获得治组时间为8个月
从入组到研究结束共18周(FPI-EOS)
预计三周
9天时间准备会议
数据审核会后数据清理预计一周后DBL
并且一致性核查完成之后,预计一个月
DBL之后即可开始准备
DBL之后即可开始准备
CSR一个月后完成 资料移交一个月(TMF纸版+电子版)
计划完成时间 20210708 20210721 2021/8/31 20210831 2021/9/14 20210714 20210716 20210721 20210728 20210811 2021/8/30 20210913 20210924 2021/10/11 2021/11/8 2022/1/22 2022/3/17 2022/7/7 2022/5/9 2022/7/9 2022/11/26 2022/12/17 2022/12/26 2023/1/2 2023/1/16 2023/1/16 2023/1/23 2023/1/30 2023/2/27 2023/3/29
药物临床试验伦理审查申请表

项目职责分工:1、项目负责人/主要研究者2、研究者3、质控员4、其他,请描述
药物临床试验伦理审查申请表
项目名称
SFDA批件号
□I期
□U期
□川期□"期
试验类别
□临床验证
□国际多中心
□研究者发起
□其他
项目立项类别
□新启动项目
□增加中心项目
主要研究者
电话
邮箱
拟承接科室
申办者
申办者联系人
电话
邮箱
CRO
CRO联系人
电话
邮箱
组长单位
组长单位项目 负责人
电话
邮箱
我已认真审阅本项目申报材料,认定各项材料符合
主要研究者简历
研究者 基本情况
姓名:
科室:
职称:
职务:
联系电话: 邮箱:
昭
八、、 片 电 子 版
工作经历
期间
单位/专业
职称或职务
技术专长
GCP培训情况
既往承担的临床 试验项目情况
研究者签名
年月日
专业组项目研究团队说明
项目名称
项目负责人
联系方式
项目组主要成员
姓名
职称
项目职责分工
负责/参与
在研项目数
联系方式
有□无口
是□否口
5
研究者履历、团队人员分工等
有□无口
是□否口
6
试验用药检测报告(试验药和对照药)
有□无口
是□否口
7
申办者单位资质证明(包括CRO资质证明)
有□无口
是□否口
8
研究病历和/或病例报告表
有□无口
是□否口
药物临床试验初始审查申请表
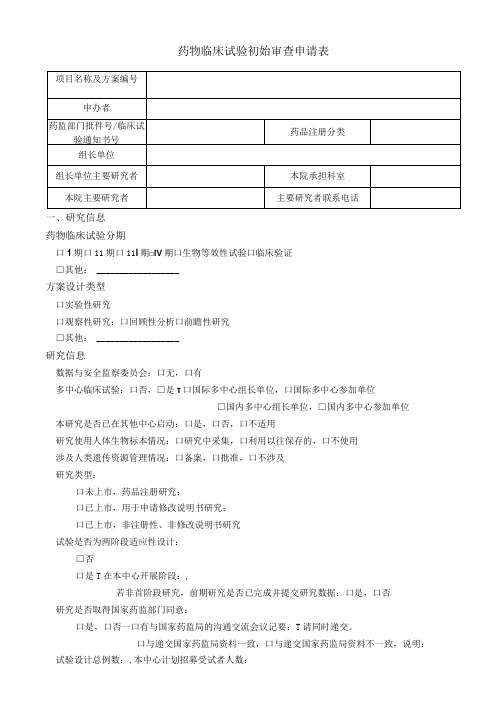
药物临床试验初始审查申请表一、研究信息药物临床试验分期口1期口11期口11I期□IV期口生物等效性试验口临床验证□其他: __________________方案设计类型口实验性研究口观察性研究:口回顾性分析口前瞻性研究□其他: __________________研究信息数据与安全监察委员会:口无,口有多中心临床试验:口否,□是τ口国际多中心组长单位,口国际多中心参加单位□国内多中心组长单位,□国内多中心参加单位本研究是否已在其他中心启动:口是,口否,口不适用研究使用人体生物标本情况:口研究中采集,口利用以往保存的,口不使用涉及人类遗传资源管理情况:口备案,口批准,口不涉及研究类型:口未上市,药品注册研究;口已上市,用于申请修改说明书研究;口已上市,非注册性、非修改说明书研究试验是否为两阶段适应性设计:□否口是T在本中心开展阶段:,若非首阶段研究,前期研究是否已完成并提交研究数据:口是,口否研究是否取得国家药监部门同意:口是,口否一口有与国家药监局的沟通交流会议记要:T请同时递交。
口与递交国家药监局资料一致,口与递交国家药监局资料不一致,说明:试验设计总例数:,本中心计划招募受试者人数:预期研究时间:年月至年月(伦理收费及批准研究时间以此为准)招募方式及人群招募者:口医生,口研究人员,口中介公司,口其他:招募方式:口发布招募广告,口临床诊疗过程,口数据库,□其他:研究是否涉及弱势人群或个体:□否口是T弱势的具体特征:________________________________________________________ 针对性的附加保护措施:__________________________________________________ 受试者补偿和支付计划口无补偿口有补偿:口货币补偿,补偿理由及数额:_____________________________________________ 口非货币补偿,说明:____________________________________________________ 支付计划(请详细描述发放周期及流程):受试者参与研究的费用谁支付研究干预和研究程序的费用:研究药物费用支付者:□申办者,口受试者,口其他:研究理化检查费用支付者:□申办者,口受试者,口其他:其他,说明:____________________________________________________________知情同意谁获取知情同意:口主要研究者,口研究者,口其他:获取知情同意地点:口私密房间/受试者接待室,口诊室,口病房,口其他:知情同意签字:口受试者签字,口监护人签字知情同意的例外口否,口是τ口申请变更知情同意,口申请豁免知情同意理由: _______________________________________________________________ 研究者其他研究工作本人在研的研究项目数:—项,其中与本项目目标疾病相同的项目数:一项。
浙江省药学会医院药学科研专项申请表

浙江省药学会
医院药学科研专项
申请表
申请编号:
项目名称:
科研专项名称:
申请单位:
(盖章)申请日期:
浙江省药学会
二O二三年制
填表说明
一、《申请书》各项内容,应实事求是地逐项认真填写。
表达要明确、严
谨,字迹要清晰。
外来语要同时用原文和中文表达。
第一次出现的缩写词须注出全称。
如无该项内容请填“无”,各栏空格不够,均可加页。
二、汉字请用国家公布的标准简化汉字,数字请用阿拉伯数字。
研究经费以万元为单位,用阿拉伯数字表示,注意小数点。
三、《申请书》一份,请采用A4规格打印及装订。
、项目情况
二、承担单位
三、项目负责人及项目组成员
四、主要研究内容和要达到的主要指标(限800字)
六、承诺书
本单位(或个人)承诺:
本申请书中所填写的内容和资料真实、有效,如存在弄虚作假和与事实相违背的内容,由本单位(个人)承担全部责任。
申报单位(盖章)
项目负责人签字:
年月日
七、单位意见
八、合作单位意见。
医院临床试验结题小结盖章申请表

关中心函已完成
是口否口不适用口
资料归档
提交资料是否齐全
是口否□不适用口
机构档案管理员签名签日期:
盖章(以上满足要求才能在小结表和总结报告上盖章并径记)
机构办主任签字盖章:
编号:RY3.0
临床试验结题(小结表盖章)申请表
信息
试验项目名称:
研究药物/医疗器械/体外诊断试剂名称
申办方/CRO:
专业:
主要研究者
临床试验批件号
本中心伦理委员会批件
号
研究时间
年月——年月
试脸计划入组受试者数
筛选人数
入组人数
完成试验人数
SAE发生
有口无口
发生SAE的药物编号/受试者编号
本中心药研经费编号(经费核查用)
统计报告中本中心有效病例数、无效病例数是否一致
是口否□不适用口
是否审签全部试脸资料并对所有文件进行自查
是口否口不适用口
1ห้องสมุดไป่ตู้
机构质控
试验过程中机构是否进行质量控制检查
是□否□不适用□
机构质量管理员签名签日期:
机构是否进行试验结束质控检查并合乎要求
是□否口不适用口
经费、关中心函
试脸经费是否全部支付
是口否口不适用口
试验过程中机构是否进行质量控制检查
是□否□不适用□
专业科室质控员签名签日期:
主要研究者
统计报告中本中心完成病例数是否一致(包括筛选、入组、脱落、剔除病例)
是□否□不适用口
主要研究者签名签日期:
统计报告中本中心不良事件的发生率是否一致
是□否□不适用口
统计报告中本中心不良事件的描述是否一致
是口否□不适用口
复方制剂立项申请书模板

尊敬的评审委员会:您好!我谨代表我们的团队向贵委员会提交本次复方制剂立项申请。
我们希望通过本次项目的研究,为我国医药事业做出贡献。
以下是我们的立项申请报告:一、项目背景与意义1. 背景随着医学科技的不断发展,药物治疗在疾病预防和治疗方面发挥着越来越重要的作用。
复方制剂作为一种新型的药物形式,将多种有效成分合理组合,以达到治疗疾病的目的。
在我国,复方制剂的研究和应用逐渐广泛,但在某些领域仍存在很大的发展空间。
2. 意义(1)提高药物治疗效果:复方制剂可以通过多种药物成分的相互作用,提高治疗效果,降低副作用。
(2)简化治疗方案:复方制剂将多种药物组合在一起,可以减少患者用药次数,提高治疗依从性。
(3)降低医疗成本:复方制剂的生产成本相对较低,可以减轻患者经济负担,提高药物可及性。
二、项目目标与研究内容1. 项目目标本次项目旨在研究一种新型复方制剂,用于治疗XX疾病,提高治疗效果,降低副作用,为患者提供更优质的药物治疗方案。
2. 研究内容(1)药物成分筛选:根据药物作用机制和临床需求,筛选出适用于复方制剂的药物成分。
(2)药物配比优化:通过实验研究,确定药物成分之间的最佳配比,以提高治疗效果和降低副作用。
(3)制剂工艺研究:探讨适用于该复方制剂的生产工艺,确保药物成分的稳定性和有效性。
(4)临床研究:进行临床试验,验证复方制剂的安全性和有效性。
三、研究计划与进度安排1. 研究阶段一(1-6个月):药物成分筛选与配比优化。
2. 研究阶段二(7-12个月):制剂工艺研究。
3. 研究阶段三(13-18个月):临床前研究。
4. 研究阶段四(19-24个月):临床试验。
四、项目预算与资金需求1. 项目总预算:人民币XX万元。
2. 资金需求说明:包括药物采购、实验材料、试验设备购置、人力资源等费用。
五、项目风险与应对措施1. 药物筛选失败的风险:通过多渠道收集药物信息,加强前期研究,降低药物筛选失败的风险。
2. 临床试验失败的风险:严格遵循临床试验规范,确保试验质量和数据可靠性,减少临床试验失败的风险。
临床试验立项申请表药物
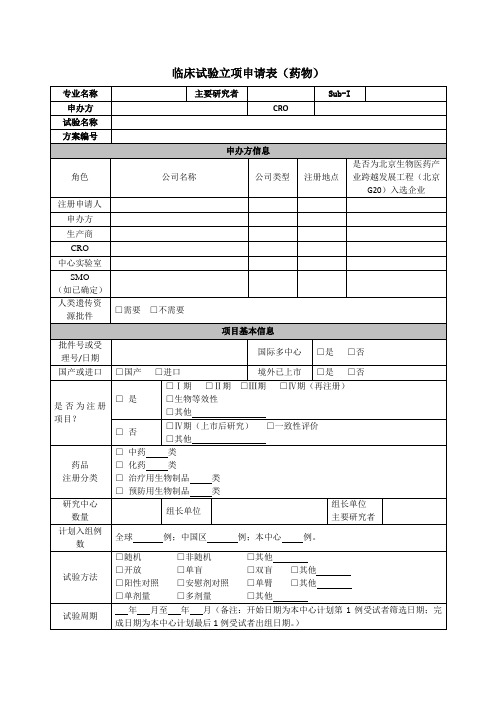
专业名称
主要研究者
Sub-I
申办方
CRO
试验名称
方案编号
申办方信息
角色
公司名称
公司类型
注册地点
是否为北京生物医药产业跨越发展工程(北京G20)入选企业
注册申请人
申办方
生产商
CRO
中心实验室
SMO
(如已确定)
人类遗传资源批件
□需要□不需要
项目基本信息
批件号或受理号/日期
国际多中心
CRO项目经理:姓名/联系电话/邮箱
SSU:姓名/联系电话/邮箱
CRA:姓名/联系电话/邮箱
CRC:姓名/联系电话/邮箱
PI:姓名/联系电话/邮箱
Sub-I:姓名/联系电话/邮箱
监查计划
次/月;天/次
申办方或CRO签字盖章/日期
主要研究者签字/日期
规格及包装规格
用法用量
储存条件
备注:药物类型填写基础用药/急救用药等。
发放周期及发放数量:
(举例:访视X:发放片或支(最小单位);访视X至访视X:发放片或支(最小单位),共次。)
是否涉及周六日或节假日发药:□是□否
是否涉及夜间发药:□是□否
联络信息及监查计划
联络信息
申办方项目经理:姓名/联系电话/邮箱
□男科中心
□药理所
□Ⅰ期临床试验研究室
受试者来源
□门诊患者
□住院患者
□住院患者+门诊随访
□健康人
试验用药物基本信息
试验药
药理学分类
目标适应症
□新靶点或新作用机制的创新药物
□罕见病品种
□优先审评审批的儿童用药品种
临床试验立项审查表
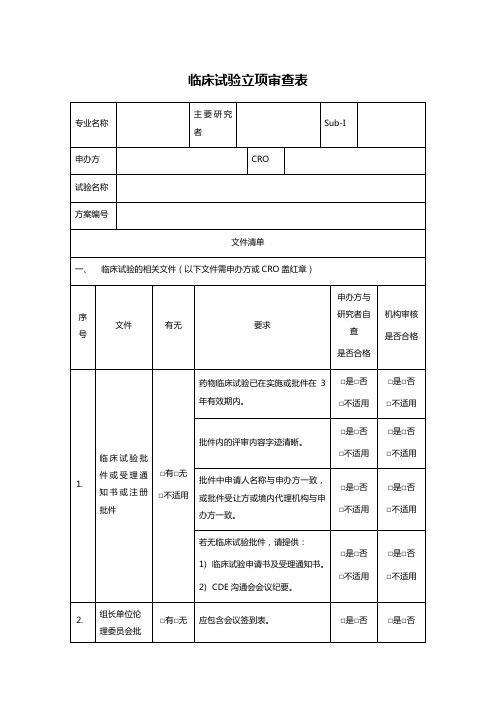
一、
简历中需体现曾经参与临床试验情况,及目前正在承担临床试验情况。
□是□否
□不适用
□是□否
□不适用
一、
简历中需体现GCP培训经历。
□是□否
□不适用
□是□否
□不适用
一、
GCP证书在有效期内(5年)。
□是□否
□不适用
□是□否
□是□否
□不适用
□是□否
□不适用
一、
需内容字迹清晰。
□是□否
□不适用
□是□否
□不适用
5.
临床试验方案
□有□无
注明版本号及版本日期。
□是□否
□不适用
□是□否
□不适用
一、
申办方签字盖章原件。
□是□否
□不适用
□是□否
□不适用
一、
主要研究者签字原件。
□是□否
□不适用
□是□否
□不适用
一、
若为参加单位,需提供组长单位主要研究者签字页复印件。
□是□否
□不适用
□是□否
□不适用
一、
在盲法试验中,试验用药品的编码系统须包括紧急揭盲程序,以便在紧急医学状态时能够迅速识别何种试验用药品,而不破坏临床试验的盲法设计。
□是□否
□不适用
□是□否
□不适用
6.
药物标签
□有□无
方案涉及的由申办方提供的试验用药品均需提供药物标签,包括研究药物、对照药品、安慰剂、基础用药、急救用药等。
□不适用
□是□否
□不适用
批件内的评审内容字迹清晰。
□是□否
□不适用
□是□否
□不适用
批件中申请人名称与申办方一致,或批件受让方或境内代理机构与申办方一致。
药物临床试验审查备案申请表

①依临床试验性质而定,试验需要则必备
②注明版本号或日期,如有英文版本需同时递送
7、研究者手册(注明版本号及日期,如有英文版本需同时递送)
8、研究原始记录(注明版本号及日期,如有英文版本需同时递送)
9、病例报告表(注明版本号及日期,如有英文版本需同时递送)
10、受试者信息卡(注明版本号及日期,如有英文版本需同时递送)
主要研究者签字:
日期:
机构办秘书签字:
日期:
注:上述资料中的某些文件不适用于本研究过程,可删除;以下各项顺序号依次提前。
18、中心伦理的批件复印件,或其他伦理委员会对申请研究项目重要决定的说明
19、试验用药品检验合格报告
20、产品说明书(阳性对照药和已上市产品需要)
21、保险合同
22、申办方相关资质证明:
①包括GMP证书及药品生产许可证等
②或/及申办方委托CRO的证明
23、研究项目经费来源说明
24、其他(如承诺书等)
药物临床试验审查(备案)申请表
项目名称
申请文件
1、CFDA药物临床试验申请受理通知书复印件;
2、关于“自受理缴费之日起60日内,未收到药审中心否定或质疑意见”的说明
3、临床试验方案摘要
4、试验方案(注明版木号及日期,申பைடு நூலகம்方及主要研究者签字或盖章,如有英文版本需同时递送)
5、知情同意书(注明版本号及日期,如有英文版本需同时递送)
11、受试者日记(注明版本号及日期,如有英文版本需同时递送)
12、临床试验小组成员名单(本院)
13、研究者履历(签名,签署日期)
14、研究者GCP证书复印件
15、主要研究者遵照GCP要求开展工作的声明(签名,签署日期)
药物临床试验立项申请表

保存
13
研究团队(含研究者资格证书、GCP证书)
保存
保存
14
其他文件
保存
保存
主要研究者签名(含时间)
机构办公室秘书(时间)
药物临床试验机构办公室意见及签名(含时间)
注意:
1.立项文件参考GCP附件2临床试验保存文件准备阶段目录;
2.“保存原件”指有鲜章或签名;
3.清单内容有修改ຫໍສະໝຸດ 请备注。保存保存3
组长单位伦理批件(含伦理委员会成员表)
保存
保存
4
申办者/CRO公司资质证明
保存
保存
5
CRA委托书和简历
保存原件
保存原件
6
研究者手册
保存
保存
7
试验方案及其修正案(已签名)
保存原件
保存原件
8
病例报告表
保存
保存
9
知情同意书
保存
保存
10
受试者招募材料
保存
保存
11
研究协议样稿(含财务规定)
保存
12
主要研究者简历
药物临床试验立项申请表
主要研究者/专业组
(科室)
项目名称
试验药品名称
申报方(含公司名称、所在地、联系人及电话)
合同研究组织(含公司名称、所在地、联系人及电话)
监查员(含姓名、公司名称、电话)
临床试验递交文件清单(含版本号及修改日期)
研究者文件夹
机构办公室
1
药物临床试验立项申请表
保存原件
2
国家食品药品监督管理局批件
医药项目立项申请报告

医药项目立项申请报告一、项目背景及意义随着人口老龄化和生活方式的改变,慢性疾病的发病率不断上升,医药市场潜力巨大。
本项目旨在开发一种新型药物,用于治疗其中一种慢性疾病,填补市场空白,提高患者的生活质量和生活预期。
该药物在前期的试验中已经显示出良好的疗效和安全性,具备市场推广的潜力。
二、项目目标1.开发一种新型药物,用于治疗其中一种慢性疾病。
2.在前期试验的基础上,进一步验证该药物的疗效和安全性。
3.尽快获得药物的上市许可,并投入市场推广。
三、项目内容及工作计划1.药物研发:通过临床试验等方式,进一步验证该药物的疗效和安全性。
2.生产设备采购:根据预计的产能需求,购买所需的生产设备。
3.临床试验申请:向相关部门提交临床试验申请,获取试验批准。
4.上市许可申请:根据临床试验结果,向药品监管部门申请上市许可。
5.市场推广:根据药物的特性和目标受众,制定市场推广策略,开展相关活动。
四、项目预期效益1.经济效益:该新药物在上市后,预计将获得可观的销售收入,为公司带来丰厚的利润。
2.社会效益:该药物的上市将提高患者的生活质量,减轻他们的病痛,同时也降低了社会的医疗负担。
五、项目投资及资金筹措1.项目总投资额预计为X万元,具体包括研发费用、生产设备采购费用、临床试验费用、市场推广费用等。
2.资金筹措方式主要包括自筹资金、银行贷款、投资者投资等。
六、项目风险及对策1.研发风险:存在开发周期延长、研发失败等风险。
应加强项目管理,提高研发效率,及时调整研发策略。
2.临床试验风险:可能出现招募困难、试验失败等风险。
应加大招募力度,确保试验的顺利进行。
3.上市许可风险:可能出现审批时间延长、不被批准等风险。
应积极配合审批部门的要求,及时提供相关材料,确保申请的顺利进行。
七、项目进度计划1.第一年:药物研发及生产设备采购。
2.第二年:临床试验及上市许可申请。
3.第三年:市场推广及销售启动。
八、可行性分析1.技术可行性:现有的前期试验结果已经证明了药物的疗效和安全性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
审核人(签名):机构办主任(签字):日期:年月日
机构意见:
机构主任(签字):日期:年月日
备注:请将申请表完成后交至机构办公室刘颖
药物临床试验立项申请表(注册类)
机构受理编号:填表日期:年月日
项目名称
方案名称/方案编号
注册分类
□药品类剂型:
项目类型
□Ⅰ期□Ⅱ期□Ⅲ期□Ⅳ期
其他:CFDA批件号:
研究范围
□国际
□国内
□本院
我院参研形式
□组长
□Байду номын сангаас立
□参加组长单位:
预计完成病例数
计划起止日期
—
科室是否有同类临床试验项目
□是□否
科室在研临床试验项目
项
申办者
联系人
联系电话
CRO
联系人
联系电话
监查员
联系电话
承担试验科室
主要研究者
联系电话
有关本研究简介:
日期:年月日
主要研究者声明:我已熟悉本试验方案及相关文件。我将根据《药物临床试验质量管理规范》等相关规定,认真履行研究者职责和遵从本试验方案的要求开展临床试验,并同意承担本试验。
主要研究者(签字):日期:年月日