General Protein Handling Guideline 之冻融蛋白的基本策略
《中国药典》2020—精蛋白重组人胰岛素混合注射液(50R)国家标准公示稿

精蛋白重组人胰岛素混合注射液(50R) Jingdanbai Chongzu Ren Yidaosu Hunhe Zhusheye (50R) Mixed Protamine Recombinant Human Insulin Injection (50R) 本品为常规重组人胰岛素与精蛋白重组人胰岛素在灌装前混合而成的预混型制剂,其中常规重组人胰岛素占 50%,精蛋白重组人胰岛素占 50%。
1基本要求
生产和检定用设施、原辅料、水、器具等应符合“凡例”的相关要求。
2制造
2.1原料
应符合“重组人胰岛素项下”的规定。
2.2半成品
2.2.1配制与除菌
按照经批准的配方进行稀释、配制,除菌过滤后即为半成品,保存于适宜的温度。
2.2.2半成品检定
按3.1 项进行。
2.3成品
2.3.1分批
应符合“生物制品分批规程”规定。
2.3.2分装
应符合“生物制品分装和冻干规程”及通则 0102 有关规定。
2.3.3规格
3ml:300 单位
2.3.4包装
应符合“生物制品包装规程”及通则 0102 有关规定。
3检定
3.1半成品检定。
GENMED 柠檬酸化学法冰冻组织抗原修复处理试剂盒 产品说明书(中文版)

GENMED SCIENTIFICS INC. U.S.A GMS40039.3 v.A GENMED柠檬酸化学法冰冻组织抗原修复处理试剂盒产品说明书(中文版)主要用途GENMED柠檬酸化学法冰冻组织抗原修复处理试剂是一种旨在通过柠檬酸缓冲系统,辅以物理加热的强化处理,进行醛类固定的冰冻组织块的抗原修复和暴露,增强免疫化学的染色强度,降低背景噪音的权威而经典的技术方法。
该技术由大师级科学家精心改良、成功实验证明的。
主要适用于醛类处理的冰冻动物组织块的各种抗原的修复,尤其是核蛋白的修复。
产品即到即用,操作简捷,性能稳定,修复显著,敏感可靠。
技术背景组织制片固定时,使用醛类固定剂(例如福尔马林等),会导致醛类分子与组织蛋白产生分子交联,遮蔽了组织抗原。
其原理是在组织蛋白的反应部位(胺、氨类基团、巯基、氢氧基团、芳香环等)形成了亚甲基桥(methylene bridges)。
通过物理热处理和化学处理方法,破坏醛类分子交联,暴露和修复抗原。
产品内容GENMED清理液(Reagent A)XX毫升GENMED固着液(Reagent A)XX毫升GENMED修复液(Reagent B)XX毫升GENMED保护液(Reagent B)XX毫升产品说明书1份保存方式保存在4℃冰箱里,有效保证6月用户自备小型染色缸:用于清理的容器200毫升烧杯:用于修复操作的容器实验步骤1.准备1个6孔细胞培养板2.放进新鲜切除的或冰冻保存的动物组织样本(注意:建议组织大小为0.5厘米厚,1厘米长;如果过大,最好刀切处理一下)3.将组织压平4.小心加入XX毫升GENMED清理液(Reagent A),清洗组织样本5.小心抽去清理液6.小心加入XX毫升GENMED固定液(Reagent B),浸没整个组织7.在4℃条件下孵育2小时8.小心抽去固定液9.小心加入XX毫升GENMED清理液(Reagent A),清洗组织样本10.小心抽去清理液11.小心加入XX毫升GENMED修复液(Reagent C),浸没整个组织12.在4℃条件下孵育16小时13.准备一个200毫升的烧杯14.加入XX毫升GENMED修复液(Reagent C)15.煮沸16.用镊子取出组织,放入到煮沸的GENMED修复液(Reagent C)中17.孵育5分钟18.用镊子取出组织,放入到预冷的XX毫升GENMED保护液(Reagent D)里,浸没整个组织19.在4℃条件下孵育,直至组织块沉入底部20.即刻包埋和冰冻21.放进-80℃冰箱保存22.进行冰冻切片23.继续后续免疫化学染色操作注意事项1.本产品为20次操作2.操作时,须戴手套3.操作时,避免污染母液,尤其是GENMED保护液(Reagent D)4.本公司提供系列组织抗原修复试剂产品质量标准1.本产品经鉴定性能稳定2.本产品经鉴定修复效果显著使用承诺杰美基因秉着“信誉至上、客户满意、质量承诺”的宗旨为我们的用户提供优质产品和服务。
Rubella IgG作业指导书

检验科免疫室分析项目作业指导书风疹IgG (Rubella IgG)第页,共页版本:A/0生效日期:2008-02-0135根据要求进行标定:如质控结果超出范围时;更换某些试剂时,根据规定进行多次标定。
5.操作步骤参见仪器标准操作规程。
6.质量控制用质控品1和质控品2,至少每24小时或每一次定标后测定一次。
质控间隔期应适用于各实验室的具体要求。
检测值应落在确定的范围内,如出现质控值落在范围以外,应采取校正措施。
7.计算方法对每一个标本,仪器会自动计算含量。
单位是IU/mL.8.结果解释阴性< 10 IU/mL阳性≥10 IU/mL9.分析性能检测范围:0.17-500 IU/mL精密度:以下列出了此试剂盒在Elecsys仪器上的表现。
每个实验室结果会不同。
重复性测定采用的是美国临床化学实验室标准委员会修订稿EP5-A标准方法进行评估,使用了Elecsys试剂及人血清池、质控进行测定:连续测定10天,每天测定6次(n=60次);E170的批内重复性共测定了21次。
结果如下:检验科免疫室分析项目作业指导书风疹IgG (Rubella IgG)第页,共页版本:A/0生效日期:2008-02-014510.干扰因素该方法不受黄疸(胆红素<30mg/dl)、溶血(血红蛋白<5.6 g/dl)、脂血(脂质<2000mg/dl)生物素<60ng/ml 等干扰,接受高剂量生物素(>5mg/天)治疗的病人,至少要等最后一次摄入生物素8小时后才能采血。
不受类风湿因子(1500U/ml)和透析的干扰。
异常病人应结合病史、临床其他检查结果综合起来进行诊断。
11.临床意义风疹病毒是德国麻疹的病原体,德国麻疹是一种通常发生在儿童时期的普通轻微皮疹。
但是,孕妇特别是妊娠早期的最初三个月的感染风疹则是一种严重疾病,风疹病毒可以通过胎盘,导致胎儿死亡或严重残障,通常称之为先天性风疹综合征(CRS),可引起失明,耳聋,先天性心脏病和智力发育迟缓。
蛋白质纯化手册【ProteinPurificationHandbook】

Protein PurificationHandbook18-1132-29Edition ABHiTrap, Sepharose, STREAMLINE, Sephadex, MonoBeads, Mono Q,Mono S, MiniBeads, RESOURCE, SOURCE, Superdex, Superose, HisTrap, HiLoad, HiPrep, INdEX, BPG, BioProcess, FineLINE, MabTrap, MAbAssistant, Multiphor, FPLC, PhastSystem and ÄKTA are trademarks of Amersham Pharmacia Biotech Limitedor its subsidiaries.Amersham is a trademark of Nycomed Amersham plcPharmacia and Drop Design are trademarks of Pharmacia & Upjohn Inc Coomassie is a trademark of ICI plcAll goods and services are sold subject to the terms and conditions of sale of the company within the Amersham Pharmacia Biotech group which supplies them. A copy of these terms and conditions of sale is available on request.© Amersham Pharmacia Biotech AB 1999-All rights reserved.Amersham Pharmacia Biotech ABSE-751 84 Uppsala SwedenAmersham Pharmacia Biotech UK Limited Amersham Place Little Chalfont Buckinghamshire England HP7 9NA Amersham Pharmacia Biotech Inc800 Centennial Avenue PO Box 1327 Piscataway NJ 08855 USAProtein Purification HandbookContents Introduction (7)Chapter 1Purification Strategies - A Simple Approach (9)Preparation (10)Three Phase Purification Strategy (10)General Guidelines for Protein Purification (12)Chapter 2 Preparation (13)Before You Start (13)Sample Extraction and Clarification (16)Chapter 3Three Phase Purification Strategy (19)Principles (19)Selection and Combination of Purification Techniques (20)Sample Conditioning (26)Chapter 4Capture (29)Chapter 5Intermediate Purification (37)Chapter 6Polishing (40)Chapter 7Examples of Protein Purification Strategies (45)Three step purification of a recombinant enzyme (45)Three step purification of a recombinant antigen binding fragment (49)Two step purification of a monoclonal antibody (54)One step purification of an integral membrane protein (57)Chapter 8Storage Conditions (61)Extraction and Clarification Procedures (62)Chapter 9Principles and Standard Conditions for Purification Techniques (73)Ion exchange (IEX) (73)Hydrophobic interaction (HIC) (79)Affinity (AC) (85)Gel filtration (GF) (88)Reversed phase (RPC) (92)Expanded bed adsorption (EBA) (95)IntroductionThe development of techniques and methods for protein purification has been an essential pre-requisite for many of the advancements made in biotechnology. This booklet provides advice and examples for a smooth path to protein purification. Protein purification varies from simple one-step precipitation procedures to large scale validated production processes. Often more than one purification step is necessary to reach the desired purity. The key to successful and efficient protein purification is to select the most appropriate techniques, optimise their performance to suit the requirements and combine them in a logical way to maximise yield and minimise the number of steps required.Most purification schemes involve some form of chromatography. As a result chromatography has become an essential tool in every laboratory where protein purification is needed. The availability of different chromatography techniques with different selectivities provides a powerful combination for the purification of any biomolecule.Recombinant DNA developments over the past decade have revolutionised the production of proteins in large quantities. Proteins can even be produced in forms which facilitate their subsequent chromatographic purification. However, this has not removed all challenges. Host contaminants are still present and problems related to solubility, structural integrity and biological activity can still exist. Although there may appear to be a great number of parameters to consider, with a few simple guidelines and application of the Three Phase Purification Strategy the process can be planned and performed simply and easily, with only a basic knowledge of the details of chromatography techniques.78Chapter 1Purification Strategies- a simple approachApply a systematic approach to development of a purification strategy. The first step is to describe the basic scenario for the purification. General considerations answer questions such as: What is the intended use of the product? What kind of starting material is available and how should it be handled? What are the purity issues in relation to the source material and intended use of the final product? What has to be removed? What must be removed completely? What will be the final scale of purification? If there is a need for scale-up, what consequences will this have on the chosen purification techniques? What are the economical constraints and what resources and equipment are available?Most purification protocols require more than one step to achieve the desired level of product purity. This includes any conditioning steps necessary to transfer the product from one technique into conditions suitable to perform the next technique. Each step in the process will cause some loss of product. For example, if a yield of 80% in each step is assumed, this will be reduced to only 20% overall yield after 8 processing steps as shown in Figure 1. Consequently, to reach the targets for yield and purity with the minimum number of steps and the simplest possible design, it is not efficient to add one step to another until purity requirements have been fulfilled. Occasionally when a sample is readily available purity can be achieved by simply adding or repeating steps. However, experience shows that, even for the most challenging applications, high purity and yield can be achieved efficiently in fewer than four well-chosen and optimised purification steps. Techniques should be organised in a logical sequence to avoid the need for conditioning steps and the chromatographic techniques selected appropriately to use as few purification steps as possible.Limit the number of steps in a purification procedure910Fig.1.Yields from multi-step purifications.PreparationThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to every purification. It is important to set objectives for purity,quantity and maintenance of biological activity and to define the economical and time framework for the work. All information concerning properties of the target protein and contaminants will help during purification development. Some simple experiments to characterise the sample and target molecule are an excellent investment. Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess its effectiveness. Sample preparation and extraction procedures should be developed prior to the first chromatographic purification step.With background information, assays and sample preparation procedures in place the Three Phase Purification Strategy can be considered.Three Phase Purification Strategy Imagine the purification has three phases Capture, IntermediatePurification and Polishing.In the Three Phase Strategy specific objectives are assigned to each step within the process:In the capture phase the objectives are to isolate, concentrate and stabilise the target product.During the intermediate purification phase the objective is to remove most of the bulk impurities such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase the objective is to achieve high purity by removing any remaining trace impurities or closely related substances.The selection and optimum combination of purification techniques for Capture,Intermediate Purification and Polishing is crucial to ensure fast method development, a shorter time to pure product and good economy.108060402012345678Number of steps 95% / step90% / step 85% / step 80% / step 75% / stepYield (%)The final purification process should ideally consist of sample preparation, including extraction and clarification when required, followed by three major purification steps, as shown in Figure 2. The number of steps used will always depend upon the purity requirements and intended use for the protein.Fig. 2.Preparation and the Three Phase Purification Strategy11Guidelines for Protein PurificationThe guidelines for protein purification shown here can be applied to any purification process and are a suggestion as to how a systematic approach can be applied to the development of an effective purification strategy. As a reminder these guidelines will be highlighted where appropriate throughout the following chapters.Define objectivesfor purity, activity and quantity required of final product to avoid over or under developing a methodDefine properties of target protein and critical impuritiesto simplify technique selection and optimisationDevelop analytical assaysfor fast detection of protein activity/recovery and to work efficientlyMinimise sample handling at every stageto avoid lengthy procedures which risk losing activity/reducing recovery Minimise use of additivesadditives may need to be removed in an extra purification step or may interfere with activity assaysRemove damaging contaminants earlyfor example, proteasesUse a different technique at each stepto take advantage of sample characteristics which can be used for separation (size, charge, hydrophobicity, ligand specificity)Minimise number of stepsextra steps reduce yield and increase time, combine steps logicallyKEEP IT SIMPLE!12Chapter 2PreparationBefore You StartThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to any purification, from preparation of an enriched protein extract for biochemical characterisation to large scale production of a therapeutic recombinant protein. It is important to set objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time. Purity requirements must take into consideration the nature of the source material, the intended use of the final product and any special safety issues. For example, it is important to differentiate between contaminants which must be removed and those which can be tolerated. Other factors can also influence the prioritisation of objectives. High yields are usually a key objective, but may be less crucial in cases where a sample is readily available or product is required only in small quantities. Extensive method development may be impossible without resources such as an ÄKTA™design chromatography system. Similarly, time pressure combined with a slow assay turnaround will steer towards less extensive scouting and optimisation. All information concerning properties of the target protein and contaminants will help during purification development, allowing faster and easier technique selection and optimisation, and avoiding conditions which may inactivate the target protein.Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess effectiveness (yield, biological activity, recovery).Define objectivesGoal:To set minimum objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time.Define purity requirements according to the final use of the product. Purity requirement examples are shown below.Extremely high > 99%Therapeutic use, in vivo studiesHigh 95- 99 %X-ray crystallography and most physico-chemicalcharacterisation methodsModerate < 95 %Antigen for antibody productionN-terminal sequencing13Identify 'key' contaminantsIdentify the nature of possible remaining contaminants as soon aspossible.The statement that a protein is >95% pure (i.e. target protein constitutes 95% of total protein) is far from a guarantee that the purity is sufficient for an intended application. The same is true for the common statement "the protein was homogenous by Coomassie™ stained SDS-PAGE". Purity of 95% may be acceptable if the remaining 5% consists of harmless impurities. However, even minor impurities which may be biologically active could cause significant problems in both research and therapeutic applications. It is therefore important to differentiate between contaminants which must be removed completely and those which can be reduced to acceptable levels. Since different types of starting material will contain different contaminant profiles they will present different contamination problems.It is better to over-purify than to under-purify.Although the number of purification steps should be minimised, thequality of the end product should not be compromised. Subsequent results might be questioned if sample purity is low and contaminants are unknown.Contaminants which degrade or inactivate the protein or interfere withanalyses should be removed as early as possible.The need to maintain biological activity must be considered at every stage during purification development. It is especially beneficial if proteases are removed and target protein transferred into a friendly environment during the first step.Economy is a very complex issue. In commercial production the time to market can override issues such as optimisation for recovery, capacity or speed. Robustness and reliability are also of great concern since a batch failure can have major consequences.It may be necessary to use analytical techniques targetted towards specific conta-minants in order to demonstrate that they have been removed to acceptable levels. 14Define properties of target protein and critical impurities Goal:To determine a 'stability window' for the target protein for easier selection and optimisation of techniques and to avoid protein inactivation during purification.Check target protein stability window for at least pH and ionic strength. All information concerning the target protein and contaminant properties will help to guide the choice of separation techniques and experimental conditions for purification. Database information for the target, or related proteins, may give size, isoelectric point (pI) and hydrophobicity or solubility data. Native one and two dimensional PAGE can indicate sample complexity and the properties of the target protein and major contaminants. Particularly important is a knowledge of the stability window of the protein so that irreversible inactivation is avoided. Itis advisable to check the target protein stability window for at least pH and ionic strength. Table 1 shows how different target protein properties can affect a purification strategy.Table 1.Protein properties and their effect on development of purification strategies. Sample and target protein properties Influence on purification strategyTemperature stability Need to work rapidly at lowered temperaturepH stability Selection of buffers for extraction and purificationSelection of conditions for ion exchange, affinity orreversed phase chromatographyOrganic solvents stability Selection of conditions for reversed phasechromatographyDetergent requirement Consider effects on chromatographic steps and the needfor detergent removal. Consider choice of detergent.Salt (ionic strength)Selection of conditions for precipitation techniques andhydrophobic interaction chromatographyCo-factors for stability or activity Selection of additives, pH, salts, buffersProtease sensitivity Need for fast removal of proteases or addition ofinhibitorsSensitivity to metal ions Need to add EDTA or EGTA in buffersRedox sensitivity Need to add reducing agentsMolecular weight Selection of gel filtration mediaCharge properties Selection of ion exchange conditionsBiospecific affinity Selection of ligand for affinity mediumPost translational modifications Selection of group specific affinity medium Hydrophobicity Selection of medium for hydrophobic interactionchromatography15Develop analytical assaysGoal:To follow the progress of a purification, to assess effectiveness (yield, biological activity, recovery) and to help during optimisation.Select assays which are fast and reliable.To progress efficiently during method development the effectiveness of each step should be assessed. The laboratory should have access to the following assays:• A rapid, reliable assay for the target protein• Purity determination• Total protein determination• Assays for impurities which must be removedThe importance of a reliable assay for the target protein cannot be over- emphasised. When testing chromatographic fractions ensure that the buffers used for separation do not interfere with the assay. Purity of the target protein is most often estimated by SDS-PAGE, capillary electrophoresis, reversed phase chromatography or mass spectrometry. Lowry or Bradford assays are used most frequently to determine the total protein.The Bradford assay is particularly suited to samples where there is a high lipid content which may interfere with the Lowry assay.For large scale protein purification the need to assay for target proteins and critical impurities is often essential. In practice, when a protein is purified for research purposes, it is too time consuming to identify and set up specific assays for harmful contaminants. A practical approach is to purify the protein to a certain level, and then perform SDS-PAGE after a storage period to check for protease cleavage. Suitable control experiments, included within assays forbio-activity, will help to indicate if impurities are interfering with results.Sample Extraction and Clarification Minimise sample handlingMinimise use of additivesRemove damaging contaminants earlyDefinition:Primary isolation of target protein from source material.Goal:Preparation of a clarified sample for further purification. Removal of particulate matter or other contaminants which are not compatible with chromatography.16The need for sample preparation prior to the first chromatographic step is dependent upon sample type. In some situations samples may be taken directly to the first capture step. For example cell culture supernatant can be applied directly to a suitable chromatographic matrix such as Sepharose™ Fast Flow and may require only a minor adjustment of the pH or ionic strength. However, it is most often essential to perform some form of sample extraction and clarification procedure.If sample extraction is required the chosen technique must be robust and suitable for all scales of purification likely to be used. It should be noted that a technique such as ammonium sulphate precipitation, commonly used in small scale, may be unsuitable for very large scale preparation. Choice of buffers and additives must be carefully considered if a purification is to be scaled up. In these cases inexpensive buffers, such as acetate or citrate, are preferable to the more complex compositions used in the laboratory. It should also be noted that dialysis and other common methods used for adjustment of sample conditions are unsuitable for very large or very small samples.For repeated purification, use an extraction and clarification techniquethat is robust and able to handle sample variability. This ensures areproducible product for the next purification step despite variability instarting material.Use additives only if essential for stabilisation of product or improvedextraction. Select those which are easily removed. Additives may need tobe removed in an extra purification step.Use pre-packed columns of Sephadex™ G-25 gel filtration media, forrapid sample clean-up at laboratory scale, as shown in Table 2.Table 2.Pre-packed columns for sample clean-up.Pre-packed column Sample volume Sample volume Code No.loading per run recovery per runHiPrep™Desalting 26/10 2.5 -15 ml7.5 - 20 ml17-5087-01HiTrap Desalting0.25 - 1.5 ml 1.0 - 2.0 ml17-1408-01Fast Desalting PC 3.2/100.05 - 0.2 ml0.2 - 0.3 ml17-0774-01PD-10 Desalting 1.5 - 2.5 ml 2.5 - 3.5 ml17-0851-01 Sephadex G-25 gel filtration media are used at laboratory and production scale for sample preparation and clarification of proteins >5000. Sample volumes of up to 30%, or in some cases, 40% of the total column volume are loaded. In a single step, the sample is desalted, exchanged into a new buffer, and low molecular weight materials are removed. The high volume capacity, relative insensitivity to sample concentration, and speed of this step enable very large sample volumes to be processed rapidly and efficiently. Using a high sample volume load results in a separation with minimal sample dilution (approximately 1:1.4). Chapter 8 contains further details on sample storage, extraction and clarification procedures.17Sephadex G-25 is also used for sample conditioning i.e. rapid adjustment of pH, buffer exchange and desalting between purification steps.Sephadex G-25 gel filtrationFor fast group separations between high and low molecular weight substances Typical flow velocity 60 cm/h (Sephadex G-25 SuperFine, Sephadex G-25 Fine), 150 cm/h (Sephadex G-25 Medium).If large sample volumes will be handled or the method scaled-up in the future, consider using STREAMLINE™ expanded bed adsorption. This technique is particularly suited for large scale recombinant protein and monoclonal antibody purification. The crude sample containing particles can be applied to the expanded bed without filtration or centrifugation. STREAMLINE adsorbents are specially designed for use in STREAMLINE columns. Together they enable the high flow rates needed for high productivity in industrial applications of fluidised beds. The technique requires no sample clean up and so combines sample preparation and capture in a single step. Crude sample is applied to an expanded bed STREAMLINE media. Target proteins are captured whilst cell debris, cells, particulate matter, whole cells, and contaminants pass through. Flow is reversed and the target proteins are desorbed in the elution buffer.STREAMLINE (IEX, AC, HIC)For sample clean-up and capture direct from crude sample.STREAMLINE adsorbents are designed to handle feed directly from both fermentation homogenate and crude feedstock from cell culture/fermentation at flow velocities of 200 - 500 cm/h, according to type and application.Particle size: 200 µmNote:cm/h: flow velocity (linear flow rate) = volumetric flow rate/cross sectional area of column.18Chapter 3Three Phase Purification StrategyPrinciplesWith background information, assays, and sample preparation and extraction procedures in place the Three Phase Purification Strategy can be applied (Figure 3). This strategy is used as an aid to the development of purification processes for therapeutic proteins in the pharmaceutical industry and is equally efficient as an aid when developing purification schemes in the research laboratory.Fig. 3.Preparation and the Three Phase Purification Strategy.Assign a specific objective to each step within the purification process.In the Three Phase Strategy a specific objective is assigned to each step. The purification problem associated with a particular step will depend greatly upon the properties of the starting material. Thus, the objective of a purification step will vary according to its position in the process i.e. at the beginning for isolation of product from crude sample, in the middle for further purification of partially purified sample, or at the end for final clean up of an almost pure product.The Three Phase Strategy ensures faster method development, a shorter time to pure product and good economy.In the capture phase the objectives are to isolate, concentrate and stabilise the target product. The product should be concentrated and transferred to an environment which will conserve potency/activity. At best, significant removal of other critical contaminants can also be achieved.19During the intermediate purification phase the objectives are to remove most of the bulk impurities,such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase most impurities have already been removed except for trace amounts or closely related substances. The objective is to achieve final purity.It should be noted that this Three Phase Strategy does not mean that all strategies must have three purification steps. For example, capture and intermediate purification may be achievable in a single step, as may intermediate purification and polishing. Similarly, purity demands may be so low that a rapid capture step is sufficient to achieve the desired result, or the purity of the starting material may be so high that only a polishing step is needed. For purification of therapeutic proteins a fourth or fifth purification step may be required to fulfil the highest purity and safety demands.The optimum selection and combination of purification techniques for Capture, Intermediate Purification and Polishing is crucial for an efficient purification process.Selection and Combination ofPurification TechniquesMinimise sample handlingMinimise number of stepsUse different techniques at each stepGoal:Fastest route to a product of required purity.For any chromatographic separation each different technique will offer different performance with respect to recovery, resolution, speed and capacity. A technique can be optimised to focus on one of these parameters, for example resolution, or to achieve the best balance between two parameters, such as speed and capacity.A separation optimised for one of these parameters will produce results quite different in appearance from those produced using the same technique, but focussed on an alternative parameter. See, for example, the results shown on page 49 where ion exchange is used for a capture and for a polishing step.20Select a technique to meet the objectives for the purification step. Capacity,in the simple model shown, refers to the amount of target protein loaded during purification. In some cases the amount of sample which can be loaded may be limited by volume (as in gel filtration) or by large amounts of contaminants rather than the amount of the target protein.Speed is of the highest importance at the beginning of a purification where contaminants such as proteases must be removed as quickly as possible. Recovery becomes increasingly important as the purification proceeds because of the increased value of the purified product. Recovery is influenced by destructive processes in the sample and unfavourable conditions on the column. Resolution is achieved by the selectivity of the technique and the efficiency of the chromatographic matrix to produce narrow peaks. In general, resolution is most difficult to achieve in the final stages of purification when impurities and target protein are likely to have very similar properties.Every technique offers a balance between resolution, speed, capacity and recovery and should be selected to meet the objectives for each purification step. In general, optimisation of any one of these four parameters can only be achieved at the expense of the others and a purification step will be a compromise. The importance of each parameter will vary depending on whether a purification step is used for capture, intermediate purification or polishing. This will steer the optimisation of the critical parameters, as well as the selection of the most suitable media for the step.Proteins are purified using chromatographic purification techniques which separate according to differences in specific properties, as shown in Table 3. Table 3.Protein properties used during purification.Protein property TechniqueCharge Ion exchange (IEX)Size Gel filtration (GF)Hydrophobicity Hydrophobic interaction (HIC),reversed phase (RPC)Biorecognition (ligand specificity)Affinity (AC)Charge, ligand specificity or hydrophobicity Expanded bed adsorption (EBA) follows theprinciples of AC, IEX or HIC21。
单克隆抗体纯化工艺流程

单克隆抗体纯化工艺流程英文回答:Single clone antibody purification process involves several steps to obtain a highly pure and concentrated antibody sample. Here, I will describe the general process in detail.1. Harvesting the cells: The first step in the purification process is to harvest the cells that produce the desired antibody. This can be done by culturing the cells in a suitable growth medium until they reach the desired density. Then, the cells are harvested by centrifugation or filtration.2. Cell lysis: Once the cells are harvested, they need to be lysed to release the intracellular components, including the antibody. This can be achieved by using detergents, sonication, or freeze-thaw cycles. The lysate is then clarified by centrifugation to remove cell debris.3. Affinity chromatography: The next step is to purify the antibody using affinity chromatography. This involves the use of a specific ligand that binds to the antibody with high affinity. For example, protein A or protein G can be used as ligands for purifying antibodies of different classes or species. The lysate is passed through a column containing the ligand, and the antibody binds to the ligand while other impurities are washed away. The bound antibody is then eluted using a low pH buffer or a competitive elution agent.4. Size exclusion chromatography: After affinity chromatography, the antibody sample may still contain some impurities such as aggregates or fragments. Size exclusion chromatography is used to separate these impurities based on their size. A gel filtration column is employed, and the antibody elutes in a separate peak while the impurities are excluded from the gel matrix.5. Concentration and buffer exchange: The purified antibody is typically in a low concentration and may be ina buffer that is not compatible with downstream applications. Therefore, concentration and buffer exchange steps are performed. This can be achieved by using centrifugal filter units or ultrafiltration devices. The concentrated antibody is then exchanged into a suitable buffer using dialysis or buffer exchange columns.6. Sterile filtration: To ensure the antibody is free from any microbial contamination, sterile filtration is performed. The purified antibody is passed through a sterilizing-grade filter with a pore size of 0.2 μm or smaller. This step is crucial for the final product to be used in therapeutic or diagnostic applications.7. Quality control: Finally, the purified antibody sample undergoes rigorous quality control testing to ensure its purity, potency, and stability. This includes assessing its binding affinity, specificity, and functionality. The sample is also tested for endotoxin levels and checked for any degradation or aggregation.中文回答:单克隆抗体纯化工艺流程涉及多个步骤,以获得高纯度和高浓度的抗体样品。
细胞因子溶解

我们知道,PeproTech的所有细胞因子和蛋白均为冻干粉,这使得运输非常便捷,只要常温即可。
而且,细胞因子和蛋白冻干粉非常稳定,在-20o C或-80o C条件下可保存数年。
冻干粉在使用前需进行溶解,然后以液体形式加到培养体系或注射入动物体内。
溶解步骤非常关键,因溶解不好会导致细胞因子或蛋白的失活,这也是很多用户在实际使用中经常遇到的问题。
那么,应该如何进行正确的溶解呢?下面我们以Recombinant Human IL-4 (重组人IL-4,产品编号:200-04)的说明书为例,对细胞因子或蛋白的溶解方法进行详细的阐述。
拿到重组人IL-4的说明书后,您会发现有一段关于Reconstitution(重悬)的叙述,这段内容含有溶解相关的所有信息。
1. Centrifuge the vial prior to opening第1步:开盖前离心试剂管PeproT ech的细胞因子或蛋白冻干粉装盛在塑料管中,为无菌包装。
冻干粉在运输过程中可能会因颠簸而漂散并粘贴于管壁或管盖上,所以在打开塑料瓶盖前,需将冻干粉通过离心收集到管底,以便用很小体积的液体即可将冻干粉完全溶解。
有很多用户会问一个问题,即应该用多少转速、多长时间离心试剂管,才能达到良好的收集效果?答:有些小型高速离心机(多为进口品牌)的面板上有一个Spin键,按了此键后,离心机会自动快速上升到其最大速度(10000rpm或12000rpm),上升到最高点后速度即刻下降,直至停止旋转,整个过程大约30s。
这个Spin键足以很好的将细胞因子或蛋白收集到管底。
但有些实验室没有这样的高速离心机,只有最高转速为4000-4500rpm的离心机。
这种情况下,需3000-3500rpm离心5min,也能达到类似的效果。
2. Reconstitute in water to a concentration of 0.1-1.0 mg/ml. Do not vortex.第2步:用无菌水重悬至0.1-1.0 mg/ml,不可振荡。
抗核抗体检测试剂盒(多重微珠免疫法) 说明 书

抗体。整个检测步骤包括两步温育过程。 1、待测血清(经过稀释)与复合悬浮微珠在孔中温育。 复合悬浮微珠为不同荧光编码的聚苯乙烯微粒( polystyrene microspheres); 不同颜色的微粒上结合有不同的抗原。如果病人 血清中含有自身抗体的话,一种或多种自身抗体就会分别和不 同颜色的微粒特异性地结合, 温育 3010 分钟后清洗微球, 去除 不反应的血清蛋白。 2、加入荧光素(Phycoerythrin,PE,藻红蛋白)标记的 羊抗人 IgG 继续进行温育。标记抗体会与通过上一步反应固定 在 微 粒 表 面 的 自 身 抗 体 结 合 。 用 AtheNA Multi-Lyte® 系 统 (Luminex®仪器)对微粒悬浮液进行分析。仪器可以辨认出不同 颜色的微粒, 并测量出每个颗粒上的荧光强度(PE)。 利用孔内校 正技术(Intra-Well Calibration TechnologyTM) ,内对照颗粒上的 荧光信号可以将读到的荧光强度转换成浓度(单位)结果。 五、 【主要组成成份】 A、活性成分: (所有活性成分都含有浓度为 0.1% w/v 的叠氮钠 作为防腐剂。 ) 1、 复合悬浮微珠。可以直接使用,5.5ml 一瓶。悬浮液中包含 有可辨识的 5.6 微米的聚苯乙烯颗粒, 表面结合有以下自身 抗原:SSA、SSB、Sm、U snRNP B/B’、U1 snRNP 68、U1 snRNP A、U1 snRNP C、Scl-70、Jo-1、Hep-2 细胞。这些 微珠可以被 AtheNA Multi-LyteTM 抗核抗体检测系统识别。 最后,微珠的混合物也包括一套能在病人标本中检测出非 特异性抗体的系统和四套校正系统。 2、 荧光素标记的羊抗人 IgG (链) 。 可以直接使用, 1 管, 15ml, 琥珀色瓶子。 3、 人阳性血清对照:3 管,0.2ml/管。 4、 人阴性血清对照:1 管,0.2ml/管。 5、 样本稀释液:50ml,1 瓶,含磷酸缓冲盐,可以直接使用。 注:样本稀释液加入血清后会变色(指示已稀释) 。 6、 10X 浓缩洗涤液:50ml,含磷酸缓冲盐。使用时需按 1 份 浓缩洗涤液加 9 份蒸馏水或去离子水稀释。 B、非活性成分: 1、 96 孔过滤板一块 2、 96 孔稀释盘一块 3、 数据标签:一张标签贴于试剂盒盖内,另一张在试剂盒内。 4、 使用说明书中文版和英文版各 1 份 5、 校正 CD 一张。包括所有批间特异性的试剂盒校正值,用 于标本分析和质量控制。 六、 【其它所需物品和仪器】 1、 能精确吸取 10 到 200ul 的移液器; 2、 能精确吸取 50-200 ul 多通道移液器(选配) ; 3、 适用于多通道移液器的贮液槽(选配) ; 4、 一次性吸头;
免疫细胞培养用澳洲胎牛血清fbs解冻方法

免疫细胞培养用澳洲胎牛血清fbs解冻方法
解冻澳洲胎牛血清(FBS)是进行免疫细胞培养的重要步骤。
以下是解冻FBS的方法:
1. 先将FBS的冻存管或瓶放入4°C的冰箱中,让其缓慢解冻。
一般来说,FBS应该以5-10°C的速度解冻。
2. 解冻前,需将FBS摇匀。
这样能确保其中的成分均匀混合。
3. 将冻存管或瓶放入37°C的水浴中,直到FBS完全解冻。
水
浴的使用可以加快解冻的速度,但需要注意不要将水浴温度过高,否则会导致FBS的变性。
4. 解冻后,将FBS用吸管转移到无菌离心管中。
注意,吸管、离心管以及其他使用的工具都应是无菌的,以确保细胞培养的纯度和无菌性。
5. 将无菌离心管中的FBS进行离心,以去除其中的异物和冰渣。
6. 将无菌离心管中的FBS分装到合适的小容器中,如10 ml的离心管,以方便后续使用。
注意,每个小容器中应当只放置足够解冻FBS的量,避免重复多次冻融。
7. 使用培养基时,将解冻后的FBS加入培养基中,使其浓度
为所需的最终浓度。
常见的细胞培养浓度为5-20%的FBS。
解冻澳洲胎牛血清的方法可以根据具体情况的需求进行调整,但在整个过程中,保持无菌性和避免过高温度是至关重要的。
同时,根据实验需要,可以进行更加精确的测量和计算,以确保所使用的FBS浓度符合实验要求。
快速冰冻免疫组化染色流程

快速冰冻免疫组化染色流程英文回答:Rapid Immunohistochemistry Staining Protocol.Immunohistochemistry (IHC) is a powerful technique for visualizing the presence and localization of specific proteins in tissue sections. The traditional IHC protocol can be time-consuming, often taking several hours or even days. Rapid IHC protocols have been developed to reduce staining time without compromising accuracy or sensitivity.Materials:Tissue sections.Primary antibody.Secondary antibody.DAB substrate.Hematoxylin counterstain.Microscope.Procedure:1. Deparaffinization and rehydration: Heat tissue sections at 60-70°C for 10 minutes. Immerse in xylene for 5 minutes, then in 100% ethanol for 3 minutes, 95% ethanol for 3 minutes, 70% ethanol for 3 minutes, and finally distilled water for 3 minutes.2. Antigen retrieval: Heat tissue sections in an appropriate antigen retrieval buffer at 95-100°C for 20 minutes. Allow to cool for 20 minutes.3. Blocking: Incubate tissue sections in 1-5% bovine serum albumin (BSA) or goat serum for 30 minutes at room temperature.4. Primary antibody incubation: Apply primary antibody to tissue sections and incubate for 30-60 minutes at room temperature or overnight at 4°C.5. Secondary antibody incubation: Apply secondary antibody to tissue sections and incubate for 15-30 minutes at room temperature.6. DAB staining: Apply DAB substrate to tissue sections and incubate for 5-10 minutes at room temperature.7. Counterstaining: Apply hematoxylin counterstain for 1-2 minutes, then wash with water.8. Dehydration and mounting: Dehydrate tissue sections by immersion in 70% ethanol for 3 minutes, 95% ethanol for 3 minutes, and 100% ethanol for 3 minutes. Mount on glass slides and allow to dry.Tips:Use high-quality antibodies and reagents.Optimize the incubation times and temperatures for the primary and secondary antibodies.Ensure complete antigen retrieval to maximize antibody binding.Use appropriate controls, such as negative controls without primary antibody and positive controls with known antigen expression, to validate staining results.中文回答:快速免疫组化染色流程。
DNA RNA 蛋白质等各种生物物质的保存

各种生物活性物质的保存Experience 2009-08-16 16:34:36 阅读336 评论0 字号:大中小订阅DNA、RNA、蛋白质、抗体和探针的保存方法:(网上搜索,仅供参考^_^)1.DNA短期保存,两年之内吧,无菌水和TE缓冲液是ok的。
如果是长期保存,比如10年20年的话,我推荐你刚提取好的DNA直接保存到乙醇里,放-20°,放10年没问题。
需要用的时候,再离心后,弃去乙醇,加水或TE溶解即可。
长期保存用TE,DNA本来就是酸性,所以需要弱碱性环境保存,如果用中性或者酸性环境保存容易降解。
保存两年应该会降解的!还是不要超过两年,再一年内都有降解的可能,所以还是要尽快的利用了。
做成干粉不太可能,哪里有那么多的量!还是在-80度以下保存吧,也要避免反复冻融。
如果DNA很纯的话,应该影响不是很大啊,有文献报道高纯DNA的最佳保存条件是4度,你也可以应证一下你的DNA有没有降解啊,跑个电泳看看啊,如果出现smear,就不要用了啊!提纯的DNA放在4度一段时间(数周-数月)都没问题。
但是长期保存建议-20度,并且浓度不要稀释的太低,否则容易降解。
2.RNARNA若要长期保存,需沉淀下来后臵于无水乙醇冻在-70度,用的时候再离心下来除去无水乙醇,用DEPC水溶解。
保存RNA应该尽量低温。
为了防止痕量RNase的污染,从富含RNase的样品(如胰脏、肝脏)中分离到的RNA需要贮存在甲醛中以保存高质量的RNA,对于长期贮存更是如此。
从大鼠肝脏中提取的RNA,在水中贮存一个星期就基本降解了,而从大鼠脾脏中提取的RNA,在水中保存3年仍保持稳定。
另外,长度大于4kb的转录本对于痕量RNase的降解比小转录本更敏感。
为了增加贮存RNA样品的稳定性,可以将RNA溶解在去离子的甲酰胺中,存于-70℃。
用于保存RNA的甲酰胺一定不能含有降解RNA的杂物。
来源于胰脏的RNA至少可以在甲酰胺中保存一年。
冻融工艺在mRNA疫苗中应用
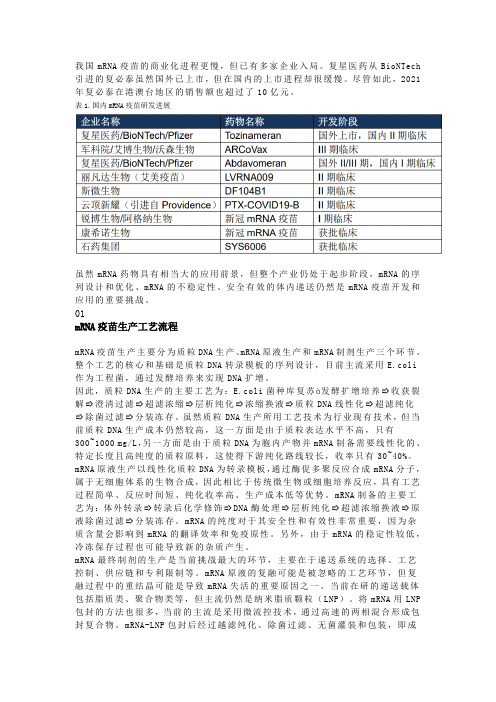
我国mRNA疫苗的商业化进程更慢,但已有多家企业入局。
复星医药从BioNTech 引进的复必泰虽然国外已上市,但在国内的上市进程却很缓慢。
尽管如此,2021年复必泰在港澳台地区的销售额也超过了10亿元。
表1.国内mRNA疫苗研发进展虽然mRNA药物具有相当大的应用前景,但整个产业仍处于起步阶段,mRNA的序列设计和优化、mRNA的不稳定性、安全有效的体内递送仍然是mRNA疫苗开发和应用的重要挑战。
01mRNA疫苗生产工艺流程mRNA疫苗生产主要分为质粒DNA生产、mRNA原液生产和mRNA制剂生产三个环节。
整个工艺的核心和基础是质粒DNA转录模板的序列设计,目前主流采用E.coli 作为工程菌,通过发酵培养来实现DNA扩增。
因此,质粒DNA生产的主要工艺为:E.coli菌种库复苏à发酵扩增培养➩收获裂解➩澄清过滤➩超滤浓缩➩层析纯化➩浓缩换液➩质粒DNA线性化➩超滤纯化➩除菌过滤➩分装冻存。
虽然质粒DNA生产所用工艺技术为行业现有技术,但当前质粒DNA生产成本仍然较高,这一方面是由于质粒表达水平不高,只有300~1000 mg/L,另一方面是由于质粒DNA为胞内产物并mRNA制备需要线性化的、特定长度且高纯度的质粒原料,这使得下游纯化路线较长,收率只有30~40%。
mRNA原液生产以线性化质粒DNA为转录模板,通过酶促多聚反应合成mRNA分子,属于无细胞体系的生物合成,因此相比于传统微生物或细胞培养反应,具有工艺过程简单、反应时间短、纯化收率高、生产成本低等优势。
mRNA制备的主要工艺为:体外转录➩转录后化学修饰➩DNA酶处理➩层析纯化➩超滤浓缩换液➩原液除菌过滤➩分装冻存。
mRNA的纯度对于其安全性和有效性非常重要,因为杂质含量会影响到mRNA的翻译效率和免疫原性。
另外,由于mRNA的稳定性较低,冷冻保存过程也可能导致新的杂质产生。
mRNA最终制剂的生产是当前挑战最大的环节,主要在于递送系统的选择、工艺控制、供应链和专利限制等。
输注冷沉淀凝血因子的流程

输注冷沉淀凝血因子的流程The process of infusing cold precipitate coagulation factors begins with a thorough assessment of the patient's condition and medical history by the healthcare team. 输注冷沉淀凝血因子的流程始于医疗团队对患者病情和医疗史的全面评估。
This assessment helps determine the specific coagulation factors needed for the patient and the dosage required. 这种评估有助于确定患者需要的特定凝血因子以及所需的剂量。
Once the specific coagulation factors and dosage have been determined, the healthcare team begins to prepare the cold precipitate coagulation factors for infusion. 一旦确定了特定的凝血因子和剂量,医疗团队就开始准备冷沉淀凝血因子供输注。
This process involves carefully handling and storing the coagulation factors to ensure their stability and effectiveness. 这个过程包括认真处理和储存凝血因子,确保它们的稳定性和有效性。
The next step in the process is the actual infusion of the cold precipitate coagulation factors into the patient's bloodstream. 过程的下一步是将冷沉淀凝血因子实际输注到患者的血液中。
蛋白质共晶冷冻技术在药物结构研究中的应用

蛋白质共晶冷冻技术在药物结构研究中的应用蛋白质是生命体内最重要的元素之一,它参与了包括代谢和免疫在内的许多生理过程。
在药物研究领域中,研究蛋白质的结构和功能变得越来越重要。
随着科技的发展,蛋白质共晶冷冻技术成为了药物结构研究的关键技术之一。
蛋白质分子结构的解析是化学、生物学等领域研究的基础之一,而共晶冷冻技术可以通过调节温度和溶液浓度来获得高质量的蛋白质晶体。
利用共晶冷冻技术,可以帮助研究者制备好质量、高分辨率的蛋白质晶体,更加深入地了解到蛋白质的结构和功能。
共晶冷冻法是一种传统的蛋白质晶体制备方法,该方法是将蛋白质和晶体原料混合在一起,然后在减压条件下冷却至低于晶体化点,从而获得晶体。
然而,传统的共晶冷冻技术面临着许多技术上的挑战,如制备时间长、晶体不易稳定、晶体质量一致性差等缺点。
在这个情境下,共晶冷冻技术被引入到了药物结构研究中。
这种技术最初用于生物晶体学领域,如研究病毒蛋白质晶体结构等,但现在已被广泛应用于药物结构研究领域,特别是在药物的配合物研究以及药物分子与蛋白质相互作用的研究中,取得了重要进展。
蛋白质-药物配合物的结构研究是药物研发过程中的重要部分之一。
不同的药物与蛋白质结合的方式、结构、规模和特性都有所不同。
对于这些配合物的高分辨率结构的解析将有助于我们了解它们对疾病治疗的作用机制,进而设计出更好的药物。
共晶冷冻技术可以制备高质量且稳定的小分子配合物结构,并通过X-射线晶体结构法对配合物的结构进行高分辨率、高精度的测定,为药物的开发提供了先决条件。
此外,蛋白质共晶冷冻和生物分子相互作用也具有重要意义。
涉及到药物分子与蛋白质结合的相互作用,通常是指药物分子与蛋白质结合的结构、作用方式、选择性和亲和力等。
X-射线晶体结构法所提供的信息可以帮助药物研究人员设计具有较高选择性的药物分子,从而获得更好的疗效。
因此,蛋白质共晶冷冻技术在药物设计和研发中具有重要的应用价值。
总之,蛋白质共晶冷冻技术在药物结构研究中正变得越来越重要,这种技术可帮助制备高质量、高分辨率的蛋白质晶体,并解析它们的结构和功能,以便进行药物设计和研发。
鱼类结冰耐受适应的分子机制

鱼类结冰耐受适应的分子机制
鱼类结冰耐受适应的分子机制是指鱼类在寒冷环境中能够适应并忍受结冰的能力。
以下是可能的分子机制:
1. 冷凝素(antifreeze proteins,AFPs):鱼类体内可能会产生一类叫做冷凝素的蛋白质,它们能够抑制水分子的结冰。
冷凝素通过与结冰发生的晶格形成相互作用,从而抑制冰晶的生长和扩展。
这样,鱼的体液就能够维持在液态状态,避免了冰晶的形成和对细胞的损害。
2. 冷麻醉抗性相关蛋白(anesthesia-related protein,ARP):一些鱼类体内可能含有冷麻醉抗性相关蛋白,这些蛋白质能够减轻或消除鱼类在寒冷环境中遭受的冷麻醉效应。
冷麻醉抗性相关蛋白可能通过调节或影响神经传导、细胞膜的渗透性以及蛋白质的构象等方式,使得鱼类能够保持正常的神经活动和生理功能。
3. 细胞膜的适应性:鱼类的细胞膜会发生结构和组成的调整,以适应寒冷环境下的冰冻和解冻。
这些调整包括增加脂肪酸链的饱和度,增强细胞膜的流动性以及调节细胞膜内部的离子通道和转运体。
4. 抗氧化机制的增强:寒冷环境下,鱼类的代谢活动可能会增加,导致细胞内产生更多的代谢产物和自由基。
为了应对这种情况,鱼类可能会增强抗氧化机制,包括提高抗氧化酶的表达和活性,以及增加细胞内抗氧化物质的浓度,来减轻自由基对细胞的损伤。
这样可以有助于提高鱼类对结冰环境的耐受性。
需要指出的是,对于鱼类结冰耐受适应的分子机制还存在着很多不确定因素,目前还需要更多的研究来完全理解这个过程。
此外,不同的鱼类可能具有不同的适应机制,因此具体的分子机制可能会因鱼类的种类而异。
冷冻细胞技术及流程

冷冻细胞技术及流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!冷冻细胞技术是一种常见的方法,用于保留细胞样本以便将来进行实验或其他应用。
注射剂低温冻融试验的出处

注射剂低温冻融试验的出处
注射剂低温冻融试验的出处
注射剂低温冻融试验,是一项针对现代医药工业中的重要领域进行的研究和开发。
该试验是为了评估注射剂在低温环境下,经历冻结和解冻过程后的性状和质量,以确保药品在运输和贮存中不会受到损害,进而保证药品的安全性和有效性。
这项试验最早可以追溯到20世纪50年代,当时美国药品管理局就已经提出了对药品进行评估的标准和方法。
在之后的几十年中,各个国家和组织都在积极地研究和开发低温冻融试验的相关技术,并不断推进试验标准的完善和统一。
当前最为广泛使用的注射剂低温冻融试验标准是根据国际药典(Ph. Eur.)、美国药典(USP)和世界卫生组织(WHO)等多个药典制定的,其中最新的版本是2019年发布的USP 43-NF 38版。
这些药典中详细说明了试验的操作流程、试验环境和相关检测指标等内容,确保了试验的规范性和可靠性。
除了官方的药典标准外,一些行业组织也制定了自己的试验标准和方法。
例如,欧洲生物技术工业协会(EBE)和国际生物制品制造商联合
会(IFPMA)等组织都制定了适合于生物制品的低温冻融试验标准。
总的来说,注射剂低温冻融试验是现代医药工业中非常重要的一个环节,它不仅能够确保药品的品质和安全性,还能够促进新药和治疗方案的研发和推广。
而相应的试验标准的不断完善和统一,也为医药工业的发展和健康保障提供了有力保障。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
General Protein Handling GuidelineBrief Guidelines for Freezing and Thawing Protein SamplesDisclaimer: These guidelines are intended for use in general with protein solutions, but the stability of individual proteins varies widely. The investigator must determine the proper storage and freeze-thaw conditions for each protein.For proteins purchased from PBL InterferonSource, refer first to the data sheet. In the absence ofwritten guidance, contact technical service (at 1 877-PBL-8881 or + 1 732-777-9123 for recommendations on handling of specific proteins.It is usually best to work with protein solutions on wet ice. The low temperature will slow inactivation of the protein.Never vigorously agitate a protein sample. The preferred method of mixing protein solutions is to gently mix with a micropipettor using a polypropylene tip. (Do not use larger pipettors with polystyrene pipettes.)An alternative for larger solutions is gentle inversion in a 15 to 50 ml capped polypropylene tube.For volumes over 50 ml, mixing using a stir bar is acceptable, but not too vigorously. Never introduce foam or air bubbles ?these denature proteins.Do not use glass or polystyrene containers or pipettes for antibodies or interferons unless these proteins are diluted in serum or albumin-containing media.Freeze/Thaw of proteins. Usually a quick freeze and a quick thaw are the best methods for retaining protein activities. Rapid freezing and thawing prevent phase partitioning of the salts or protein. If a protein solution is found to be cloudy upon thawing, the bioactivities of the protein are likely to have been adversely affected.Freezing methodPreferred:Be aware that glass tubes can crack, leak, or explode upon snap freezing or thawing of solutions. Polypropylene tubes are recommended for snap freezing.Prior to freezing, make sure the solution identification number is written on the top of each tube and/or on a freezer-safe label using an alcohol-resistant marker.Always take appropriate precautions when working with dry ice. Handle only with appropriate tools and gloves. Never touch dry ice or its solutions with bare hands. Refer to the MSDS sheet for dry ice/solid CO2. Also, when working with dry ice and its solutions, be certain to work in a well ventilated area, preferably in a chemical fume hood where CO2 gas will not accumulate.Set up a tube rack in an ice bucket.In the ice bucket, surround the rack with dry ice and then add sufficient isopropanol to saturate the dry ice. Cover with an ice bucket lid.Allow the liquid to cool until it has a syrupy consistency.Place tubes in the rack allowing them to snap freeze in the covered ice bucket.Once tube contents are frozen, rapidly wipe the liquid off (blot, do not wipe the label?alcohol can solubilize ink), transfer tubes to a pre-chilled box (possibly in another bucket with dry ice (no liquid!), then place tubes in a -80ºC freezer.After the freeze, allow all the dry ice to evaporate in the fume hood and the alcohol to warm to room temperature.Decant the remaining isopropanol into a bottle labeled according to OSHA and State labeling standards with the additional qualifier of “Freezing Isopropanol.?nbsp; This isopropanol can be reused for freezing many times.Alternative:A slightly less effective way to freeze the samples is simply to prepare a bucket of dry ice, then insert the tubes into the dry ice until they are almost buried. This will freeze the small aliquots sufficiently quickly, but freezing of larger volumes/tubes will take longer, allowing loss of homogeneity in the samples.Thawing methodThe preferred method of thawing protein samples is to place the tube in cold tap water. Remove the tube from the freezer, wipe off the rime and then place it in a beaker with some cold tap water. “Floaties?used in the water baths work well for multiple samples.Once thawed, gently mix the sample using a micropipettor to make sure the solution is homogeneous. Again, be careful not to introduce bubbles into the solution.Place the thawed tube on wet ice.Check the protein solution for cloudiness.Very small samples such as 10 μl aliquots can be thawed by holding the tube in your hand and monitoring thawing.Avoid introducing bubbles or foam into any protein solution!Most proteins are unstable at very dilute concentrations in the absence of carrier. Unless formulation work has been done to verify stability, do not dilute carrier-free solutions to protein concentrations below 100 μg/ml. This is only a guideline and needs to be verified for each protein.Be aware that all interferons will stick somewhat to plastics. When performing serial or multiple dilutions, change pipette tips between each dilution step. Rinsing the pipette tip is insufficient to prevent carryover between wells of tubes. Interferon beta proteins tend to exhibit greater hydrophobicity than most interferon alpha proteins, thus more prone to "stickiness".。