全基因组关联分析共44页文档
全基因组关联分析

单倍体型分析
LCYE associations across seasons
Mixed Model Environment Avg, Observation No. 157 2003 154 Ratio Across Environments** 2002 44 2003 156 2004 154 2005 156 2003
0.5
Ear diameter (Low population structure)
a.
0.4 Simple Q 0.3 K Q+K 0.3
b.
0.4
Simple Q 0.4
c.
Q GC Q+K 0.3 Simple
Cumulative P
K
0.2 0.2 GC 0.1 GC 0.1 0.1 0.2 Q+K
6.02
HYD1 HYD2 IspFg ZDS
7.02
8.02
9.02
DXSe
10.02
6.03 IPP1 IPP2 6.04
7.03
8.03
9.03
10.03
7.04
8.04
பைடு நூலகம்9.04
10.04
DXSc 6.05
7.05
LYCe 8.05
9.05
10.05
δ- Carotene LCY-b α- Carotene HYD-e Lutein
0 0 (0) 0.2 (0.8) 0.4 (3.3) 0.6 (7.1) 0.8 (11.9) 1 (17.4)
0
Genetic effect (Phenotypic variation explained in %)
全基因组关联分析的原理和方法

全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中数以百万计的单核苷酸多态性(single nucleotide ploymorphism ,SNP)为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。
随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。
近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。
全基因组关联方法首先在人类医学领域的研究中得到了极大的重视和应用,尤其是其在复杂疾病研究领域中的应用,使许多重要的复杂疾病的研究取得了突破性进展,因而,全基因组关联分析研究方法的设计原理得到重视。
人类的疾病分为单基因疾病和复杂性疾病。
单基因疾病是指由于单个基因的突变导致的疾病,通过家系连锁分析的定位克隆方法,人们已发现了囊性纤维化、亨廷顿病等大量单基因疾病的致病基因,这些单基因的突变改变了相应的编码蛋白氨基酸序列或者产量,从而产生了符合孟德尔遗传方式的疾病表型。
复杂性疾病是指由于遗传和环境因素的共同作用引起的疾病。
目前已经鉴定出的与人类复杂性疾病相关联的SNP位点有439 个。
全基因组关联分析技术的重大革新及其应用,极大地推动了基因组医学的发展。
(2005年, Science 杂志首次报道了年龄相关性视网膜黄斑变性GWAS结果,在医学界和遗传学界引起了极大的轰动, 此后一系列GWAS陆续展开。
2006 年, 波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的GWAS结果(Herbert 等. 2006);2007 年, Saxena 等多个研究组联合报道了与2 型糖尿病( T2D ) 关联的多个位点, Samani 等则发表了冠心病GWAS结果( Samani 等. 2007); 2008 年, Barrett 等通过GWAS发现了30 个与克罗恩病( Crohns ' disrease) 相关的易感位点; 2009 年, W e is s 等通过GWAS发现了与具有高度遗传性的神经发育疾病——自闭症关联的染色体区域。
生物大数据技术的全基因组关联分析方法

生物大数据技术的全基因组关联分析方法近年来,随着生物大数据技术的快速发展,全基因组关联分析方法已成为生物学、医学研究领域中的重要工具。
全基因组关联分析(GWAS)是一种寻找基因与某一特定性状或疾病之间相互关联的分析方法。
本文将介绍全基因组关联分析的原理和方法,并探讨其在研究中的应用和挑战。
全基因组关联分析的基本原理是将多个个体的基因组数据与其具体的性状或疾病状态进行比较,寻找基因位点与性状或疾病之间的关联。
这种分析方法的关键在于基因型-表型关联的检测。
在全基因组关联分析中,研究对象通常是单核苷酸多态性(SNP)位点,因为SNP是个体基因组中最常见的变异类型。
全基因组关联分析方法通常包括以下几个步骤。
首先,收集研究对象的基因组数据和相关性状或疾病的表型数据。
其次,通过基因组测序技术或芯片技术对个体的基因组进行分析,得到其SNP位点的基因型数据。
然后,通过统计学方法计算基因型与表型之间的关联。
最后,对这些关联进行统计分析,判断是否存在显著的关联信号。
在全基因组关联分析中,常用的统计学方法包括卡方检验、线性回归分析和逻辑回归分析等。
卡方检验适用于疾病的风险和基因型之间的关联分析;线性回归和逻辑回归分析则适用于连续性和二分性表型特征的关联分析。
不同的统计方法适用于不同的研究问题和数据类型。
全基因组关联分析方法在生物学、医学研究中的应用广泛。
它可以揭示基因变异与疾病发生发展之间的关系,有助于发现潜在的疾病风险基因和药物靶标。
全基因组关联分析还可以帮助了解个体在药物代谢、药物反应和药物副作用方面的差异,实现个体化医疗的目标。
此外,全基因组关联分析还可以为遗传病的早期筛查和诊断提供重要依据。
然而,全基因组关联分析也存在一些挑战。
首先,全基因组关联分析需要大样本量来获得可靠的结果,并且需要考虑到样本的种族和人口结构,以避免虚假关联的出现。
其次,全基因组关联分析结果需要进行复制实验来验证其确切性。
此外,全基因组关联分析还需要解决对多个检验进行校正和纠正,以降低虚假关联的发生概率。
全基因组关联

LOGO
全基因组关联分析(GWAS)
实验设计方案
实验设计
1.研究背景 2.方法与原理 3.步骤 4.结果分析 5.讨论
Company Logo
研究背景:原发性肝癌是常见恶性肿瘤之一,在恶性肿
瘤中其占世界范围年发病率占第五位,死亡率占第三位。 每年新发病例约600000例,其中约78%的病例是亚洲人 ,约54%病例是中国人。乙型肝炎病毒慢性感染是致病最 重要的风险因子,但对于同样暴露人群,只有部分人发生 肝癌,以及具有家族聚集性,这表明遗传背景在乙型肝炎 病毒相关性肝癌发病中起重要作用。对肝癌相关基因复杂 性疾病的发生,发展的遗传学机制我们知之甚少。因此通 过GWAS寻找肝癌相关基因对于疾病的诊断和治疗具有重 要意义。 人类基因组计划(HGP)和人类单倍型图谱(Haplotype map )更强大了GWAS的研究。
肝右叶巨块型原发性肝癌
原发性肝癌适形放射治疗剂量分布
方法
本研究采用病例—对照的研究方法,GWAS目前 主要分单阶段和两阶段或多阶段研究设计方法。 1.单研究阶段即选择足够的病例和对照样本,一次 性在所有研究对象中对选中的SNP进行基因分型, 然后分析每个SNP与疾病的关联,分别计算关联强 度和OR(早期GWAS多使用该法) 2.多阶段研究即在单阶段研究用覆盖全基因组范围 的SNP进行病例—对照关联分析,统计分析后筛选 出较少数量的阳性SNP,然后采用更大样本的病 例—对照样本人群进行基因分型,然后结合两或多 阶段的结果进行分析。(目前多采用该方法)
Company Logo
该设计策略需要保证第一阶段筛选与疾病或ห้องสมุดไป่ตู้型 关联SNP的敏感性和特异性,尽量减少分析的假 阳性和假阴性的发生,并在第二阶段应用大样本 人群,甚至在多种族人群中进行基因分型验证。
全基因组关联分析的原理和方法

全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中数以百万计的单核苷酸多态性(single nucleotide ploymorphism,SNP)为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。
随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。
近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。
全基因组关联方法首先在人类医学领域的研究中得到了极大的重视和应用,尤其是其在复杂疾病研究领域中的应用,使许多重要的复杂疾病的研究取得了突破性进展,因而,全基因组关联分析研究方法的设计原理得到重视。
人类的疾病分为单基因疾病和复杂性疾病。
单基因疾病是指由于单个基因的突变导致的疾病,通过家系连锁分析的定位克隆方法,人们已发现了囊性纤维化、亨廷顿病等大量单基因疾病的致病基因,这些单基因的突变改变了相应的编码蛋白氨基酸序列或者产量,从而产生了符合孟德尔遗传方式的疾病表型。
复杂性疾病是指由于遗传和环境因素的共同作用引起的疾病。
目前已经鉴定出的与人类复杂性疾病相关联的SNP位点有439个。
全基因组关联分析技术的重大革新及其应用,极大地推动了基因组医学的发展。
(2005年, Science杂志首次报道了年龄相关性视网膜黄斑变性 GWAS结果,在医学界和遗传学界引起了极大的轰动,此后一系列GWAS陆续展开。
2006年, 波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的 GWAS结果 (Herbert等. 2006);2007年, Saxena等多个研究组联合报道了与 2型糖尿病( T2D )关联的多个位点, Samani等则发表了冠心病 GWAS结果( Samani 等. 2007); 2008年, Barrett等通过 GWAS发现了 30个与克罗恩病( Crohns ' disrease)相关的易感位点; 2009年, W e is s等通过 GWAS发现了与具有高度遗传性的神经发育疾病——自闭症关联的染色体区域。
全基因组关联分析-基于全基因组重测序
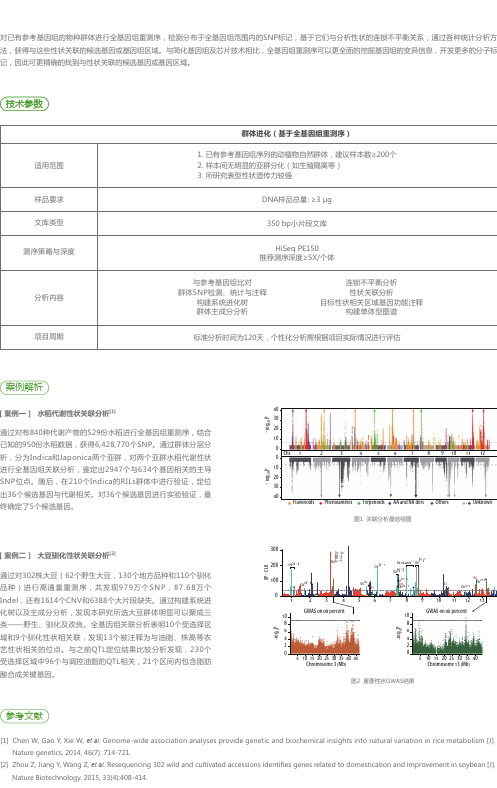
图2 重要性状GWAS结果
参考文献
[1] Chen W, Gao Y, Xie W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism [J]. Nature genetics, 2014, 46(7): 714-721.
对已有参考基因组的物种群体进行全基因组重测序,检测分布于全基因组范围内的SNP标记,基于它们与分析性状的连锁不平衡关系,通过各种统计分析方 法,获得与这些性状关联的候选基因或基因组区域。与简化基因组及芯片技术相比,全基因组重测序可以更全面的挖掘基因组的变异信息,开发更多的分子标 记,因此可更精确的找到与性状关联的候选基因或基因区域。
ቤተ መጻሕፍቲ ባይዱ
与参考基因组比对 群体SNP检测、统计与注释
构建系统进化树 群体主成分分析
连锁不平衡分析 性状关联分析
目标性状相关区域基因功能注释 构建单体型图谱
标准分析时间为120天,个性化分析需根据项目实际情况进行评估
案例解析
[案例一] 水稻代谢性状关联分析[1]
通过对有840种代谢产物的529份水稻进行全基因组重测序,结合 已知的950份水稻数据,获得6,428,770个SNP。通过群体分层分 析,分为Indica和Japonica两个亚群,对两个亚群水稻代谢性状 进行全基因组关联分析,鉴定出2947个与634个基因相关的主导 SNP位点。随后,在210个Indica的RILs群体中进行验证,定位 出36个候选基因与代谢相关。对36个候选基因进行实验验证,最 终确定了5个候选基因。
全基因组关联分析的方法与应用

全基因组关联分析的方法与应用全基因组关联分析(GWAS)是一种采用大样本数量和高密度的基因检测技术,通过寻找基因和表型之间的关联,发现对人类疾病表型贡献的基因变异。
GWAS是人类遗传学和疾病学领域中的一个重大发现,为基因疾病学、基因组医学、以及个性化治疗提供了可靠的理论基础。
GWAS的实验方法是对多个样本进行基因测序,通过对数据进行比对,从数百万个基因中筛选出与表型相关的基因变异。
GWAS的数据处理往往需要使用多个算法,将数据整合,以便得到最准确的结果。
对于GWAS定位到的基因变异,研究人员通常会运用其他实验技术进一步验证其功能和生物学意义,并探究其与特定表型之间的关系。
GWAS的应用领域非常广泛,包括心血管疾病、糖尿病、癌症、眼科疾病、免疫系统疾病和神经系统疾病。
其中,心血管疾病是GWAS最早的应用领域之一。
例如,GWAS研究发现了在心血管疾病中具有风险地位的基因,例如APOE、TCF7L2 和CETP脂蛋白。
目前,疾病治疗中根据基因组数据设计的个性化治疗方案已经被广泛应用。
GWAS研究的终极目标是了解基因变异如何导致疾病,探索更好的治疗方法。
GWAS的发现使得医学迈向了基于基因组的个性化治疗时代,而不是以往的基于症状诊断的治疗方式。
例如,在药物治疗领域,通过GWAS发现在药物代谢途径中的基因多态性,医生可以预测患者对药物的响应和耐受性,并制定更准确的个性化治疗方案,有效提高疗效并降低不良反应的风险。
然而, GWAS也存在一些局限性和挑战。
首先,GWAS需要大量标本和高通量技术、较长时间和高昂经费,因此 GWAS 研究的费用非常昂贵。
其次,许多具有重要生物学意义的基因变异并没有被 GWAS 研究所涵盖,这些基因变异往往具有较低的频率和较小的效应大小,无法被当前的 GWAS 技术所检测。
最后,GWAS所找到的相关位点与表型间的相关并不意味着直接的因果关系,GWAS只能揭示关系,实际具体机制需要进一步研究和探索。
全基因组关联分析剖析

对家系数据进行检查,排 除样本混淆、亲子关系 错误等问题,控制家系关 系的正确性。
全基因组关联分析的结果验证
验证检查
对于全基因组关联分析的结果,需要进行严格的验证检查,以确保结果的可靠性和重复性。
重复实验
在不同的人群或样本中重复实验,比较结果是否一致进一步的功能实验,探讨基因变异与表型之间的机制。
全基因组关联分析的统计方法
统计分析
全基因组关联分析通常采用统计模型对遗传标记与表型之间的关联进行测试,如线性回归、logistic 回归等。
多重检验校正
由于基因组级别的大量比较检验,需要采用Bonferroni、FDR等方法进行多重检验校正,以控制I型错 误风险。
机器学习方法
近年来,全基因组关联分析也开始采用机器学习技术,如Ridge回归、Lasso回归等方法,以提高检测 能力。
全基因组关联分析的研究 热点
1 复杂疾病研究
全基因组关联分析被广 泛应用于探索复杂疾病 如糖尿病、心血管疾病 、肿瘤等的遗传学基础 。
3 交互作用研究
多基因、基因-环境等交 互作用的研究是全基因 组关联分析的重要方向 。
2 药物反应预测
全基因组分析有助于识 别影响药物反应的基因 变异,助力个体化精准医 疗。
生物学解释
从统计上显著关联的遗 传位点到生物学功能解 释存在鸿沟,需要更深入 的研究。
跨人群适用性
现有大多数研究集中于 欧美人群,如何推广到其 他人群是一大挑战。
全基因组关联分析的研究进 展
多组学整合
研究者正在探索将全基因组 关联分析与转录组学、表观 遗传学等多种组学数据相结 合的方法,以更全面地了解 复杂疾病的遗传学机制。
新型统计方法
学者们不断开发基于机器学 习、贝叶斯统计等的创新分 析方法,以提高检测复杂遗 传变异和基因-环境相互作 用的能力。