[3]Protein structure determination from NMR
蛋白质化学4高级结构
• An actual physical model is then constructed so as to place the nuclei of each of the non-hydrogen atoms in the center of one of each of the areas of high electron density (originally done by hand is now done by computer).
- sheet 模式之一
- sheet 模式之一
- sheet 模式之二
- sheet
模式之三
细胞核抗原的结构
纤溶酶原的结构
锌指结构 (螺旋折叠-折叠模序)
转录因子MyoD的 螺旋-环-螺旋模序
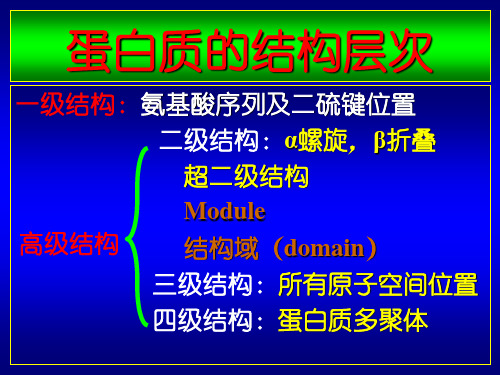
结构域 (Domain)
也称辖区,空间上相对独立,是进化过程中 不同基因融合的结果。结构域是在二级结构或超 二级结构的基础上形成的三级结构的局部折叠区, 一条多肽链在这个域范围内来回折叠,但相邻的 域常被一个或两个多肽片段连结。通常由50-300 个氨基酸残基组成,其特点是在三维空间可以明 显区分和相对独立,并且具有一定的生物学功能, 如结合小分子。模体或基序(motif)是结构域 的亚单位,通常由2-3个二级结构单位组成,一 般为α螺旋、β折叠和环(loop)。
维持蛋白质三级结构的作用力
X-射线(光)衍射
(X-Ray Diffraction, XRD)
X-射线衍射,通过 对材料进行X-射线 衍射,分析其衍射 图谱,分析材料的 成分等。
(高考生物)生物化学名词解释

(生物科技行业)生物化学名词解释1.peptideunit肽单元参与肽键的6个原子C1、C、O、N、H、C2位于同一平面,C1和C2在平面上所处的位置为反式(trans)构型,此同一平面上的6个原子构成了所谓的肽单元(peptideunit)2.motif模序在蛋白质分子中,可发现二个或三个具有二级结构的肽段,在空间上相互接近,形成一个特殊的空间构象,并具有相应的功能,被称为模序。
3.proteindenature蛋白质变性。
在某些理化因素作用下,致使蛋白质的空间构象破坏,从而改变蛋白质的理化性质和生物活性,称为蛋白质变性。
4.glutathione谷胱甘肽由谷氨酸、半胱氨酸和甘氨酸组成的三肽,半胱氨酸的巯基是该三肽的功能基团。
它是体内重要的还原剂,以保护体内蛋白质或酶分子等中的巯基免遭氧化。
5.β-pleatedsheet在多肽链β折叠结构中,每个肽单元以Cα为旋转点,依次折叠成锯齿状结构,氨基酸残基侧链交替地位于锯齿状结构的上下方。
两条以上肽链或一条肽链内的若干肽段的锯齿状结构可平行排列,其走向可相同,也可相反。
并通过肽链间的肽键羰基氧和亚氨基氢形成氢键从而稳固β-折叠结构。
6.chaperon分子伴侣是一类帮助新生多肽链正确折叠的蛋白质。
它可逆的与未折叠肽段的疏水部分结合随后松开,如此重复进行可以防止错误的聚集发生,使肽链正确折叠。
分子伴侣对于蛋白质分子中二硫键的正确形成起到重要作用。
7.proteinquaternarystructure蛋白质的四级结构数个具有三级结构的多肽链,在三维空间作特定排布,并以非共价键维系其空间结构稳定,每一条多肽链称为亚基。
这种蛋白质分子中各个亚基的空间排布及亚基间的相互作用,称为蛋白质的四级结构。
8.结构域蛋白质的三级结构常可分割成1个和数个球状区域,折叠得较为紧密,各行其能,称为结构域。
9.蛋白质等电点在某一pH溶液中,蛋白质分子所带的正电荷和负电荷相等,净电荷为零,此溶液的pH值,即为该蛋白质的等电点。
生化自找名词解释

核酸分子杂交:两条不同来源的单链DNA,或一条单链DNA,一条RNA,只要它们有大部分互补的碱基顺序,也可以复性,形成一个杂合双链,此过程称杂交。
蛋白质的二级结构(secondary structure):是指蛋白质分子中某一段肽链的局部空间结构,也就是该段肽链主链骨架原子的相对空间位置,并不涉及氨基酸残基侧链的构象。
蛋白质的三级结构(protein tertiary structure):是指整条肽链中全部氨基酸残基的相对空间位置,也就是整条肽链所有原子在三维空间的排布位置。
主要是靠氨基酸侧链之间的疏水相互作用,氢键,范德华力和盐键维持的。
酮体(acetone body):在肝脏中由乙酰CoA合成的燃料分子(β羟基丁酸、乙酰乙酸和丙酮)。
在饥饿期间酮体是包括脑在内的许多组织的燃料,酮体过多将导致中毒。
底物水平磷酸化(substrate phosphorylation):ADP或某些其它的核苷-5ˊ-二磷酸的磷酸化是通过来自一个非核苷酸底物的磷酰基的转移实现的。
这种磷酸化与电子传递链无关。
氧化脱氨(oxidative deamination):α-氨基酸在酶的催化下脱氨生成相应α-酮酸的过程。
氧化脱氨过程实际上包括脱氢和水解两个步骤。
操纵子(operons):是由一个或多个相关基因以及调控它们转录的操纵基因和启动子序列组成的基因表达单位。
等电点(pI,isoelectric point):使分子处于兼性分子状态,在电场中不迁移(分子的净电荷为零)的pH值。
DNA变性(DNA denaturation):DNA双链解链分离成两条单链的现象。
半保留复制(semiconservative replication):DNA复制的一种方式。
每条链都可用作合成互补链的模板,合成出两分子的双链DNA,每个分子都是由一条亲代链和一条新合成的链组成。
同工酶(isoenzyme或isozyme):催化同一化学反应而化学组成不同的一组酶。
蛋白组和蛋白组学

Protein function
Molecular function • What what kind of molecule is this? Biological process • Why why is this being done? Cellular compartment • Where where is this located?
of the same gene (alternative splicing or one more transcriptional initiation sites) • Post-translation modification (phosphorylation, ubiquitination, methylation, acetylation,glycosylation, oxidation nitrosylation ) • The variable, distinct sets of proteins through cell spans
Proteomics, in simple, is to study on a variety of properties of all the proteins in proteomes decoded from its species’ genomes, but it is of difficult…because
knockin • Protein-protein interaction & Circuit via IP, Y2H, FRET • Protein 3D structure (crystals/X-ray & nuclear magnetic
resonance) • Informatics on proteomes • Mimic biological function in a assemble artificial system
昆明理工大学生物工程专业生物化学课程考试各章名词解释

昆明理工大学生物工程专业《生物化学》名词解释(黑体部分为重点)第一章1.氨基酸(amino acid):是含有一个碱性氨基和一个酸性羧基的有机化合物,氨基一般连在α-碳上。
2.必需氨基酸(essential amino acid):指人(或其它脊椎动物)(赖氨酸.苏氨酸等)自己不能合成.需要从食物中获得的氨基酸。
3.非必需氨基酸(nonessential amino acid):指人(或其它脊椎动物)自己能由简单的前体合成不需要从食物中获得的氨基酸。
4.等电点(pI.isoelectric point):使分子处于兼性分子状态.在电场中不迁移(分子的静电荷为零)的pH值。
5.茚三酮反应(ninhydrin reaction):在加热条件下.氨基酸或肽与茚三酮反应生成紫色(与脯氨酸反应生成黄色)化合物的反应。
6.肽键(peptide bond):一个氨基酸的羧基与另一个的氨基的氨基缩合.除去一分子水形成的酰氨键。
7.肽(peptide):两个或两个以上氨基通过肽键共价连接形成的聚合物。
8.蛋白质一级结构(primary structure):指蛋白质中共价连接的氨基酸残基的排列顺序。
9.层析(chromatography):按照在移动相和固定相(可以是气体或液体)之间的分配比例将混合成分分开的技术。
10.离子交换层析(ion-exchange column)使用带有固定的带电基团的聚合树脂或凝胶层析柱11.透析(dialysis):通过小分子经过半透膜扩散到水(或缓冲液)的原理.将小分子与生物大分子分开的一种分离纯化技术。
12.凝胶过滤层析(gel filtration chromatography):也叫做分子排阻层析。
一种利用带孔凝胶珠作基质.按照分子大小分离蛋白质或其它分子混合物的层析技术。
13.亲合层析(affinity chromatograph):利用共价连接有特异配体的层析介质.分离蛋白质混合物中能特异结合配体的目的蛋白质或其它分子的层析技术。
生化:名词解释大全

【生化:名词解释大全】第一章蛋白质1.两性离子(dipolarion)2.必需氨基酸(essential amino acid)3.等电点(isoelectric point,pI)4.稀有氨基酸(rare amino acid)5.非蛋白质氨基酸(nonprotein amino acid) 6.构型(configuration)7.蛋白质的一级结构(protein primary structure)8.构象(conformation)9.蛋白质的二级结构(protein secondary structure)10.结构域(domain)11.蛋白质的三级结构(protein tertiary structure)12.氢键(hydrogen bond)13.蛋白质的四级结构(protein quaternary structure)14.离子键(ionic bond)15.超二级结构(super-secondary structure) 16.疏水键(hydrophobic bond)17.范德华力( van der Waals force) 18.盐析(salting out)19.盐溶(salting in)20.蛋白质的变性(denaturation)21.蛋白质的复性(renaturation)22.蛋白质的沉淀作用(precipitation) 23.凝胶电泳(gel electrophoresis)24.层析(chromatography)第二章核酸1.单核苷酸(mononucleotide)2.磷酸二酯键(phosphodiester bonds)3.不对称比率(dissymmetry ratio)4.碱基互补规律(complementary base pairing)5.反密码子(anticodon)6.顺反子(cistron)7.核酸的变性与复性(denaturation、renaturation)8.退火(annealing)9.增色效应(hyper chromic effect)10.减色效应(hypo chromic effect)11.噬菌体(phage)12.发夹结构(hairpin structure)13.DNA 的熔解温度(melting temperature T m)14.分子杂交(molecular hybridization)15.环化核苷酸(cyclic nucleotide)第三章酶与辅酶1.米氏常数(K m 值)2.底物专一性(substrate specificity)3.辅基(prosthetic group)4.单体酶(monomeric enzyme)5.寡聚酶(oligomeric enzyme)6.多酶体系(multienzyme system)7.激活剂(activator)8.抑制剂(inhibitor inhibiton)9.变构酶(allosteric enzyme)10.同工酶(isozyme)11.诱导酶(induced enzyme)12.酶原(zymogen)13.酶的比活力(enzymatic compare energy)14.活性中心(active center)第四章生物氧化与氧化磷酸化1.生物氧化(biological oxidation)2.呼吸链(respiratory chain)3.氧化磷酸化(oxidative phosphorylation)4.磷氧比P/O(P/O)5.底物水平磷酸化(substrate level phosphorylation)6.能荷(energy charg第五章糖代谢1.糖异生(glycogenolysis)2.Q 酶(Q-enzyme)3.乳酸循环(lactate cycle)4.发酵(fermentation)5.变构调节(allosteric regulation)6.糖酵解途径(glycolytic pathway)7.糖的有氧氧化(aerobic oxidation)8.肝糖原分解(glycogenolysis)9.磷酸戊糖途径(pentose phosphate pathway) 10.D-酶(D-enzyme)11.糖核苷酸(sugar-nucleotide)第六章脂类代谢1.必需脂肪酸(essential fatty acid)2.脂肪酸的α-氧化(α- oxidation)3.脂肪酸的β-氧化(β- oxidation)4.脂肪酸的ω-氧化(ω- oxidation)5.乙醛酸循环(glyoxylate cycle)6.柠檬酸穿梭(citriate shuttle)7.乙酰CoA 羧化酶系(acetyl-CoA carnoxylase)8.脂肪酸合成酶系统(fatty acid synthase system)第八章含氮化合物代谢1.蛋白酶(Proteinase)2.肽酶(Peptidase)3.氮平衡(Nitrogen balance)4.生物固氮(Biological nitrogen fixation)5.硝酸还原作用(Nitrate reduction)6.氨的同化(Incorporation of ammonium ions into organic molecules)7.转氨作用(Transamination)8.尿素循环(Urea cycle)9.生糖氨基酸(Glucogenic amino acid)10.生酮氨基酸(Ketogenic amino acid)11.核酸酶(Nuclease)12.限制性核酸内切酶(Restriction endonuclease)13.氨基蝶呤(Aminopterin)14.一碳单位(One carbon unit)第九章核酸的生物合成1.半保留复制(semiconservative replication)2.不对称转录(asymmetric trancription)3.逆转录(reverse transcription)4.冈崎片段(Okazaki fragment)5.复制叉(replication fork)6.领头链(leading strand)7.随后链(lagging strand)8.有意义链(sense strand)9.光复活(photoreactivation)10.重组修复(recombination repair)11.内含子(intron)12.外显子(exon)13.基因载体(genonic vector)14.质粒(plasmid)第十一章代谢调节1.诱导酶(Inducible enzyme)2.标兵酶(Pacemaker enzyme)3.操纵子(Operon)4.衰减子(Attenuator)5.阻遏物(Repressor)6.辅阻遏物(Corepressor)7.降解物基因活化蛋白(Catabolic gene activator protein)8.腺苷酸环化酶(Adenylate cyclase)9.共价修饰(Covalent modification)10.级联系统(Cascade system)11.反馈抑制(Feedback inhibition)12.交叉调节(Cross regulation)13.前馈激活(Feedforward activation)14.钙调蛋白(Calmodulin)第十二章蛋白质的生物合成1.密码子(codon)2.反义密码子(synonymous codon) 3.反密码子(anticodon)4.变偶假说(wobble hypothesis)5.移码突变(frameshift mutant)6.氨基酸同功受体(isoacceptor)7.反义RNA(antisense RNA)8.信号肽(signal peptide)9.简并密码(degenerate code)10.核糖体(ribosome)11.多核糖体(poly some)12.氨酰基部位(aminoacyl site)13.肽酰基部位(peptidy site)14.肽基转移酶(peptidyl transferase) 15.氨酰- tRNA 合成酶(amino acy-tRNA synthetase)16.蛋白质折叠(protein folding)17.核蛋白体循环(polyribosome) 18.锌指(zine finger)19.亮氨酸拉链(leucine zipper)20.顺式作用元件(cis-acting element) 21.反式作用因子(trans-acting factor) 22.螺旋-环-螺旋(helix-loop-helix)第一章蛋白质1.两性离子:指在同一氨基酸分子上含有等量的正负两种电荷,又称兼性离子或偶极离子。
第四章 蛋白质-蛋白质复合物结构的预测及分析

第四章蛋白质-蛋白质复合物结构的预测及分析(一)一、分子对接原理二、蛋白质复合物结构预测的意义三、蛋白质复合物结构预测的方法四、ZDOCK的使用五、蝎毒素-钾通道的对接六、正在发展的蛋白质对接技术分子对接的原理1、分子对接的理论基础2、分子对接方法的分类3、分子对接方法中的重要问题4、小分子-蛋白质对接1、分子对接的理论基础最初思想---“锁和钥匙”的关系:一把钥匙开一把锁空间形状上要互相匹配分子对接的要求形状的匹配能量的匹配两个重要的原则互补性:空间结构和电学性质的互补性预组织:受体与配体在识别前,将受体中容纳配体的环境组织得愈好,其溶剂化能力愈低,则识别效果愈佳,形成的复合物愈稳定。
分子对接的原理1、分子对接的理论基础2、分子对接方法的分类3、分子对接方法中的重要问题4、小分子-蛋白质对接分子对接方法的分类刚性对接(蛋白质-蛋白质)半柔性对接(小分子柔性)柔性对接(计算时间昂贵)刚性对接研究体系的构象不发生变化,常用于较大的研究体系(蛋白质-蛋白质、蛋白质-核酸)半柔性对接在对接过程中,研究体系尤其是小分子配体的构象允许在一定范围内变化(小分子柔性,大分子刚性。
在药物设计及数据库搜索中常用)柔性对接在对接过程中,研究体系的构象是可以自由变化的(一般用于精确考察分子之间的识别情况。
由于在计算过程中体系的构象是可自由变化的,因此柔性对接过程需要耗费较长的计算时间)。
分子对接的原理1、分子对接的理论基础2、分子对接方法的分类3、分子对接方法中的重要问题4、小分子-蛋白质对接分子对接方法中的重要问题分子对接的目的:配体-受体的最佳结合位置问题:如何找到最佳的结合位置及如何评价对接分子之间的结合强度?问题1:如何找到最佳的结合位置广泛采用优化算法:遗传算法、模拟退火、人工神经网络、付里叶变换等。
问题2:如何评价对接分子之间的结合强度?= △H gas-T△S-△G A solv-△G B solv+ △G AB solv △Gbind△H:分子力学的方法gas△G solv: 准确计算还存在一定的问题T△S:最难的问题(normal-mode analysis---正交模式分析,1955---费时)分子对接的原理一、分子对接的理论基础二、分子对接方法的分类三、分子对接方法中的重要问题四、小分子-蛋白质对接小分子-蛋白质对接Docking SoftwaresDOCK: (Kuntz et al. 1982)-----Widely usedDOCK 4.0 (Ewing & Kuntz 1997)------Widely used AutoDOCK(Goodsell& Olson 1990)AutoDOCK3.0 (Morris et al. 1998)FlexX: (Rarey et al. 1996)GLIDE: (Friesner et al. 2004)CDOCKER (Wu et al. 2003)CombiDOCK(Sun et al. 1998)DIVALI (Clark & Ajay 1995)GEMDOCK (Yang & Chen 2004)约10种程序已商业化优点先导药物的高通量虚拟筛选先导药物的计算机辅助设计与改造药物与靶标相互作用分子机制的阐明一、分子对接原理二、蛋白质复合物结构预测的意义三、蛋白质复合物结构预测的方法四、蛋白质复合物对接研究进展五、ZDOCK的原理与使用六、案例:蝎毒素-钾通道的对接蛋白质组学研究目标人类蛋白质-蛋白质相互作用网络(Cell, 2005)Motivation•Biological activity depends on the specific recognition of proteins.•Understand protein interaction networks in a cell •Yield insight to thermodynamics of molecular recognition•The experimental determination of protein-protein complex structures remains difficult.蛋白质-蛋白质复合物空间结构预测四大优点预测活性区域解释实验现象加速蛋白质结构与功能关系研究阐明分子机制合理实验设计一、分子对接原理二、蛋白质复合物结构预测的意义三、蛋白质复合物结构预测的方法四、蛋白质复合物对接研究进展五、ZDOCK的原理与使用六、案例:蝎毒素-钾通道的对接般流程一、分子对接原理二、蛋白质复合物结构预测的意义三、蛋白质复合物结构预测的方法四、蛋白质复合物对接研究进展五、ZDOCK的原理与使用六、案例:蝎毒素-钾通道的对接重点:蛋白质的柔性希望更多的人从事蛋白质对接方法的应用研究!一、分子对接原理二、蛋白质复合物结构预测的意义三、蛋白质复合物结构预测的方法四、蛋白质复合物对接研究进展五、ZDOCK的原理与使用六、案例:蝎毒素-钾通道的对接Main referencesY C o r r e l a t i o n X1.准备两个蛋白质的空间结构(PDB文件)2.人工删除PDB文件中所有氢原子的坐标(能否编个小程序自动删除?)3. 分别对两个蛋白PDB文件进行处理命令格式:./mark_sur PDB new_PDB电荷半径面积ACE类型ACE: atomic contact energy。
《生物化学》常用名词解释(二)

《生物化学》常用名词解释(二)1.构型(configuration):一个有机分子中各个原子特有的固定的空间排列。
这种排列不经过共价键的断裂和重新形成是不会改变的。
构型的改变往往使分子的光学活性发生变化。
2.构象(conformation):指一个分子中,不改变共价键结构,仅单键周围的原子旋转所产生的原子的空间排布。
一种构象改变为另一种构象时,不要求共价键的断裂和重新形成。
构象改变不会改变分子的光学活性。
3.肽单位(peptideunit):又称之肽基(peptidegroup),是肽链主链上的重复结构。
是由参与肽键形成的氮原子和碳原子和它们的4个取代成分:羰基氧原子、酰胺氢原子和两个相邻的α-碳原子组成的一个平面单位。
4.蛋白质二级结构(proteinsecondarystructure):在蛋白质分子中的局部区域内氨基酸残基的有规则的排列,常见的二级结构有α-螺旋和β-折叠。
二级结构是通过骨架上的羰基和酰胺基团之间形成的氢键维持的。
5.蛋白质三级结构(proteintertiarystructure):蛋白质分子处于它的天然折叠状态的三维构象。
三级结构是在二级结构的基础上进一步盘绕、折叠形成的。
三级结构主要是靠氨基酸侧链之间的疏水相互作用、氢键范德华力和盐键(静电作用力)维持的。
6.蛋白质四级结构(quaternarystructure):多亚基蛋白质的三维结构。
实际上是具有三级结构的多肽链(亚基)以适当方式聚合所呈现出的三维结构。
7.α-螺旋(α-helix):蛋白质中常见的一种二级结构,肽链主链绕假想的中心轴盘绕成螺旋状,一般都是右手螺旋结构,螺旋是靠链内氢键维持的。
每个氨基酸残基(第n个)的羰基氧与多肽链C端方向的第4个残基(第n+4个)的酰胺氮形成氢键。
在典型的右手α-螺旋结构中,螺距为0.54nm,每一圈含有3.6个氨基酸残基,每个残基沿着螺旋的长轴上升0.15nm。
8.β-折叠(β-sheet):是蛋白质中的常见的二级结构,是由伸展的多肽链组成的。
keil rules for miscleavage by trypsin的具体内容 -回复

keil rules for miscleavage by trypsin的具体内容-回复Keil rules for mis-cleavage by trypsin are guidelines that provide insight into the preferential sites where trypsin cleaves proteins. These rules were established by Wolfgang Keil and are widely used to predict the probable cleavage sites within a protein sequence. In this article, we will explore the specific details of Keil rules for mis-cleavage by trypsin and provide a step-by-step explanation.1. Introduction to Trypsin Cleavage:Trypsin is a serine protease that plays a crucial role in protein digestion. It cleaves peptide bonds on the C-terminal side of lysine (K) and arginine (R) residues. This specific cleavage pattern makes trypsin one of the most commonly used proteases in protein research.2. Keil Rules for Mis-Cleavage:- Rule 1: Avoiding Proline (P):Trypsin rarely cleaves peptide bonds adjacent to proline residues. This is primarily because proline has a unique cyclic structure that makes it difficult for trypsin to access the peptide bond.- Rule 2: Cleavage After Lysine (K) or Arginine (R):Trypsin preferentially cleaves peptide bonds after lysine or arginine residues. However, it occasionally mis-cleaves before lysine or arginine residues, which led to the establishment of these rules for mis-cleavage.- Rule 3: Selective Mis-Cleavage at Lysine (K):Mis-cleavage occurs more frequently after lysine residues than arginine residues. This is due to the smaller size and different structure of lysine compared to arginine, making it easier for trypsin to mis-cleave after lysine.- Rule 4: Selective Mis-Cleavage at Arginine (R):Mis-cleavage can occur before arginine residues, predominantly when the following amino acid is glycine (G) or alanine (A). This rule is based on the observation that the shorter side chains of glycine and alanine allow trypsin to access the peptide bond before arginine.- Rule 5: Cleavage is More Probable in Low Molecular Weight Residues:Trypsin favors cleavage in low molecular weight residues, such as alanine and glycine. This preference is likely due to the ease of accessing peptide bonds in smaller amino acids.3. Application of Keil Rules:The Keil rules for mis-cleavage by trypsin have proven to bevaluable in many areas of protein research, including proteomics and protein structure determination.- Peptide Mapping: By applying these rules, researchers can predict the potential cleavage sites of trypsin within a protein sequence. This information is crucial for peptide mapping experiments, which help identify the protein's primary structure.- Proteomics: The Keil rules assist in the analysis of complex protein mixtures in proteomics research. By understanding where trypsin is more likely to mis-cleave, researchers can improve protein identification and quantification.- Protein Structure Determination: Mis-cleavages by trypsin can sometimes hinder protein structure determination. By knowing the preferred sites of mis-cleavage, researchers can design truncated protein constructs that avoid those vulnerable regions. This approach aids the crystallization and subsequent determination ofa protein's three-dimensional structure.4. Limitations and Future Perspectives:While the Keil rules provide valuable insights, it is important to note that trypsin cleavage is influenced by various factors such as pH, temperature, and local protein structure. Additionally, some trypsin variants may differ in their cleavage preferences. Therefore, researchers should consider other experimental factors and consultthe appropriate literature before applying the Keil rules.In conclusion, Keil rules for mis-cleavage by trypsin provide guidelines to predict the probable cleavage sites within a protein sequence. These rules help researchers in peptide mapping, proteomics, and protein structure determination. By understanding the preferential sites where trypsin mis-cleaves, scientists can optimize their experimental design and data analysis, leading to more efficient protein research.。
生物物理学课件讲义——第01讲+蛋白质结构基础

40
嗜热菌蛋白酶与人碳酸酐酶的结构图
41
研究蛋白质三级结构的方法
X射线晶体衍射(X-ray crystallography) 多维核磁共振( multi-dimensional NMR) 三维电子显微镜技术(3-dimensiional EM) 扫描探针显微术( Scanning Probe Microscopy ,SPM)
Tertiary Structure (三级结构) Quaternary Structure (四级结构)
2
3
Primary Structure (一级结构)
4
Side chain
Backbone
Peptide bond
5
Peptide torsion angles (扭转角)
phi (F), psi (Y), and omega (W)
26
Prediction of secondary structure (预测)
(a). Homology. If sequence >25-30%, structure similarity (b). Statistical. Chou & Fasman (1978). (c). Stereochemical Schiffer and Edmundson (1967)
37
Tuberculosis (肺结核) RNA Polymerase active site Hinge region (铰链区)
38
Protein Tertiary Structure (蛋白的三级结构)
实验报告生物大分子的结构测定

实验报告生物大分子的结构测定实验报告实验目的:探究生物大分子的结构测定方法及其应用。
实验方法:1. 准备样品:选择一种生物大分子,如蛋白质,DNA或多糖。
获取样品并进行纯化和浓缩处理。
2. 红外光谱技术:运用红外光谱仪测量样品,记录红外光谱图谱,通过分析谱图中吸收峰的位置和强度,确定样品中化学键的类型和存在的官能团。
3. 核磁共振技术:运用核磁共振仪测量样品,记录核磁共振谱图,通过分析谱图中峰的位置和相对积分峰面积,确定样品中各原子核的数量、化学位移和耦合关系。
4. X射线晶体学方法:通过将样品制备成晶体,进行X射线衍射的测量,得到衍射数据,再利用计算机程序分析,获得样品的三维结构信息。
5. 分子对接方法:通过计算机模拟方法,利用已知的生物大分子结构,预测与之相关的其他小分子结构,以预测其相互作用及模拟药物设计等。
实验结果与讨论:针对不同的生物大分子,实验结果如下:1. 蛋白质结构测定:采用X射线晶体学方法,成功获得了蛋白质的三维结构。
通过分析蛋白质的结构,可以推测其功能和相应的生物活性部位,为药物设计和疾病治疗提供重要依据。
2. DNA结构测定:通过核磁共振技术,确定了DNA双螺旋结构中碱基的化学位移和耦合关系。
这些信息对于理解DNA的复制、转录和修复过程至关重要。
3. 多糖结构测定:利用红外光谱技术,分析多糖样品中特定官能团的吸收峰,确定多糖的组成和结构。
这对于多糖的应用研究和相关领域的发展具有重要意义。
实验结论:生物大分子的结构测定方法多种多样,包括红外光谱技术、核磁共振技术、X射线晶体学方法和分子对接方法等。
不同方法的选择取决于待测样品的性质和研究目的。
通过结构测定,可以更好地理解生物大分子的功能和作用机制,为相关领域的研究与应用提供基础支持。
致谢:感谢实验中的指导老师对本实验的指导和支持。
感谢实验中的团队成员的协助和合作。
此外,还要感谢实验室提供的设备和相关实验条件。
参考文献:1. Smith A, et al. (2018). Protein structure determination using X-ray crystallography. Journal of Molecular Biology, 429(3), 375-386.2. Zhang H, et al. (2017). Nuclear magnetic resonance spectroscopy in protein structure determination. Archives of Biochemistry and Biophysics, 628, 33-46.3. Lu F, et al. (2019). Infrared spectroscopy in carbohydrate research. Carbohydrate Research, 472, 15-27.4. Wang C, et al. (2018). Molecular docking and drug discovery in structure-based virtual screening. Nature Protocols, 13(6), 1332-1347.注意:这是一份实验报告的示例,具体内容和格式要根据实际实验情况进行调整和修改。
蛋白质的红外特征峰

蛋白质的红外特征峰引言:蛋白质是生命体中最基本的分子之一,它们不仅构成了细胞的基本结构,还参与了几乎所有生物学过程。
了解蛋白质的结构和功能对于揭示生命的奥秘至关重要。
而红外光谱技术作为一种非常有效的分析手段,可以提供关于蛋白质结构和组成的重要信息。
本文将重点介绍蛋白质红外光谱中的特征峰及其意义。
一、背景介绍红外光谱是一种分析样品分子振动和转动状态的技术。
在蛋白质红外光谱中,存在一些特征峰,这些峰代表了蛋白质分子内部的振动和转动模式。
通过对这些特征峰的分析,可以得到有关蛋白质的结构和组成的重要信息。
二、α-螺旋结构特征峰α-螺旋是蛋白质中最常见的二级结构之一,具有稳定的氢键和内部氨基酸侧链的排列方式。
在红外光谱中,α-螺旋结构通常表现为两个特征峰:1650 cm^-1和1550 cm^-1。
1650 cm^-1峰对应于α-螺旋中氨基酸C=O的拉伸振动,1550 cm^-1峰则对应于α-螺旋中氨基酸C-N的拉伸振动。
三、β-折叠结构特征峰β-折叠是蛋白质中另一种常见的二级结构,由多肽链的平行或反平行排列形成稳定的氢键。
在红外光谱中,β-折叠结构通常表现为一个特征峰:1630 cm^-1。
这个峰对应于β-折叠中氨基酸C=O的拉伸振动。
四、无规卷曲结构特征峰无规卷曲结构是蛋白质中没有明确二级结构的部分,通常由蛋白质的尾部或连接不规则的氨基酸序列组成。
在红外光谱中,无规卷曲结构通常表现为一个特征峰:1670 cm^-1。
这个峰对应于无规卷曲结构中氨基酸C=O的拉伸振动。
五、胺基酸侧链特征峰蛋白质中的胺基酸侧链对其结构和功能起着重要作用。
在红外光谱中,不同的胺基酸侧链会产生特定的振动峰。
例如,芳香族侧链(如苯丙氨酸和酪氨酸)通常具有一个特征峰在1450 cm^-1附近,甲硫氨酸的侧链则表现为一个特征峰在2560 cm^-1附近。
六、酰胺I和酰胺II特征峰酰胺I和酰胺II是蛋白质中骨架的两个重要部分,它们分别代表了氨基酸的C=O和N-H的振动。
结构生物学技术综述|MicroED、冷冻电镜SPA与X射线晶体学是如何优势互补

结构生物学技术综述|MicroED、冷冻电镜SPA与X射线晶体学是如何优势互补结构生物学是以分子生物学,生物化学和生物物理学为基础的学科,研究对象为生物大分子,如蛋白质和核酸等。
目的是利用生物或物理手段来看清楚生物大分子的精细形态,以阐述生物大分子行驶功能的机制,帮助生物学家更好的认识生命活动的过程。
而且,很多药物分子的作用靶点大多为蛋白质,所以结构生物学在药物研发中起到重要指导作用,为机理研究到药物开发带来清晰的认识。
针对生物大分子中的蛋白质的研究,主要的研究手段有:核磁共振(nuclear magnetic resonance,NMR)、X射线晶体学(X-ray crystallography)和冷冻电子显微镜(cryo-electron microscope, cryo-EM)。
核磁共振主要针对溶液中的、分子量很小(约20kDa)的样品类型,最近几年已使用不多。
近年来,也出现了一些新技术,微晶电子衍射(MicroED)是一种利用冷冻电镜解析微小晶体结构的技术,由于电子束与物质的作用远强于X射线,可以解析X射线晶体学难以处理的纳米晶体结构。
特别是对于以往难以培养单晶的蛋白样品来说,MicroED为结构生物学家提供了一个前景广阔的新工具,弥补现有技术的不足。
该技术被《Science》杂志评为2018年十大突破技术之一。
X射线衍射——最富传统意义的结构解析方式早在1895 年,伦琴(W. C. Roentgen)就发现了X射线。
X射线的发现推动了现代生物学的发展,甚至可以说对整个科学技术领域产生了极为深刻的影响。
1912 年,劳厄(M. V. Laue)与弗里德里希(W. Friedrich)以及伦琴的博士研究生克里平(P. Knipping)利用X 射线对硫酸铜晶体进行衍射实验,并在底片上得到了一些粗大的、椭圆形的斑点,发现了X 射线晶体衍射。
而后,他们又对ZnS、PbS、NaCl 等晶体进行X 射线衍射实验,得到了清晰的四重对称衍射图。
生物化学名词解释大全

学习必备欢迎下载【生物化学:名词解释大全】第一章蛋白质1.两性离子(dipolarion)2.必需氨基酸(essential amino acid)3.等电点(isoelectric point,pI)4.稀有氨基酸(rare amino acid)5.非蛋白质氨基酸(nonprotein amino acid) 6.构型(configuration)7.蛋白质的一级结构(protein primary structure)8.构象(conformation)9.蛋白质的二级结构(protein secondary structure)10.结构域(domain)11.蛋白质的三级结构(protein tertiary structure)12.氢键(hydrogen bond)13.蛋白质的四级结构(protein quaternary structure)14.离子键(ionic bond)15.超二级结构(super-secondary structure) 16.疏水键(hydrophobic bond)17.范德华力( van der Waals force) 18.盐析(salting out)19.盐溶(salting in)20.蛋白质的变性(denaturation)21.蛋白质的复性(renaturation)22.蛋白质的沉淀作用(precipitation) 23.凝胶电泳(gel electrophoresis)24.层析(chromatography)第二章核酸1.单核苷酸(mononucleotide)2.磷酸二酯键(phosphodiester bonds)3.不对称比率(dissymmetry ratio)4.碱基互补规律(complementary base pairing)5.反密码子(anticodon)6.顺反子(cistron)7.核酸的变性与复性(denaturation、renaturation)8.退火(annealing)9.增色效应(hyper chromic effect)10.减色效应(hypo chromic effect)11.噬菌体(phage)12.发夹结构(hairpin structure)13.DNA 的熔解温度(melting temperature T m)14.分子杂交(molecular hybridization)15.环化核苷酸(cyclic nucleotide)第三章酶与辅酶1.米氏常数(K m 值)2.底物专一性(substrate specificity)3.辅基(prosthetic group)4.单体酶(monomeric enzyme)5.寡聚酶(oligomeric enzyme)6.多酶体系(multienzyme system)7.激活剂(activator)8.抑制剂(inhibitor inhibiton)9.变构酶(allosteric enzyme)10.同工酶(isozyme)11.诱导酶(induced enzyme)12.酶原(zymogen)13.酶的比活力(enzymatic compare energy)14.活性中心(active center)第四章生物氧化与氧化磷酸化1.生物氧化(biological oxidation)2.呼吸链(respiratory chain)3.氧化磷酸化(oxidative phosphorylation)4.磷氧比P/O(P/O)5.底物水平磷酸化(substrate level phosphorylation)6.能荷(energy charg第五章糖代谢1.糖异生(glycogenolysis)2.Q 酶(Q-enzyme)3.乳酸循环(lactate cycle)4.发酵(fermentation)5.变构调节(allosteric regulation)6.糖酵解途径(glycolytic pathway)7.糖的有氧氧化(aerobic oxidation)8.肝糖原分解(glycogenolysis)9.磷酸戊糖途径(pentose phosphate pathway) 10.D-酶(D-enzyme)11.糖核苷酸(sugar-nucleotide)第六章脂类代谢1.必需脂肪酸(essential fatty acid)2.脂肪酸的α-氧化(α- oxidation)3.脂肪酸的β-氧化(β- oxidation)4.脂肪酸的ω-氧化(ω- oxidation)5.乙醛酸循环(glyoxylate cycle)6.柠檬酸穿梭(citriate shuttle)7.乙酰CoA 羧化酶系(acetyl-CoA carnoxylase)8.脂肪酸合成酶系统(fatty acid synthase system)第八章含氮化合物代谢1.蛋白酶(Proteinase)2.肽酶(Peptidase)3.氮平衡(Nitrogen balance)4.生物固氮(Biological nitrogen fixation)5.硝酸还原作用(Nitrate reduction)6.氨的同化(Incorporation of ammonium ions into organic molecules)7.转氨作用(Transamination)8.尿素循环(Urea cycle)9.生糖氨基酸(Glucogenic amino acid)10.生酮氨基酸(Ketogenic amino acid)11.核酸酶(Nuclease)12.限制性核酸内切酶(Restriction endonuclease)13.氨基蝶呤(Aminopterin)14.一碳单位(One carbon unit)第九章核酸的生物合成1.半保留复制(semiconservative replication)2.不对称转录(asymmetric trancription)3.逆转录(reverse transcription)4.冈崎片段(Okazaki fragment)5.复制叉(replication fork)6.领头链(leading strand)7.随后链(lagging strand)8.有意义链(sense strand)9.光复活(photoreactivation)10.重组修复(recombination repair)11.内含子(intron)12.外显子(exon)13.基因载体(genonic vector)14.质粒(plasmid)第十一章代谢调节1.诱导酶(Inducible enzyme)2.标兵酶(Pacemaker enzyme)3.操纵子(Operon)4.衰减子(Attenuator)5.阻遏物(Repressor)6.辅阻遏物(Corepressor)7.降解物基因活化蛋白(Catabolic gene activator protein)8.腺苷酸环化酶(Adenylate cyclase)9.共价修饰(Covalent modification)10.级联系统(Cascade system)11.反馈抑制(Feedback inhibition)12.交叉调节(Cross regulation)13.前馈激活(Feedforward activation)14.钙调蛋白(Calmodulin)第十二章蛋白质的生物合成1.密码子(codon)2.反义密码子(synonymous codon) 3.反密码子(anticodon)4.变偶假说(wobble hypothesis)5.移码突变(frameshift mutant)6.氨基酸同功受体(isoacceptor)7.反义RNA(antisense RNA)8.信号肽(signal peptide)9.简并密码(degenerate code)10.核糖体(ribosome)11.多核糖体(poly some)12.氨酰基部位(aminoacyl site)13.肽酰基部位(peptidy site)14.肽基转移酶(peptidyl transferase) 15.氨酰- tRNA 合成酶(amino acy-tRNA synthetase)16.蛋白质折叠(protein folding)17.核蛋白体循环(polyribosome) 18.锌指(zine finger)19.亮氨酸拉链(leucine zipper)20.顺式作用元件(cis-acting element) 21.反式作用因子(trans-acting factor) 22.螺旋-环-螺旋(helix-loop-helix)第一章蛋白质1.两性离子:指在同一氨基酸分子上含有等量的正负两种电荷,又称兼性离子或偶极离子。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Recent advances in the analysis of chemical shifts have enabled their values to be used increasingly successfully to obtain information about a number of specific features of protein conformations, notably dihedral angles (2), in some cases with high accuracy. Such measurements do not, however, enable the high-resolution structures of proteins to be defined without the use of extensive additional information, such as that provided, for example, by NOEs or RDC (2, 23), because even small errors in dihedral angles give rise to progressive error accumulation.
It has been demonstrated recently that strategies in which experimental data are used as restraints in molecular dynamics simulations can lead to a description of the structures of proteins, at least in outline, even in highly heterogeneous states such as those adopted by natively unfolded polypeptide molecules (19, 20). Such techniques have also been shown to be able to describe, simultaneously and with high accuracy, the structures and dynamics of native states of globular proteins by using both distance information (NOEs) and NMR order parameters (21) or RDC (22). In approaches of this type, the experimental information is used essentially to complement standard force fields and to guide the sampling of conformational space toward regions consistent with experimental observations related to the specific state under investigation (19). In a similar spirit, we describe here how NMR chemical shifts can be used to define the structures of the native states of proteins at high resolution without the requirement of any additional experimental measurements.
NMR spectroscopy ͉ structural biology
C hemical shifts are exquisitely sensitive probes of molecular structure (1–4). Indeed, it is this characteristic that is the origin of their unique value in probing in atomic detail the properties of systems ranging from simple organic and inorganic compounds to complex biological macromolecules, because it enables the resolution of distinct signals from even chemically identical groups when located in different local or global environments. In structural biology, chemical shifts are most often used to predict regions of secondary structure in native and nonnative states of proteins (2, 5), to aid in the refinement of complex structures (6), and for the characterization of conformational changes associated with partial unfolding (7) or binding (8, 9). It has also been recognized that chemical shifts can aid in the determination of the tertiary structure of proteins when used in combination with other NMR probes that report on interproton distances (NOEs) and the relative orientations of the different nuclei in a protein structure [residual dipolar couplings (RDC)] (3, 6, 10). In many important cases, however, chemical shifts are the only NMR parameters that can be obtained on a given state of a protein with any degree of completeness (7, 8, 11–15), prompting us to explore the extent to which these quantities alone can be used to determine high-resolution structures.
Protein structure determination from NMR chemical shifts
Andrea Cavalli, Xavier Salvatella, Christopher M. Dobson, and Michele Vendruscolo*
Department of Chemistry, Cambridge University, Cambridge CB2 1EW, United Kingdom
Edited by David Baker, University of Washington, Seattle, WA, and approved April 23, 2007 (received for review November 21, 2006)
NMR spectroscopy plays a major role in the determination of the structures and dynamics of proteins and other biological macromolecules. Chemical shifts are the most readily and accurately measurable NMR parameters, and they reflect with great specificity the conformations of native and nonnative states of proteins. We show, using 11 examples of proteins representative of the major structural classes and containing up to 123 residues, that it is possible to use chemical shifts as structural restraints in combination with a conventional molecular mechanics force field to determine the conformations of proteins at a resolution of 2 Å or better. This strategy should be widely applicable and, subject to further development, will enable quantitative structural analysis to be carried out to address a range of complex ical problems not accessible to current structural techniques.
The unique fingerprints of proteins provided by their NMR spectra suggest that chemical shifts inherently carry sufficient information to determine their structures at high resolution, as indeed is often the case for molecules of low molecular weight (4). The structural information contained in the chemical shifts is, however, very different in nature from that provided by NOEs, because the latter report on pairwise distances between specific protons and can thus provide unequivocal information about the relative spatial locations of different residues in a protein sequence (1). The chemical shift associated with a specific atom, by contrast, is a summation of many contributing factors (16–18) so that the reliable identification of interaction partners is very difficult, even though they may be substantially influenced by contacts between residues, such as hydrogen bonding and proximity to aromatic rings, that are at very different locations in the protein sequence. If such effects could be interpreted in depth, therefore, they would enable the char-