调血脂药物阿伐他汀的合成路线
他汀类药物的生物技术合成以及应用

他汀类药物的生物技术合成以及应用摘要:他汀类药物是一类可以非常显著降低血液中胆固醇含量的药物;还可以减少中风或其他疾病的风险。
近几年来报道,他汀类药物还有其他的生物活性以及许多潜在的治疗用途。
天然他汀类药物有洛伐他汀和康帕丁,而普伐他汀是通过生物转化形成的。
辛伐他汀,是领先市场的第二他汀类药物,是一种洛伐他汀的半合成衍生物。
洛伐他汀主要是由Aspergillus terrus(土曲霉)菌株合成的,而康帕丁是由penicillium citrinum菌株合成。
洛伐他汀和康帕丁是通过液体深层发酵进行工业生产,但也固态发酵进行生产,这种新的生产方式具有一定的优势。
洛伐他汀在生物化学和遗传学上的一些研究进展让辛伐他汀在新的生产方式方面得以发展。
这种洛伐他汀衍生物可以通过monacolin J(无侧链洛伐他汀)过程有效合成,这个过程是一个酰基转移酶LovD进行的。
利用基因lovF 的组合生物合成,可以通过一种不同的方法设计土曲霉,从而使聚酮合成酶在体内合成2,2- dimethylbutyrate(simvastatin的侧链)。
这样产生的转化菌株能通过直接发酵产生辛伐他汀而非洛伐他汀。
关键字:他汀类药物生物合成和遗传学生物技术生产简介:据世界卫生组织的报道,心血管疾病是威胁健康的主导因素。
2005年,约有1750万人死于这些疾病,死亡率占全球约30%。
这种疾病是由于血浆中的胆固醇含量提高而致,因而胆固醇血症成为了动脉粥样硬化和冠状动脉疾病(Kannel等人,1961年)的主要危险因素。
一般来说,人体中的胆固醇只有三分之一是从饮食取得的,而其三分之二是由肝脏合成,还有一小部分是由其他器官合成的(Furberg 1999年;Alberts等人1980年)。
出于这个原因,抑制胆固醇的生物合成来控制其含量成为一个重要策略,以降低胆固醇在血液中浓度,如manzoni和Rollini(2002)关于他汀类的一篇报道中如是说。
他汀类药物合成工艺原理

HMG-CoA还原酶抑制剂
体内胆固醇的合成
HMG-CoA还原酶
4
发现
1976年日本科学家从桔青霉菌的培养提取物中发现 了康帕定(Compactin)及美伐他汀(Mevastatin) 抑制HMGCoA还原酶,能明显降低血浆胆固醇
H HO O O H H O H O
美伐他汀(Mevastatin)
15
阿托伐他汀钙药理作用
抑制HMG一CoA还原酶 增加低密度脂蛋白受体 抑制极低密度脂蛋白胆固醇合成 抗动脉粥样硬化作用
16
阿托伐他汀钙临床应用 阿托伐他汀钙被认为是至今最好的调血脂药物 之一 降低LDL胆固醇治疗方面优于其它他汀类药物, 疗效比普伐他汀疗效高47%;而且不良反应较少, 与辛伐他汀相比要低24%。 唯一被证明减少心血管事件优于血管重建术的 调脂药物 对骨质疏松症、老年痴呆症、心脏病、器官移 植、中风和糖尿病都有一定的疗效。
这也是很多仿制品药物与原药的差异所在。
FDA, ANDAs: Pharmaceutical Solid Polymorphism (Dec. 2004) (Draft Guidance)
市场前景 世界上最畅销的药是辉瑞公司的降胆固醇药阿 托伐他汀(atorvastatin,立普妥,Lipitor)。 这只由前华纳-兰伯特科学家20多年前研发的 “重磅之王”,是医药界的商业奇迹, 先后为 辉瑞带来了1000多亿美元的销售额。 他汀类药是占中国调血脂市场份额最大的药品 类别之一,拥有非常广阔的市场空间
30
汇聚合成方法1:
Paal一Knorr缩合
31
汇聚合成方法2:
“aza一wittig”反应
32
路线选择
阿托伐他汀

阿托伐他汀立普妥(阿托伐他汀钙片,atorvastatin)是一种合成的降脂药物。
为3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂。
这种酶能催化HMG-CoA向甲羟戊酸转变,这是胆固醇生物合成过程前期的一个限速步骤。
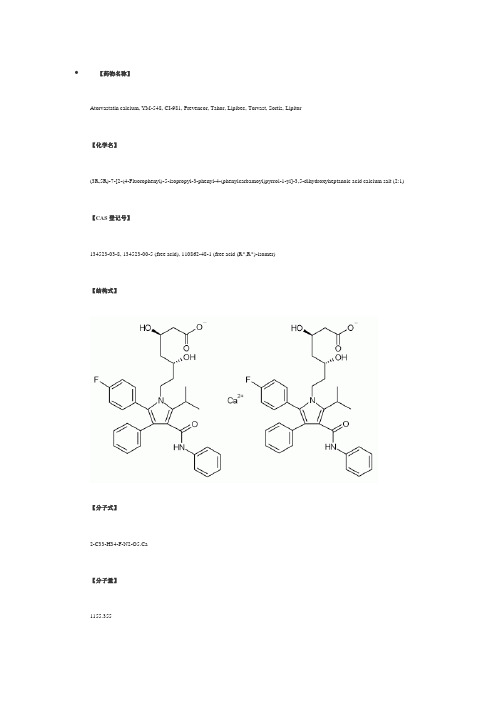
一、概述立普妥是阿托伐他汀[R-(R,R)]-2-(4-氟苯基)-β,δ-二羟基-5-(1-甲基乙基)-3-苯基-4-[(苯氨基)羰基]-1H-吡咯-1-庚酸:钙盐(2:1)的三水化物。
阿托伐他汀钙的分子式为(C33H34FN2O5)2Ca 3H2O,其分子量是1209.42。
其结构式为:阿托伐他汀钙为白色或类白色结晶性粉末,不溶于pH≤4的水溶液。
阿托伐他汀钙能微溶于蒸馏水、pH为7.4的磷酸盐缓冲液、乙腈,轻度溶于乙醇,易溶于甲醇。
口服的立普妥(阿托伐他汀钙片)分别含阿托伐他汀10、20或40mg,及下列无活性成分:碳酸钙(美国药典,USP);小烛树蜡(食品化学物质法规,FCC);羟脯氨酰纤维素(国家处方集,NF);单水乳糖(NF);硬脂酸镁(NF);微晶纤维素(NF);白色Opadry,YS-1-7040(羟丙基甲基纤维素钠、聚乙烯醇、滑石、二氧化钛);聚山梨酯80(NF),二甲基硅油乳胶等。
二、作用机理阿托伐他汀是一种选择性、竞争性HMG-CoA还原酶抑制剂。
HMG-CoA还原酶是3-羟基-3-甲基戊二酰辅酶A向甲羟戊酸转化的限速酶,而甲羟戊酸是固醇包括胆固醇的前体。
胆固醇和甘油三酯在血液中循环,作为参与载脂蛋白复合物的组成成分,对载脂蛋白进行超速离心,可使其离心成为HDL(高密度脂蛋白)、IDL(中密度脂蛋白)、LDL(低密度脂蛋白)和VLDL(极低密度脂蛋白)等成分。
在肝脏中,胆固醇和甘油三酯被合成为VLDL并被释放入血浆、运输至周围组织。
LDL是由VLDL生成,并通过与其高亲和性LDL受体的结合而被分解的。
临床和病理研究都显示,总胆固醇(Total-C)、低密度脂蛋白胆固醇(LDL-C)和载脂蛋白B(apo-B)升高都能促进人类动脉粥样硬化的形成,是心血管疾病发生的危险因素,而高密度脂蛋白胆固醇(HDL-C)的升高则能降低心血管病的危险性。
调血脂药阿托伐他汀钙的合成研究进展

随着 对 高 脂 血 症 的 不 断 研 究 和综 合 治 疗 经 验 的 积 累 , 疗 高 治
几 得 到 关 键 中 间 体 2一( 3一[ 4一氟 苯 基 ) 一异 丙 基 一 苯 基 一 一5 3一
+
F
脂血症的药物品种及其使用情况也在不断地发生变化…。 羟基 一 3一
3一甲基 戊 二 酰 辅 酶 A H ( MG— o 还 原 酶 抑 制 剂 , 称 他 汀 类 C A) 简 (a n) 物 , s t s药 ti 由于 作 用 机 制 新 颖 , 用 范 围 较为 是 目前 最 为 经典 和有 效 的 调血 脂 药 - 。 耐 被 3 ]
a i s u t e i o te r g 1 er snh s ) T e oh ro e i t pe ae ci l3 5一 i —d y r y h pa o c rg e tte f m te c t c r n h i ( n a y tei . h te n s o rp r hr , es i do e t i ai f m n,h o h d r u t n i s a h x n c d a r
线提 供 参 考 。 关键 词 : 血 脂 药 : 调 阿托 伐 他 汀钙 : 成 合
中图 分 类号 : 7 6 T 6 . 1 R92 . ; Q40 3 文 献标 识 码 : A 文章 编 号 :0 6— 9 1 2 1 )1— 0 4 3 1 0 4 3 (0 0 2 0 0 —0
药学专论
21 年第 l 卷第 2 期 00 9 1
调血 脂 药 阿托伐 他 汀 钙 的合成 研 究 进展
张宜凡 虞心红 ,
(.上 海 医药高等 专科 学校 药学系 , 海 1 上 2 11 ; 2 0 3 8 .华东理 工 大学 药学院 , 上海 20 3 ) 0 1 7
阿托伐他汀侧链中间体的合成

阿托伐他汀侧链中间体的合成阿托伐他汀侧链中间体的合成摘要:阿托伐他汀是HMG-CoA还原酶的选择性、竞争性抑制剂,能有效地降低血脂。
本文着重对其活性中心侧链中间体的合成进行了专题分析,以寻求较好的合成路线。
关键词:阿托伐他汀侧链中间体合成中图分类号:文献标志码:文章编号:阿托伐他汀是目前全球处方量最多的降胆固醇药物。
由于其含有(3R,5S)-双羟基的侧链,并且要求e.e.≥99.5%,因此合成此侧链的中间体很有挑战性,人们通过各种方法合成,得到的侧链中间体也就有所差别,本文就介绍一下几种中间体的合成路线。
1 中间体TBIA(tert-Butyl isopropylidene amine)的合成主要是通过DERA(EC4.1.2.4)突变体Ser238Asp催化的醛醇缩合反应得到内酯化合物,再经过一系列的反应得到TBIA,具体的合成步骤如下:N31O+O+ODERA6d2OHBr2,BaCO3N3OOMeONa,MeOH,83%t-BuOK,t-BuOH,72%[1]OHN3OHOORcamphorsulfonic acid2,2-dimethoxypropane,76%N3OOOOMeMeOH-H2OLiOH,83: R=Me3b R=t-BuON35O4OOHBoc2ODMPA,86%N37536OOPh3P3d,72%OOOH2NOOOTBIA2 阿托伐他汀钙中间体ATS-8的合成ATS-8的中文名称为6-氰甲基-2,2-二甲基-1,3-二氧戊环-乙酸叔丁酯,英文名称为(4R,6R)-1,1-dimethylethyl-6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate,其合成以高烯丙醇为原料生成碘代的内酯,具体合成步骤如下:OOHBuLi/THF,CO2,I291%1I2ONC4t-BuOH,DCC,DMAP/CH2Cl2,rtOH6NCATS-8[2]OOp-TsOH,acetone,rt90%IOO3ONC5OCH O°KCN/DMSO,40 C75~80%OOsO4-NaIO4/dioxane-H2O orO3,Me2S65~70%OO°CrO3-H2SO4/acetone,0 C70%NCOOOOO3 阿托伐他汀侧链的1,3-二醇中间体的合成该方法主要是通过L-脯氨酸催化正丁醛发生α-氨基氧化以及碘发生的分子内亲电子[3]环化反应,具体的合成路线如下:OHBnO1CHOaBnO2 R=H3 R=MsORbBnO3OcOOOI9ORBnO4 R=H5 R=BocdXOBoceN36 X=OH7 X=OMs8 X=N3OHOgN310OHOHCN11 (1,3-diol)fN3°Reagents and conditions:(a)(i)PhNO,L-proline(25mol%),CH3CN,-20 C,24h then MeOH,NaBH4;(ii)CuSO4(30mol%),°MeOH,0 C,10h,87%(qver twosteps);(b)(i)MsCl,Et°N,CHCl,0C,15min,92%;(ii)KCO,MeOH,rt,1h,95%;(c)vinylma32223gnesium bromide,THF,CuI,-40 C,1h,92%;(d)(i)(Boc)2O,DMAP,CH3CN,rt,5h,95%;(ii)DDQ,CH2Cl2:H2O(2:1),rt,20h,°°85%,(e)(i)MsCl,Et3N,CH2Cl2,0 C,30min,94%;(ii)NaN°3,DMF,60C,2h,83%;(iii)NIS,CH3CN,-40 to 0 C,20h,87%;(f)°K2CO3,MeOH,0 C tort,2h,96%;(g)NaCN,Ti(OPr)°4,n-Bu4NI,DMSO,70 C,6h,80%.4 中间体R(-)-4-氰基-3-羟基丁酸乙酯的合成以环氧氯丙烷为原料得到2-羟基-1,3-二氰基丙烷,此合成的关键步骤就是利用腈水解酶催化该化合物发生不对称反应,从而得到目的产物。
阿伐他汀中间体手性合成研究进展

阿伐他汀手性中间体合成研究进展摘要: 综述了近年来阿伐他汀手性中间体的合成研究进展,从手性池反应、不对称合成、外消旋体拆分三个方面介绍了阿伐他汀手性中间体合成的工艺路线和研究水平,对其工业化前景进行了展望。
他汀类药物是目前世界上十分畅销的调血脂药物,是HMGCoA 还原酶抑制剂,对心血管疾病分子水平的研究表明,HMGCoA 还原酶是胆固醇生物合成的限速酶,任何影响此酶合成或功能表达的因素都能有效抑制胆固醇的合成。
他汀类药物正是通过抑制HM GCoA 还原酶与底物的结合来抑制胆固醇的合成。
同时,他汀类药物还可以降低低密度脂蛋白(LDL )和甘油三酯( TG)、升高高密度脂蛋白( HDL ),从而对动脉粥样硬化和冠心病的防治有重要意义。
其中,由辉瑞公司开发的阿伐他汀,自1997年在美国和德国上市以来,销售额逐年攀升,2004 年达到106.8 亿美元, 成为首个年销售额突破百亿美元的处方药 1 [2] 。
阿伐他汀专利保护期到2009 年9 月止 [3] ,目前国内调血脂药市场仍以辛伐他汀为主,然而,阿伐他汀因其适应症更广、耐受性和安全性更好已经引起国内各制药企业的广泛关注,学术界也对其生产工艺特别是重要中间体的合成展开了多方面的研究,作者在此综述了近几年国内外在这一领域的研究进展。
阿伐他汀结构式如下: 其手性侧链有两个羟基,是与HMGCoA 还原酶识别的药效团,合成该侧链涉及的关键中间体主要有( S) -4氯-3-羟基丁酸乙酯(4Chloro3hydroxybutanoate esters , CHBE) ( R) 4-氰基-3-羟基丁酸乙酯和(3R ,5S) 6氯二羟基己酸叔丁酯, 结构式如下:按照合成过程中引入手性的方法来分类,合成阿伐他汀手性中间体主要包括: 手性池反应、不对称合成和外消旋体拆分三种途径,分述如下。
1、手性池反应手性池反应本身并不涉及手性的改变,而是通过从手性池中选择合适的手性化合物作为原料制备目标产物。
以缬氨酸为原料合成阿托伐他汀钙的路线

以缬氨酸为原料合成阿托伐他汀钙的路线下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!以缬氨酸为原料合成阿托伐他汀钙的路线在药物合成领域,阿托伐他汀钙(Atorvastatin Calcium)是一种广泛使用的降脂药,用于治疗高胆固醇和高脂血症等疾病。
阿托伐他汀合成图解
【药物名称】Atorvastatin calcium, YM-548, CI-981, Prevencor, Tahor, Lipibec, Torvast, Sortis, Lipitor【化学名】(3R,5R)-7-[2-(4-Fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxyheptanoic acid calcium salt (2:1) 【CAS登记号】134523-03-8, 134523-00-5 (free acid), 110862-48-1 (free acid (R*,R*)-isomer)【结构式】【分子式】2-C33-H34-F-N2-O5.Ca【分子量】1155.355【原研厂家】Jouveinal (Originator), Pfizer (Originator), Almirall Prodesfarma (Licensee), Syncro (Licensee), Yamanouchi (Licensee), Stanford University (Codevelopment)【作用类别】Alzheimer's Dementia, Treatment of , CARDIOVASCULAR DRUGS, Cognition Disorders, Treatment of, Immunologic Neuromuscular Disorders, Treatment of, Lipoprotein Disorders, Treatment of , METABOLIC DRUGS, Multiple Sclerosis, Agents for, NEUROLOGIC DRUGS, Treatment of Disorders of the Coronary Arteries and Atherosclerosis, HMG-CoA Reductase Inhibitors, TNFSF6 Expression Inhibitors【研发状态】Launched-1997【合成情况】NB2〖来源〗Drugs Fut〖合成路线〗〖标题〗Atorvastatin Calcium〖合成方法〗1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl 2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 C in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4-diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX), which is finally treated first with NaOH in methanol/water, and then with CaCl2 or calcium acetate.〖作者〗Graul, A.; Casta馿r, J.〖参考〗Graul, A.; Casta馿r, J.; Atorvastatin Calcium. Drugs Fut 1997, 22, 9, 956〖出处〗Drugs Fut1997,22,(9):956〖备注〗Synthesis Atorvastatin calcium has been obtained by several different ways: 1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 癈in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4 -diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI) (1, 2), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX) (2, 3), which is finally treated first with NaOH inmethanol/water, and then with CaCl2 or calcium acetate (3, 4). 2) The condensation of the already described aldehyde (XI) with(S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S) (2, 3). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2 (2-4). 3) The condensation of4-cyano-3(R)-hydroxybutyric acid ethyl ester (XXV) with N,N-diphenylacetamide (R1 = R2 = Ph in XXVI) by means of LDA in THF gives 6-cyano-5(R)-hydroxy-3-oxo-N,N-diphenylhexanamide (XXVII), which is reduced with diethylmethoxyborane and NaBH4 in THF yielding 6-cyano-3(R),5(R)-dihydroxy-N,N-diphenylhexanamide (XXVIII). The protection of the two OH groups of (XXVIII) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XXX), which by reduction of its CN group by hydrogenation with H2 over RaNi in methanol/liquid ammonia gives (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]-N,N -diphenylacetamide (XXXI). The cyclization of (XXXI) with 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) (its synthesis is in section 6, Scheme 4) in refluxing toluene yields the protected dihydroxyheptanamide (XXXIII), which is deprotected with HCl in methanol to afford (3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N -phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxy-N,N-diphenylheptanamide (XXXIV). Finally, this compound is hydrolyzed with NaOH and treated with calcium acetate in water (5). Scheme 18007203a. 4) The preceding reaction pathway can be repeated using other substituents for R1 and R2 in acetamide (XXVI) such as R1 = R2 = CH2Ph; R1 = R2 = Et; R1 = Bu, R2 = Me; R1 = t-Bu, R2 = CH2Ph; R1,R2 = -(CH2)5- (5). 5) The hydrolysis of methyl (Et or Bu)3(R)-(tert-butyldimethylsilyloxy)-4-cyanobutyrate (XXV) with NaOH gives the corresponding free acid (XXXVI), which is condensed with malonic acid mono-tert-butyl ester magnesium salt (XXXVII) by means of carbonyldiimidazole (CDI) yielding tert-butyl5(R)-(tert-butyldimethylsilyloxy)-6-cyano-3-oxohexanoate (XXXVIII). The desilylation of (XXXVIII) with tetrabutylammonium fluoride in acetic acid affords the expected hydroxylated ketoester (XXXIX), which is reduced with diethylmethoxyborane and NaBH4 in methanol giving tert-butyl 6-cyano-3(R),5(R)-dihydroxyhexanoate (XL). The protection of the two OH groups of (XL) with acetone dimethylketal(XXIX) and methanesulfonic acid affords the 1,3-dioxane (XLI) (6), which by reduction of its CN group by hydrogenation with H2 overPd/C gives intermediate (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLII). The cyclization of (XLII) with 4-(4-fluorophenyl-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) in refluxing toluene yields the protected dihydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water (7). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 6) The (4R,6R)-2-[6-(cyanomethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLI) can also be obtained by reaction of (4R,6R)-2-[6-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLVIII) with tosyl chloride to give the corresponding tosylate (XVIX), which is then treated with NaCN (6). 7) The tert-butyl6-cyano-5(R)-hydroxy-3-oxohexanoate (XXXIX) can also be obtained by condensation of methyl 4-cyano-3(R)-hydroxybutyrate (L) with tert-butyl acetate (XXIII) by means of LDA in THF (6). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 9) The cyclization of (XXXII) with intermediate (XLII) (preceding synthesis) in refluxing toluene yields the protected dehydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water. References 1. Roth, B.D. (Warner-Lambert Co.). Trans-6-[2-(3- or 4-carboxamido-substd.pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis. EP 247633, US 4681893. 2. Roth, B.D., Blankley, C.J., Chucholowski, A.W., Ferguson, E., Hoefle, M.L., Ortwine, D.F., Newton, R.S., Sekerke, C.S., Sliskovic, D.R., Stratton, C.D., Wilson, M.W. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem 1991, 34: 357-66. 3. Roth, B.D. (Warner-Lambert Co.). (R-(R*R*)-2-(4-Fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl-3-phenyl-4-[(phenylamino)-carbonyl]-1H-pyrrole-1-heptanoic acid, its lactone form and salts thereof. EP 409281, JP 91058967, US 5273995.4. Milb, N., Muhammad, N.A., Weiss, J., Nesbitt, R.U. (Warner-Lambert Co.). Stable oral CI-981 formulation and process for preparing same. EP 680320, JP 96505640, WO 9416693.5. Butler, D.E., Le, T.V., Nanninga, T.N. (Warner-Lambert Co.). Process fortrans-6-[2-(substd.-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis. US 5298627. 6. Brower, P.L., Butler, D.E., Deering, C.F., Le, T.V., Millar, A., Nanninga, T.N., Roth, B.D. The synthesis of (4R-cis)-1,1-dimethylethyl6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate, a key intermediate for the preparation of CI-981, a highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2279-82. 7. Baumann, K.L., Butler, D.E., Deering, C.F., Mennen, K.E., Millar, A., Nanninga, T.N., Palmer, C.W., Roth, B.D. The convergent synthesis of CI-981, an optically active, highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2283-4. 8. McKenzie, A.T. (Warner-Lambert Co.). Form III crystalline(R-(R*,R*))-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methyl-ethyl) -3-phenyl-4-((phenylamino)carbonyl)-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703958. 9. Lin, M., Schweiss, D. (Warner-Lambert Co.). Novel process for the production of amorphous [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid calcium salt (2:1). WO 9703960. 10. Briggs, C.A., Jennings, R.A., Wade, R.A., Harasawa, K., Ichikawa, S., Minohara, K., Nakagawa, S. (Warner-Lambert Co.). Crystalline[R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl) -3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703959.〖来源〗J Med Chem〖合成路线〗〖标题〗Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus〖合成方法〗1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl 2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 C in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4-diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX), which is finally treated first with NaOH in methanol/water, and then with CaCl2 or calcium acetate.〖作者〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.〖参考〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.; Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)eth IĶ 셈睋서睋쓰睎ℐ ul> li>a href=쓜睎 Ʈ 0 ๙ιᇐ ꀀÑ밸-00AA004CLSID\{0E59F1D5-1FBE-11D0-8FF2-00A0D10038BC}e Ǔ〖出处〗J Med Chem1991,34,(1):357-66〖备注〗〖来源〗Drugs Fut〖合成路线〗〖标题〗Atorvastatin Calcium〖合成方法〗2) The condensation of the previously described aldehyde (XI) with (S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2.〖作者〗Graul, A.; Casta馿r, J.〖参考〗Graul, A.; Casta馿r, J.; Atorvastatin Calcium. Drugs Fut 1997, 22, 9, 956〖出处〗Drugs Fut1997,22,(9):956〖备注〗Synthesis Atorvastatin calcium has been obtained by several different ways: 1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 癈in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4 -diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolanegroup of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI) (1, 2), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX) (2, 3), which is finally treated first with NaOH inmethanol/water, and then with CaCl2 or calcium acetate (3, 4). 2) The condensation of the already described aldehyde (XI) with(S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S) (2, 3). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2 (2-4). 3) The condensation of4-cyano-3(R)-hydroxybutyric acid ethyl ester (XXV) with N,N-diphenylacetamide (R1 = R2 = Ph in XXVI) by means of LDA in THF gives 6-cyano-5(R)-hydroxy-3-oxo-N,N-diphenylhexanamide (XXVII), which is reduced with diethylmethoxyborane and NaBH4 in THF yielding 6-cyano-3(R),5(R)-dihydroxy-N,N-diphenylhexanamide (XXVIII). The protection of the two OH groups of (XXVIII) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XXX), which by reduction of its CN group by hydrogenation with H2 over RaNi in methanol/liquid ammonia gives (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]-N,N -diphenylacetamide (XXXI). The cyclization of (XXXI) with 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) (its synthesis is in section 6, Scheme 4) in refluxing toluene yields the protected dihydroxyheptanamide (XXXIII), which is deprotected with HCl in methanol to afford(3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N -phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxy-N,N-diphenylheptanamide (XXXIV). Finally, this compound is hydrolyzed with NaOH and treated with calcium acetate in water (5). Scheme 18007203a. 4) The preceding reaction pathway can be repeated using other substituents for R1 and R2 in acetamide (XXVI) such as R1 = R2 = CH2Ph; R1 = R2 = Et; R1 = Bu, R2 = Me; R1 = t-Bu, R2 = CH2Ph; R1,R2 = -(CH2)5- (5). 5) The hydrolysis of methyl (Et or Bu)3(R)-(tert-butyldimethylsilyloxy)-4-cyanobutyrate (XXV) with NaOH gives the corresponding free acid (XXXVI), which is condensed with malonic acid mono-tert-butyl ester magnesium salt (XXXVII) by means of carbonyldiimidazole (CDI) yielding tert-butyl5(R)-(tert-butyldimethylsilyloxy)-6-cyano-3-oxohexanoate (XXXVIII). The desilylation of (XXXVIII) with tetrabutylammonium fluoride in acetic acid affords the expected hydroxylated ketoester (XXXIX), which is reduced with diethylmethoxyborane and NaBH4 in methanol giving tert-butyl 6-cyano-3(R),5(R)-dihydroxyhexanoate (XL). The protection of the two OH groups of (XL) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XLI) (6), which by reduction of its CN group by hydrogenation with H2 overPd/C gives intermediate (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLII). The cyclization of (XLII) with 4-(4-fluorophenyl-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) in refluxing toluene yields the protected dihydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water (7). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 6) The (4R,6R)-2-[6-(cyanomethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLI) can also be obtained by reaction of (4R,6R)-2-[6-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLVIII) with tosyl chloride to give the corresponding tosylate (XVIX), which is then treated with NaCN (6). 7) The tert-butyl6-cyano-5(R)-hydroxy-3-oxohexanoate (XXXIX) can also be obtained by condensation of methyl 4-cyano-3(R)-hydroxybutyrate (L) with tert-butyl acetate (XXIII) by means of LDA in THF (6). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means oftriethylamine in hot ethanol (7). 9) The cyclization of (XXXII) with intermediate (XLII) (preceding synthesis) in refluxing toluene yields the protected dehydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water. References 1. Roth, B.D. (Warner-Lambert Co.). Trans-6-[2-(3- or 4-carboxamido-substd.pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis. EP 247633, US 4681893. 2. Roth, B.D., Blankley, C.J., Chucholowski, A.W., Ferguson, E., Hoefle, M.L., Ortwine, D.F., Newton, R.S., Sekerke, C.S., Sliskovic, D.R., Stratton, C.D., Wilson, M.W. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem 1991, 34: 357-66. 3. Roth, B.D. (Warner-Lambert Co.). (R-(R*R*)-2-(4-Fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl-3-phenyl-4-[(phenylamino)-carbonyl]-1H-pyrrole-1-heptanoic acid, its lactone form and salts thereof. EP 409281, JP 91058967, US 5273995.4. Milb, N., Muhammad, N.A., Weiss, J., Nesbitt, R.U. (Warner-Lambert Co.). Stable oral CI-981 formulation and process for preparing same. EP 680320, JP 96505640, WO 9416693.5. Butler, D.E., Le, T.V., Nanninga, T.N. (Warner-Lambert Co.). Process fortrans-6-[2-(substd.-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis. US 5298627. 6. Brower, P.L., Butler, D.E., Deering, C.F., Le, T.V., Millar, A., Nanninga, T.N., Roth, B.D. The synthesis of (4R-cis)-1,1-dimethylethyl6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate, a key intermediate for the preparation of CI-981, a highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2279-82. 7. Baumann, K.L., Butler, D.E., Deering, C.F., Mennen, K.E., Millar, A., Nanninga, T.N., Palmer, C.W., Roth, B.D. The convergent synthesis of CI-981, an optically active, highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2283-4. 8. McKenzie, A.T. (Warner-Lambert Co.). Form III crystalline(R-(R*,R*))-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methyl-ethyl) -3-phenyl-4-((phenylamino)carbonyl)-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703958. 9. Lin, M., Schweiss, D. (Warner-Lambert Co.). Novel process for the production of amorphous [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid calcium salt (2:1). WO 9703960. 10. Briggs, C.A., Jennings, R.A., Wade, R.A., Harasawa, K., Ichikawa, S., Minohara, K., Nakagawa, S. (Warner-Lambert Co.). Crystalline[R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl) -3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703959.〖来源〗J Med Chem〖合成路线〗〖标题〗Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus〖合成方法〗2) The condensation of the previously described aldehyde (XI) with (S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2.〖作者〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.〖参考〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.; Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)eth IĶ 셈睋서睋쓰睎쓜睎 Ʈ 0 ๙ιᇐ ꀀÑ밸-00AA004CLSID\{0E59F1D5-1FBE-11D0-8FF2-00A0D10038BC}e Ǔ ℐ ul> li>a href=〖出处〗。
调血脂药物阿伐他汀的合成路线_杨宽

2 不对称还原羰基法
在合成阿伐他 汀 的 工 艺 研 究 过 程 中 , 研究者
[6] 其中 B 提出的不 们又开发了多条路线 , u l t e r等 1
过大量的实验完善这条 路 线 , 将 M- 4的合成产率 提高至 7 9% 。 1. 2 A T S 9 的合成 -
[2 ] 1 3 - 以 R( 1 9 9 2 年, B u t l e r等 1 4 3 -) - - 氰 基- -羟
1 5] 。 一些有机酸或更温和酸的使用 [
总的来说 , P a a l n o r r法 合 成 阿 伐 他 汀 合 成 -K
图 2 中间体 M 4 的合成路线 -
反应条件温和 , 成本较低 , 品质可控 , 且 工艺简单 , 因此是 目 前 阿 伐 他 汀 原 料 药 合 成 的 最 产率较高 , 主要的路线 。
行了优化 , 得 出 该 步 合 成 的 最 佳 工 艺 为: 正庚 V( 烷) 四氢呋喃 ) 甲 苯) ∶ V( ∶ V( =4∶1. 1∶1 的 混 反应 温 度 为 9 反 应 时 间 ≥2 合溶剂 , 0 ℃, 0h。 优 化后这 一 步 的 收 率 可 达 到 4 5% 。 对 关 环 产 物 2 进行 酸 脱 保 护 , 水 解, 反 复 重 结 晶, 成钙盐等步骤 合成最终产 物 1。 研 究 发 现 , 酸解步骤对产物的 最终产率有一定的影响 , 通常采用的酸为强酸 ( 盐 , 硫酸 ) 该步反 应 过 程 中 存 在 着 反 应 可 控 性 不 酸、 环 境 不 友 好 等 问 题, 因 此, 研究者们开始关注 强,
这 现产物中 n( t r a n s 构型 ) ∶n( c i s 构型 ) =9∶1, 一步是该 条 路 线 的 关 键 步 骤 。3 1在甲苯中回流 发生内酯化 反 应 , 得到阿伐他汀的内酯3 内酯 2, 3 2 是c i s 构 型 和t r a n s 构 型 的 混 合 物。为 了 提 高 产物 中t 需要对其进行重结晶 r a n s 构 型 的 比 率, 处理 , 经 过 一 次 重 结 晶 后 可 将 单 一 的t r a n s型内 酯提高至 9 7% 。 对纯化后的产物 3 具 2 还需要进行手性拆分 , 体方法如 下 : 以( R) 2 - - 甲 基 -苄 胺 为 手 性 拆 分 剂 , 将其 与 3 2 反 应 生 成 酰 胺 后 分 离 纯 化。并 需 要 在 碱性的条件下脱去拆分剂后 环 合 得 2 最后进行 1, 水解成盐 。 本路 线 采 用 的 是 消 旋 体 拆 分 的 方 法 , 从产率上比较相 对 较 差 , 且在反应的过程中涉及 了拆分剂的脱除 , 因此原子经济性较差 , 因此该方 案在工业上没有较大的使用前景 。
阿托伐他汀钙合成工艺

阿托伐他汀钙合成工艺
阿托伐他汀钙合成工艺反应原理:
N
F
O O
O
H
2
O O O
N
F
O
N
O O O +
AT-9M-4AT-10
N
F
O N
O O O
N
F
O
N
OH OH O
N
F
O N
OH OH O
N
F
O
N O
OH OH O
2
Ca++
AT-10AT-11
AT-11
AT
精
制
精品阿托伐他汀钙
AT-10的合成
在有回流冷凝器的园底烧瓶中,加入41.7克(0.1mol)M-4,28.7克(0.1mol)AT-9,0。
002mol 的对甲苯磺酸,200ml甲苯,加热回流反应,并用分水器将反应生成的水分掉,6小时后,降温,
减压整除甲苯,加入100ml叔丁基甲基醚(MTBE)分散,过滤出固体,水洗,直接用于下一步反应。
AT的合成。
园底烧瓶中,加入上一步物料,甲醇40ml,氢氧化钠(0。
105mol)的水溶液200毫升,加热到55度水解1.5小时至溶液彻底变澄清,降至室温,用80*2叔丁基甲基醚洗涤;水溶液(即AT-11)重新加热到55度,搅拌滴加一水合醋酸钙(0.53mol)的水溶液,并在此温搅拌两个小时,冷却至20度,过滤,水洗得产物,70度减压干燥,得白色固体。
总收率85%。
AT 的精制:
用甲醇水加热回流脱色,冷却析晶,过滤得白色晶体。
70度以下减压干燥。
收率94% .。
阿托伐他汀的全合成研究进展

2007年第15卷合成化学Vol .15,2007 第5期,519~527Chinese Journal of Synthetic Che m istry No .5,519~527 ・综合评述・阿托伐他汀的全合成研究进展3王继宇1,2,沈健芬1,2,王立新1,王 文1,蔡泽贵1,杜振军1(1.中国科学院成都有机化学研究所,四川省不对称合成重点实验室,四川成都 610041;2.中国科学院研究生院,北京 100039)摘要:介绍了近20年来利用消旋体拆分法、非对映选择性醇醛缩合法、Paal 2Knorr 反应、环加成法、双羰基不对称还原法等,完成阿托伐他汀全合成的研究进展。
分析了各条路线的关键步骤,讨论了各方法的特点。
参考文献27篇。
关 键 词:阿托伐他汀;全合成;综述中图分类号:R914.5;O621.3文献标识码:A文章编号:100521511(2007)0520519209Progress i n Tot al Synthesis of Atrovast ati nWANG J i 2yu1,2, SHEN J ian 2fen1,2, WANG L i 2xin 1,WANG W en 1, CA I Ze 2gui 1, DU Zhen 2jun1(1.Key Laborat ory f or A sy mmetric Synthesis and Chir otechnol ogy of Sichuan Pr ovince,Chengdu I nstitute of O rganic Che m istry,Chinese Acade my of Sciences,Chengdu 610041,China;2.Graduate University of Chinese Acade my of Sciences,Beijing 100039,China )Abstract:Recent p r ogress in t otal synthesis of A tr ovastatin were revie wed .The key reacti on step s and characteristics of each i m portant synthetic r oute were analyzed and discussed .27Referenceswere cited .Keywords:A t ovastatin;t otal synthesis;review 阿托伐他汀(1,Chart 1)的化学名为(3R,5R )272[22(42氟苯基)252异丙基232苯基242(苯氨基甲酰基)吡咯212基]23,52二羟基庚酸钙盐,是由美国W arner 2La mbert 公司和辉瑞(Pfizer )公司共同开发的他汀类血脂调节药,1997年在英国率先上市。
立普妥的合成方法

课题名称立普妥的合成方法班级学号姓名黄山学院化学化工学院2012年5月立普妥的合成方法立普妥简介:立普妥又称阿托伐他汀钙为一种新型3-羟基-3甲基戊二酰辅酶A还原酶抑制剂,化学名为:[R,(R﹡,R﹡)]-2-(4-氟苯基 )-α,β-二羟基-5-(1-甲基乙基)-3-苯基-[(苯胺基)-羟基]-1H-吡咯-1-庚酸钙盐,临床用其三水化合物,具有同时降低血清胆固醇和三酰甘油的作用,调脂作用高于其他HMG-CoA还原酶抑制剂,不良反应小,属于第三代全合成的阿托伐他汀类调血脂药化学结构式(Chemical Structure):其合成路线如下:合成路线图解说明:化合物(一)2 - (1,3 - 二氧戊环 -2-基)乙基二乙胺与化合物(二)溴-2 - (4 - 氟苯基)乙胺在乙腈醋酸条件下缩合生成化合物(三)甲酸乙酯2 - [2 - (1,3-二氧戊环-2-基)乙基] -2 - (4 - 氟苯基)酯,这是与化合物异丁酰氯,并产生相应的化合物(四)酰胺二氯甲烷乙胺,然后酰化。
酯与甲醇/水的NaOH皂化化合物(五)能提供的游离酸(六),与化合物(七)得到反应化合物(八)与苯胺(九环化)的二环己基碳二亚胺(DCC)的手段,通过加热在90℃醋酸酐生成化合物(十)。
水解产生的(十)与盐酸二氧类化合物生成相应的醛(十一),将得到的醛浓缩与乙酰乙酸甲酯(十二)在四氢呋喃中反应并提供的NaH和酸甲酯生成化合物(十三)。
减少羰基类化合物与三丁基硼和NaBH4在THF条件下与化合物(十三)反应生成(3R,5R)-羟基酯(十四),然后用NaOH皂化产生相应的游离酸(十五)的水溶液。
甲苯回流加热内酯化合物(十五)可得到(的R *,R*)-内酯(十六),将化合物(十六)分离结晶与(R)-1 - 苯乙胺反应得到的产物利用光学分辨率可获得纯的(R,R)对映体酰胺(十七)。
将化合物(十七)用甲苯回流加热,用NaOH酰胺(十八)水解得到(R) - 内酯(十九),最后用甲醇/水碱洗涤,然后用氯化钙或醋酸钙干燥可得到阿托伐他汀钙。
阿托伐他汀侧链结构式

阿托伐他汀侧链结构式
阿托伐他汀(Atorvastatin)是一种降脂药物,属于他汀类药物,用于治疗高胆固醇血症和预防心血管疾病。
其侧链结构是该分子中与羟基苯甲酸酯基团相连的部分,对于药物的吸收、分布、代谢和排泄具有重要作用。
阿托伐他汀的侧链结构包括一个2-[4-氟苯基]-β,δ-二羟基戊酸酯基团。
这个侧链通过酯键与阿托伐他汀分子的主体部分相连。
具体的化学结构如下:
OH
│
HOOC-CH=CH-CH2-CH2-O-CO-R
│
OCH2CH2NHCO
│
R'-COOH
其中,R代表阿托伐他汀分子的主体部分,包含一个内酯环和一个噻唑烷酮环;R'则是指与酯基相连的烃基,在阿托伐他汀中,R'为一个氢原子。
请注意,这个侧链结构式是一个简化的表示,实际的分子结构可能更为复杂,并且需要考虑立体化学因素。
在药物设计和合成中,侧链的精确结构对药物的活性、稳定性和生物利用度都至关重要。
高等有机-阿托伐他汀的合成

阿托伐他汀的合成
阿托伐他汀(1, Chart1)的化学名为(3R,5R)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-(苯氨基甲酰基)吡咯-1-基]-3,5-二羟基庚酸钙盐,是由美国Warner-Lambert公司和辉瑞(Pfizer)公司共同开发的他汀类血脂调节药, 1997年在英国率先上市。
1能强力抑制HMG-CoA还原酶的活性,阻断HMG-CoA还原成羟甲戊酸,大大降低总胆固醇和低密度脂蛋白的含量由于1的活性优于在它之前的所有他汀类药物,且毒副作用小,因此一经上市就表现出不同凡响的上升势头, 2000年后一跃成为全球销售额最高的药物,巨大的经济效益刺激着人们长期关注其合成方法的研究,有关其全合成的研究不断被报道。
一、消旋体拆分法
在20世纪80年代Parke-Davis小组[2, 3]首次开发了1的化学小规模合成,先是合成外消旋体,然后再对非对映体进行拆分。
消旋体拆分法由于必须通过拆分才能得到最终产物,因此,产率不高。
从原子经济性考虑是不可取的。
其意义在于建立了合成1对映体的方法,给后续对映选择性合成方法的开辟指明了方向。
二、环加成法
除上述两种方法之外,还有非对映选择性醇醛缩合法,Paal-Knorr反应合成法,双羰基不对称还原法等方法。
该化合物很有合成价值。
左广征,1101111145,应化11-1。
他汀类药物的生物技术合成以及应用
他汀类药物的生物技术合成以及应用摘要:他汀类药物是一类可以非常显著降低血液中胆固醇含量的药物;还可以减少中风或其他疾病的风险。
近几年来报道,他汀类药物还有其他的生物活性以及许多潜在的治疗用途。
天然他汀类药物有洛伐他汀和康帕丁,而普伐他汀是通过生物转化形成的。
辛伐他汀,是领先市场的第二他汀类药物,是一种洛伐他汀的半合成衍生物。
洛伐他汀主要是由Aspergillus terrus(土曲霉)菌株合成的,而康帕丁是由penicillium citrinum菌株合成。
洛伐他汀和康帕丁是通过液体深层发酵进行工业生产,但也固态发酵进行生产,这种新的生产方式具有一定的优势。
洛伐他汀在生物化学和遗传学上的一些研究进展让辛伐他汀在新的生产方式方面得以发展。
这种洛伐他汀衍生物可以通过monacolin J(无侧链洛伐他汀)过程有效合成,这个过程是一个酰基转移酶LovD进行的。
利用基因lovF 的组合生物合成,可以通过一种不同的方法设计土曲霉,从而使聚酮合成酶在体内合成2,2- dimethylbutyrate(simvastatin的侧链)。
这样产生的转化菌株能通过直接发酵产生辛伐他汀而非洛伐他汀。
关键字:他汀类药物生物合成和遗传学生物技术生产简介:据世界卫生组织的报道,心血管疾病是威胁健康的主导因素。
2005年,约有1750万人死于这些疾病,死亡率占全球约30%。
这种疾病是由于血浆中的胆固醇含量提高而致,因而胆固醇血症成为了动脉粥样硬化和冠状动脉疾病(Kannel等人,1961年)的主要危险因素。
一般来说,人体中的胆固醇只有三分之一是从饮食取得的,而其三分之二是由肝脏合成,还有一小部分是由其他器官合成的(Furberg 1999年;Alberts等人1980年)。
出于这个原因,抑制胆固醇的生物合成来控制其含量成为一个重要策略,以降低胆固醇在血液中浓度,如manzoni和Rollini(2002)关于他汀类的一篇报道中如是说。
阿托伐他汀钙母核的合成及工艺研究

阿托伐他汀钙母核的合成及工艺研究
阿托伐他汀钙,是美国Warner-Lambert公司(现与Pfizer公司合并)在1985年8月合成的一种羟基甲基戊二酰辅酶A(HMG-Co A)还原酶抑制剂。
它能同时降低总胆固醇、低密度脂蛋白胆固醇、载脂蛋白B和甘油三酯的水平,对高血脂症治疗效果显著,可降低心脑血管疾病的患病率和死亡率,也对心脑血管疾病的一
级和二级预防起着重要作用。
4-氟-α-(2-甲基-1-氧丙基)-γ-氧-N,β-二苯基苯丁酰胺(1)是合成阿托
伐他汀钙的母核结构,本文主要对其的合成及工艺进行了系统的研究,建立了以
甲基异丙基甲酮和苯乙酸为起始原料,经过酯缩合(收率88%)、N-酰基化(收率91.9%)、酰氯化(收率97%)、Friedel-Crafts酰基化(收率92%)、溴化(收率88%)、亲核取代反应(收率80%)6步反应制得化合物1的合成路线,并对路线中的每步反应进行了优化,在优化条件下,化合物1的总产率达62.8%,高于文献报道的51.4%的收率。
在研究过程中,发现在第一步缩合反应过程中,存在甲基异丙基甲酮的羟醛缩合副产物,所得结构经GC-MS验证,可通过适当控制甲基异丙基甲酮与氢化
钠的搅拌时间,减少副反应的发生,并在反应后处理中使用干燥的HCl气体代替
冰乙酸,操作简单,易于工业化生产。
在第二步反应中,使用三乙胺代替乙二胺,减少了副反应,并通过蒸除反应生成的甲醇,缩短了反应时间。
建立了氧化溴化法,提高溴化过程中溴原子的利用率。
在最后一步反应中,发现了一个溴化物脱溴的副产物,经确认为4-氟苯基苯
乙酮,对该副产物的产生进行了研究,并通过避光减少了副反应的发生,最终产品中可通过重结晶的方法除去该副产物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
伐 他汀 的合 成路 线 , 为 其 进 一 步研 究 与 开 发提 供 依据。 文 献调 研显示 , 阿伐他 汀合成 路线 较 为丰富 , 其 中较 为经 典 的合成 方 法 有 P a a l — Kn o r r法 、 不对 称 还原 羰基 法 、 消旋体 苯 乙胺 拆分 法 、 选 择性 醇醛
以 M- 4和 ATS - 9为 底 物 , P a a l — Kn o r r 法 合 成 阿伐他 汀 的工 艺 成熟 , 且 具 有 较 高 的产 率 E { ] 。
*基 金 项 目: 西 安 医学 院 博 士科 研 启 动 基 金 ( 2 0 1 1 I  ̄ ) C 0 5 ) ; 陕西 省卫 生厅 科研 资助项 目( 2 0 1 2 D1 4 ) ; 西安 医 学院科研计划 扶植 项 目( 1 2 F Z 0 4 ) ; 西安 医学 院大学 生科 研项 目( 1 2 DX S 0 1 ) ;陕 西 省 教 育 厅 科 研 计 划 项 目
缩合法等。 1 P a a l — K n o r r 法
P a a l — Kn o r r 法[ 6 是 目前 阿伐他 汀原料 药生 产 过 程 中最 为 常见 的一种 方法 。P a a l — Kn o r r 合成 法 以 中 间 体 M_ 4和 ATS( a t o r v a s t a t i n i n t e r me d i — a t e s , 阿伐 他 汀 中 间体 ) 为原 料 , 通过 P a a l — Kn o r r
综 述 专 论
S C I E N C E & T E C H N O L O G Y 化 I N 工 C 科 H 技 E M , 2 0 I C 1 A 3 , L 2 1 I ( N 4 D ) U : 7 S 1 T ~ R 7 Y 5
调 血脂 药 物 阿伐 他 汀 的合 成 路 线 *
( 2 01 3 J K0 7 5 9 )
在该合成路线 中, 中间体 M- 4和 A T S - 9的合 成
研 究 受 到 了较 为 广泛 的关 注 , 如何 有 效 提高 M一 4
・
7 2 ・
第2 1 卷
和 AT 9的最 终产 率 , 以提 高合 成 过 程 中 的原 子 经济性 , 是 研究 者 和生 产者 关 注 的重点 。
间体 。
№
S c h 6 n i n g等 l 9 ] 以苯 乙 酰氯 和 氟苯 为原 料 , 在 A1 C 1 。 的催 化下 发生 傅瑞 德尔 一 克拉 夫茨 法得 到 化
阿伐 他汀 作为 降脂 药具 有较 为 广泛 的适 应症 , 如, 原发性高胆固醇血症 、 混合型高脂血症 、 高胆 固醇 血症 、 动脉 粥样 硬 化等 l 4 ] 。此外 , 阿伐 他 汀 的毒 副
作用小 , 并 具 有 良好 的耐 受 性 和 安 全性 , 是强 效 、 安 全 的一 线 降脂 药 物 l 5 ] 。据 此 , 近年 来 , 关 于 阿 伐
杨 宽 , 余丽丽一 , 姚 琳, 白林奎 , 刘 娅 , 贺 宇
( 西安医学院 药学 院, 陕西 西安 7 1 0 0 2 1 )
摘
要: 阿伐他 汀是一个 高效 、 安全的调血脂 药物 , 具有 广阔的 市场前景 。作 者综述 了其经典 的合
成 方法有 P a a l — Kn o r r 法、 不对称还原羰基 法、 消旋体 苯 乙胺拆分 法、 选择 性醇 醛缩合 法等方法 。为 阿伐
1 . 1 M一 4的合 成
詈 N c
N c \人 O H人 O H儿 0
大
一 。
文献 调研 显 示 目前 有 2条 较 为 成 熟 的 M_ 4
N c \ 人 人 / 儿 、 / 上 < 一O A O /
-
—
-
"  ̄
.
合成 路 线 , 均 需 通 过 异 丁 酰 乙 酰 苯 胺 8这 一 中
F
F
图 1 P a a l — K n o r r 法合 成 路 线
收 稿 日期 : 2 0 1 3 - 0 5 0 8
作者简介 : 杨 宽( 1 9 9 1 一) , 男, 陕西咸 阳人 , 西安 医学 院 本科生 , 从 事化药合成 的研究 。
* *通 讯 联 系人 。
他 汀的工业化发展和创新 打下基础 。 关键词 : 阿伐他汀 ; 合 成路 线 ; 降血 脂 药 ; 研 究进 展
中图分类号 : R 9 6 9ቤተ መጻሕፍቲ ባይዱ
文献标识码 : A
文章编 号 : 1 0 0 8 — 0 5 1 1 ( 2 0 1 3 ) 0 4 — 0 0 7 1 — 0 5
阿伐 他 汀 ( a t o r v a s t a t i n ) 商 品 名 为 立 普 妥 ( L i p i t o r ) , 由辉 瑞 ( P f i z e r ) 公 司 和 Wa me r — L a m— b e r t 公 司共 同开 发 , 并于 1 9 9 7年 经美 国 F DA 批 准上 市 的一 种 新 型 3 一 羟基 一 3 一 甲基 戊 二 酰 辅 酶 A ( HMG- C o A) 还原酶抑制剂 , 是 一 种 全 新 的他 汀 类 降血脂 药 物 _ 1 ] 。阿伐 他 汀是 人 工 全 合 成 的 他 汀类 药物 之一 , 与微 生 物 发 酵 和 化 学 半 合 成 的 他 汀类 药物 相 比 , 具 有 更 优 的药 效 和 临 床 作 用 … 3 ] 。
他 汀合 成 工 艺 的 优 化 受 到 了 研 究 者 们 的广 泛 关 注 。作 者 根据 国内外 相关研 究 进展 , 简 要综 述 阿
缩 合反应 汇 聚式合 成得 到含 吡咯 环 的中间化合 物 2 , 再经过水解 , 脱保 护, 成 盐 得 到 最 终 目标 产 物 1 , 具体合 成 路线见 图 1 ( M- 4和 AT 9 ) 。