动物体内转染实验(Entranster)重复性如何?
动物体内转染(Entranster)答疑

动物体内转染答疑----用RNA或DNA直接注射动物完成干扰和表达动物体内转染,简单地说,就是用RNA和DNA直接打动物完成干扰和表达。
再通俗地说,用合成的(RNA)或者提取的核酸(DNA),就可以完成以前的动物转基因或者基因敲除的实验,无需再用病毒或者基因敲除动物。
实验周期可以缩短为几天,花费几千元即可进行实验。
动物体内转染技术的出现,让广大生物医学研究者,轻松进行动物的基因干扰、导入等操作。
尤其是临床医学工作者,可以在很少工作量较少经费的情况下,直接针对研究的疾病进行动物实验,发表高水平文章。
比如在英格恩客户已发表的文章中,有尾静脉注射DNA研究治疗病毒性心肌炎,有皮下肿瘤注射miRNA 研究治疗结肠癌,有脑室注射siRNA研究脑缺血机理,有皮肤涂抹siRNA治疗皮肤瘢痕等。
这些研究都非常有临床和现实意义。
由于动物体内转染技术应用的时间不长,对这种崭新的技术人们还不太了解。
此次受丁香园邀请,特开此动物体内转染相关实验技术答疑专帖。
对站友们提出的问题给予解答,希望能够和大家相互学习,共同进步。
任何与动物体内转染有关的问题(包括实验设计、产品、实验过程、结果分析、文献等问题),大家尽管提出,我们会尽力解答,也欢迎站友们参加讨论。
技术资料目录:1.体内转染试剂的原理和方法1).动物体内转染技术可以做什么?2).体内转染的原理3).体内转染的过程4).体内转染需要的实验条件5).体内转染适合进行怎样的实验6).体内转染可以在哪些组织器官进行7).应用体内转染试剂发表的部分文献8).动物体内转染和病毒感染的比较9).动物体内转染和基因敲除的比较2.体内转染实验的设计1).需要的材料2).需要的时间3).需要的费用4).常见的结果检测方法3.体内转染过程相关问题及解答1).体内转染试剂对动物有什么影响?2).注射后,试剂是如何在体内分布的,有靶向性吗?3).转染试剂和核酸需要使用多少?提问与解答:1、如何技术上解决(排除)RNAi的非特异性?是否需要复原实验(Rescue Experiment)。
体内基因转染—我与小动物的相爱相杀

体内基因转染—我与小动物的相爱相杀说起生物医药科研界的让人撕心裂肺的实验,动物实验肯定是其中一大块。
裸鼠成瘤,各种疾病造(zuo)模(nie),让我们饱尝与鼠小弟的相爱相杀。
既然有牺牲,还得有价值。
如何让鼠小弟们的牺牲换来高逼格的实验数据?自然少不了时下最火爆的动物体内基因转染技术。
所谓基因转染,通俗一点说,主要指基因上调或下调(即Gain of function OR loss of function)。
动物体内基因的上下调,其实现方法主要包括:基因工程小鼠及病毒载体介导动物体内基因转染。
基因工程小鼠最经典传统的动物体内基因干预,当属基因工程小鼠了。
基因工程小鼠包括转基因小鼠,(TG mouse,transgenetic mouse),敲除小鼠(KO mouse,Konckout mouse),敲入小鼠(KI mouse,Knockin mouse)。
在引入Cre-loxP系统后,又发展了组织特异性TG和KO小鼠。
基因工程小鼠对生物医药研究与研发发挥了巨大的作用,但也存在不少的限制因素:1)从受精卵开始的基因干预,经过发育的长时间复杂代偿,所表现出来的功能表型和实际的基因功能表型可能存在巨大的偏差。
(2015年nature杂志发表了Didier Stainier研究小组发现受精卵敲除egfl7基因和成年后用RNA干扰阻断egf17的表达,模式生物所得到的表型完全不一样。
)2)基因工程小鼠周期长,成本相对高。
是否还记得,在动物房里伺候老鼠的那些日子,一把屎一把尿拉扯它长大,给它找对象,还得帮它养孩子……病毒载体介导的动物体内基因转染目前最主流的病毒载体工具有三种:腺病毒(adenovirus),慢病毒(lentivirus),腺相关病毒(AAV,adenovirus associated virus)。
相比较基因工程小鼠的局限性,病毒载体工具有如下优势:1)对成年的动物直接进行急性的基因干预,避免了发育代偿的问题;2)病毒载体相对与基因工程小鼠来说,周期、成本要小得多;3)病毒载体介导的基因干预可以在动物造模后进行,作为一个基因治疗干预手段,与临床更加接近,而不仅仅是完成基因功能研究。
动物体内转染(Entranster)答疑

动物体转染答疑----用RNA或DNA直接注射动物完成干扰和表达动物体转染,简单地说,就是用RNA和DNA直接打动物完成干扰和表达。
再通俗地说,用合成的(RNA)或者提取的核酸(DNA),就可以完成以前的动物转基因或者基因敲除的实验,无需再用病毒或者基因敲除动物。
实验周期可以缩短为几天,花费几千元即可进行实验。
动物体转染技术的出现,让广大生物医学研究者,轻松进行动物的基因干扰、导入等操作。
尤其是临床医学工作者,可以在很少工作量较少经费的情况下,直接针对研究的疾病进行动物实验,发表高水平文章。
比如在英格恩客户已发表的文章中,有尾静脉注射DNA研究治疗病毒性心肌炎,有皮下肿瘤注射miRNA研究治疗结肠癌,有脑室注射siRNA研究脑缺血机理,有皮肤涂抹siRNA治疗皮肤瘢痕等。
这些研究都非常有临床和现实意义。
由于动物体转染技术应用的时间不长,对这种崭新的技术人们还不太了解。
此次受丁香园邀请,特开此动物体转染相关实验技术答疑专帖。
对站友们提出的问题给予解答,希望能够和大家相互学习,共同进步。
任何与动物体转染有关的问题(包括实验设计、产品、实验过程、结果分析、文献等问题),大家尽管提出,我们会尽力解答,也欢迎站友们参加讨论。
技术资料目录:1.体转染试剂的原理和方法1).动物体转染技术可以做什么?2).体转染的原理3).体转染的过程4).体转染需要的实验条件5).体转染适合进行怎样的实验6).体转染可以在哪些组织器官进行7).应用体转染试剂发表的部分文献8).动物体转染和病毒感染的比较9).动物体转染和基因敲除的比较2.体转染实验的设计1).需要的材料2).需要的时间3).需要的费用4).常见的结果检测方法3.体转染过程相关问题及解答1).体转染试剂对动物有什么影响?2).注射后,试剂是如何在体分布的,有靶向性吗?3).转染试剂和核酸需要使用多少?提问与解答:1、如何技术上解决(排除)RNAi的非特异性?是否需要复原实验(Rescue Experiment)。
RNA转染试剂,高效率转染

Entranster TM-R4000(用于将RNA转染入动物细胞)使用手册一产品介绍本品推荐用于将siRNA、microRNA(miRNA)、mimic、inhibitor等小片断RNA转染入动物细胞(包括各种细胞系、原代细胞、悬浮细胞、昆虫细胞等)。
本品在多种细胞系的验证中均表现了很好的RNA转染效率,并有很低的细胞毒性,而较低的细胞毒性对RNAi实验尤其重要。
二重要提示1.由于本品细胞毒性很低,所以采用本品进行转染的细胞数量相对较少,这有助于提高转染效率。
2.采用本品进行转染,RNA用质量(ug)进行计量,对21nt双链的siRNA来说,1OD=3.0nmol=40ug。
3.siRNA等较短RNA请参照表1中siRNA用量,mRNA、shRNA等较长RNA请参照表1中mRNA用量。
4.如转染后需要用Trizol提取RNA,建议转染后6小时换液。
三实验过程(以24孔板siRNA转染为例)1.提前1天细胞种植贴壁细胞:提前一天将细胞种植在24孔板中,以转染时细胞汇合度(Confluence)在30%左右为宜,转染前全培养基总量为0.45ml。
悬浮细胞:采用对数生长期的细胞,数量为常规培养细胞数的1/3进行转染实验。
如某细胞常规培养的细胞数是6×105,那么就用2×105的细胞进行转染。
2.转染过程⑴取0.67ug(50pmol)的siRNA,加入一定量无血清稀释液,充分混匀,制成RNA稀释液,终体积为25μl。
注意:无血清稀释液建议采用OPTI-MEM、无血清DMEM或1640。
⑵取1ul的Entranster TM-R4000,然后加入24ul无血清稀释液体,充分混匀,制成Entranster TM-R4000稀释液,终体积为25μl。
室温静置5分钟。
⑶将Entranster TM-R4000稀释液和RNA稀释液充分混合(可用振荡器振荡或用加样器吹吸10次以上)混合,室温静置15分钟。
转基因动物的技术方法

转基因动物的技术方法根据外源基因导入的方法和对象的不同,转基因动物的技术方法主要有显微注射法、反转录病毒法、胚胎干细胞(embryonic stem cell,ES细胞)法、电脉冲法、精子载体导入法等。
1、显微注射是最常用且成功率较高的方法。
基因显微注射法的特点是外源基因的导入整合效率较高,不需载体,直接转移目的基因,目的基因的长度可达lOOkb(10万个碱基对)。
它可直接获得纯系,所以实验周期短。
但需要贵重精密仪器,技术操作难度大,并且外源基因的整合位点和整合的拷贝数都无法控制,易造成宿主动物基因组的插入突变,引起相应的性状改变,重则致死。
2、反转录病毒感染法该法整合率较高,目的基因不易破坏,多是单拷贝、单位点整合,适合于难以观察到原核的禽类受精卵。
由于病毒衣壳大小的限制,目的基因不宜超过10kb,否则影响活性和稳定。
此外,病毒DNA可能影响外源基因在宿主动物的表达。
3、胚胎干细胞法胚胎干细胞(ES细胞)指从囊胚期的内细胞团中分离出来的尚未分化的胚胎细胞,具有发育全能性,能进行体外培养;扩增、转化和制作遗传突变型等遗传操作。
本法外源基因整合率高,植入囊胚前筛选合适的转化的ES细胞,克服了以前只能在子代选择的缺点,并能充分利用分子生物学发展起来的各种先进方法,是很有前途的技术。
缺点是不易建立ES细胞系。
并且由于通过嵌合体途径,所以实验周期长。
4、电脉冲法电脉冲法(electroporation)又称电穿孔法,是将供体DNA与受体细胞充分混匀,在外界的高电压短脉冲下改变细胞膜结构,使细胞膜产生瞬间可逆性电穿孔,从而使一定大小的DNA可以通过细胞膜进入细胞,运送到细胞核。
5、精子导入法利用精子作为外源基因载体,借助受精作用把外源基因导入受精卵,整合到受精卵的基因组中,称之为精子载体导入法,是构建转基因动物的一种新尝试。
该法简单、方便,依靠生理受带过程,免去了对原核的损伤。
但在实践中成功率较低,对于精子是否可作为外源DNA 载体也存在争论。
体内转染(Entranster),不同的组织和注射方法,如何评估干扰效率?

不同的组织和注射方法,如何评估干扰效率?
运用体内试剂Entranster进行体内转染实验后,针对具体的组织器官,不同的注射方法会明显影响效果。
比如,大脑神经细胞的体内转染,可以采用尾静脉注射和侧脑室注射,用侧脑室的方法就好得多。
再比如,肺部的体内转染,用气管灌注就比用尾静脉注射的方法好得多。
体内转染试剂和核酸的混合物与靶器官的接触越直接越充分,效果越好。
干扰效率可以理解为干扰的细胞数量和全部细胞的数量的比例。
在体外细胞实验中,这2个数字可以精确计算。
但在动物体内无法精确统计。
一般评估效果,我们建议在分子水平,用RT-PCR,在细胞水平,用Western-Blot,在组织水平,用病理生理等方法。
体内转染(Entranster)研究脓毒症,方法简单

2 but also includes Schistosome eggs, T. muris, or Hymenolepis diminuta which have been shown to protect rats or mice from autoimmune diseases like IBD and asthma [8]. The mechanism is that helminth or their antigens or excretory-secretory antigens have been shown to induce host T helper (Th) 2 immune response and produce anti-inflammatory factors (IL-4, IL-5, and IL-13) and regulatory factors (IL-10, TGF������1), which could inhibit Th1 inflammatory factors (e.g., TNF-������, IL-1������, and IL-6) and Th17 immune response [9, 10]. In vitro, T. spiralis excretory-secretory antigens (TsES) significantly reduce TNF-������, IL-1������, IL-6, and IL-12 by inhibiting NF-������B and mitogen-activated protein kinase (MAPK) in LPS-stimulated macrophages [11]. TsES also reduce proinflammatory factors in LPS-stimulated dendritic cells (DC) [12]. Therefore, TsES have an immunomodulatory effect on T cells, macrophages, and DC. TsES have been characterized as cystatins, serpins, glycans, mucins, lectins, or cytokine homologs which could be potential immunomodulators [13]. TsES are rich in oligomannose residues and contain various carbohydrates and glycoproteins that can potential ligands for various C-type lectin receptors (CLR) such as mannose receptor (MR) [14]. The MR is a pattern recognition receptor of the innate immune system expressed on the surface of macrophages, DC, and some epithelial cells, that binds to microbial structures bearing mannose to mediate endocytosis and phagocytosis, as well as activation of macrophages and DC and antigen presentation [15, 16]. Studies have shown that T. spiralis can recognize MR and affect IL-6 and NO expression in macrophages [14]. In addition, MR recognition of T. muris components produces an effective immune response that leads to macrophage activation [17]. Because of the anti-inflammatory role of TsES and their potential importance as MR ligands, we hypothesized that TsES might control SIRS during polymicrobial sepsis. To investigate this, we established a polymicrobial sepsis model in mice using cecal ligation and puncture (CLP). We found that treatment with TsES improved the survival rate of septic mice. In addition, TsES inhibited the TLR signaling pathway MyD88 and NF-������B and regulated the production of pro- and anti-inflammatory factors via MR in vitro and in vivo. The results of this study suggest that TsES should be investigated as a novel therapeutic strategy for sepsis.
体内转染(Entranster)与海马神经研究

R E S E A R C H A R T I C L EWnt/β‐catenin signalling pathway mediated aberranthippocampal neurogenesis in kainic acid ‐induced epilepsyZhengyi Qu 1|Fang Su 1|Xueting Qi 2|Jianbo Sun 2|Hongcai Wang 1|Zhenkui Qiao 1|Hong Zhao 1|Yulan Zhu21Department of Neurology,The Fourth Affiliated Hospital of Harbin Medical University,Harbin,Heilongjiang,China 2Department of Neurology,The Second Affiliated Hospital of Harbin Medical University,Harbin,Heilongjiang,ChinaCorrespondenceYulan Zhu ,Department of Neurology,The Second Affiliated Hospital of Harbin Medical University,246Xuefu Road,Harbin 150086,Heilongjiang,China.Email:ylz1111@Temporal lobe epilepsy is a chronic disorder of nerve system,mainly characterized by hippocam-pal sclerosis with massive neuronal loss and severe gliosis.Aberrant neurogenesis has been shown in the epileptogenesis process of temporal lobe epilepsy.However,the molecular mecha-nisms underlying aberrant neurogenesis remain unclear.The roles of Wnt signalling cascade have been well established in neurogenesis during multiple aspects.Here,we used kainic acid ‐induced rat epilepsy model to investigate whether Wnt/β‐catenin signalling pathway is involved in the aberrant neurogenesis in temporal lobe epilepsy.Immunostaining and western blotting results showed that the expression levels of β‐catenin,Wnt3a,and cyclin D1,the key regulators in Wnt signalling pathway,were up ‐regulated during acute epilepsy induced by the injection of kainic acids,indicating that Wnt signalling pathway was activated in kainic acid ‐induced temporal lobe epilepsy.Moreover,BrdU labelling results showed that blockade of the Wnt signalling by knocking down β‐catenin attenuated aberrant neurogenesis induced by kainic acids injection.Altogether,Wnt/β‐catenin signalling pathway mediated hippocampal neurogenesis during epilepsy,which might provide new strategies for clinical treatment of temporal lobe epilepsy.Temporal lobe epilepsy is a chronic disorder of nerve system,mainly characterized by hippocam-pal sclerosis.Aberrant neurogenesis has been shown to involve in the epileptogenesis process of temporal lobe epilepsy.In the present study,we discovered that Wnt3a/β‐catenin signalling pathway serves as a link between aberrant neurogenesis and underlying remodelling in the hippo-campus,leading to temporal lobe epilepsy,which might provide new strategies for clinical treat-ment of temporal lobe epilepsy.KEYWORDSkainic acids,neurogenesis,temporal lobe epilepsy,Wnt signalling pathway,β‐catenin1|INTRODUCTIONTemporal lobe epilepsy,one of the most common chronic diseases of the nerve system,remains a disease with extremely deficient clinical therapies worldwide.1,2Temporal lobe epilepsy is a very common formof partial or localization related epilepsy and is frequently resistant to medications and associated with a particular finding on MRI.This find-ing is called hippocampal sclerosis,characterized by massive neuronal loss and severe gliosis.3-6Aberrant neurogenesis andneuronal circuitry reorganization in the hippocampus generates an epileptic focus of spontaneous recurrent seizures and was found to involve in the epileptogenesis process of temporal lobe epilepsy.7-10Although recent data showed that aberrant neurogenesis was induced by acute seizures or precipitating insults,however,the mechanisms underlying aberrant neurogenesis in temporal lobe epilepsy are still elusive.Wnt/β‐cateninsignallingpathwayiswidelyinvolvedinembryonic development,cell migration,and cell proliferation in different cell types.11-13In the central nervous system,it also plays important roles in regulating neurogenesis,neural differentiation,synapse development,and plasticity.Recently,collective data have shown that Wnt/β‐catenin signalling pathway may mediate aberrant neurogenesis and be involved in the epileptogenesis in some epilep-tic animal models and also human patients.14In the present study,to examine the potential role of Wnt signalling pathway in hippocampal neurogenesis,we used the rat model of temporal lobe epilepsy byZhengyi Qu and Fang Su contributed equallyReceived:12July 2017Revised:12September 2017Accepted:17September 2017DOI:10.1002/cbf.3306Cell Biochem Funct .2017;1–5.Copyright ©2017John Wiley &Sons,Ltd./journal/cbf1intracerebral injection of kainic acid,a well‐established model with similar neuropathological and electroencephalographic features that are seen in patients with temporal lobe epilepsy.15Our results indicated that the Wnt pathway was activated after kainic acid injection,which may contribute to the structural and functional abnormality of hippocampus in temporal lobe epilepsy.Rescuing this pathway by knocking downβ‐catenin attenuated aberrant neurogenesis induced by kainic acid injection,which may provide a promising therapeutic target for the treatment of temporal lobe epilepsy.2|MATERIAL AND METHODS2.1|Animals and reagentsAdult Wistar rats(male,250g±10g)were used in these experiments. All animals were housed in an environment of12/12hours light/dark cycle and controlled ambient temperature(24±1°C).Water and food are freely available.All animal experiments were approved by the Animal Care and Use Committee of China Medical University.The kainic acid was purchased from Sigma;siRNA oligos were synthesized by GenePharma;the in‐vivo RNA transfection reagent was from Engreen Biosystem Co.Ltd;theβ‐catenin and cyclin D1 antibodies were purchased from Cell Signalling Technology;the doublecortin(DCX)antibody was purchased from Immuno Way;the β‐actin antibody was from ZSGB bio.2.2|Model of kainic acid‐induced epilepsyThe model of temporal lobe epilepsy was established by intracerebral injection of kainic acid.The appropriate dose of kainic acid was determined according to the severity of epileptic seizures and the frequency of spontaneous seizures,as well as the loss of neurons and the budding of mossy fibres.Rats in class3or higher(based on the Race classification)and with convulsions for more than2hours were chosen for further experiments.For the rats that were strongly reacted with drugs,were intraperitoneally injected with chloral hydrate,to avoid death.Behavioural observations were recorded, and the rats were sacrificed at day7,day14,day28,or day35after injection.2.3|Histology,immunohistochemistry,and western blotRat temporal lobes were fixed with4%buffered paraformaldehyde, embedded in paraffin,and4‐μm‐thick sections were prepared for staining with haematoxylin‐eosin and Timm.Immunohistochemical analyses were performed using anti‐β‐catenin,anti‐cyclin D1,or anti‐Wnt3a antibodies.Image Pro‐plus was used to analyse the immunohistochemistry data as other groups used.16,17The parameter of integrated optical density(IOD)represented the expression levels ofβ‐catenin,cyclin D1,and wnt3a.For western blot analysis,tissue lysates from rat temporal lobes were extracted with buffer containing phosphatase inhibitors(Roche)for SDS polyacrylamide electrophore-sis.Blotting and antibody exposure were done by standard procedures.2.4|RNA interferenceSynthetic siRNA oligos targetingβ‐catenin(5′‐GCCUUAGUAAACAU AAUGATT‐3′)or negative control(5′UUCUCCGAACGUGUCACGU TT‐3′)were mixed with in‐vivo transfection agents and were injected into each hippocampus.Ten minutes later,we established the model of temporal lobe epilepsy by intracerebral injection of kainic acid. Every48hours,siRNA oligos were injected into each hippocampus again to obtain better knockdown effect.All the rats were sacrificed at day14,and the hippocampus was collected,and expression of β‐catenin,cyclin D1,and doublecortin(DCX)was analysed by western blot.The neurogenesis was analysed by BrdU incorporation assay.2.5|Statistical analysisThe experimental data were expressed as mean±standard deviation, the model group,and the control group compared with the2samples t test,group design multiple sample meanings with single factor analysis of variance(ANOVA),test level P<0.05.Statistical analysis and chart drawing were done with SPSS17.0statistical software.3|RESULTS3.1|Intracerebral injection of kainic acids induced significant epilepsyIn this study,epilepsy was induced by intracerebral injection of kainic acids.In the kainic acid‐treatment group,rats displayed significant phenotype,with average seizures reaching grade III or above based on the Race classification,then relieving within3to4hours.Our results showed that spontaneous seizures were observed at approxi-mately10days after injection,manifested as double upper limb clonus, blink,hind limb erect,standing instability,fall,and body rhythm clonus (recovered within approximately5–6seconds).In the tissue level,haematoxylin and eosin(HE)staining showed that in the sham group,the neuronal cells in the hippocampus CA3 region were well arranged(Figure1A.B).However,in the kainic acid‐treatment group,the neuronal cells in the same region were significantly absent(Figure1C–F),manifested as irregularly arranged neuronal cells,increased cell gap,degeneration,necrosis,disintegra-tion,cell body shrinkage,cytoplasmic staining,and ill‐defined nucleus. Altogether,the behavioural and histological data agreed well with other groups'studies15,indicating that the kainic acid‐induced epi-lepsy model is established successfully in our study.3.2|Increased expression of Wnt3a,cyclin D1,and β‐catenin and aberrant neurogenesis after kainic acid injectionThe expression levels of Wnt3a,cyclin D1,andβ‐catenin,key regulators in Wnt3a/β‐catenin pathway,were estimated by both immunohistochemistry and western blotting.Figure2is the immuno-histochemistry result,showing thatβ‐catenin was mainly expressed within dentate gyrus,CA1and pared with the sham group,the expression level ofβ‐catenin was up‐regulated in day7after kainic acid injection and reached the peak in day 14,then returned to the baseline level in day 35(Figure 2A).Cyclin D1is mainly expressed in the CA3region.The expression of cyclin D1was also changed in a time ‐dependent manner,which was elevated in day 14after kainic acid injection (Figure 2B).Similarly,Wnt3a is mainly expressed in CA1and CA3regions (Figure 2C)with highest expression in day 14.Western blot results also showed similar expression pattern of Wnt3a,cyclin D1,and β‐catenin (Figure 3A,B).Based on the results of immunohistochemistry and western blotting,the expression levels of β‐catenin,cyclin D1and Wnt3a all reached the peak in day 14after kainic acid injection.Therefore,we chose this time point to detect the neuronal cell proliferation usingBrdU incorporation assay.We found that BrdU ‐positive cells signifi-cantly increased after kainic acid treatment (Figure 4C,D),indicating that aberrant neurogenesis was induced by kainic acid,which may be mediated by the activation of Wnt3a/β‐catenin signal pathway.3.3|β‐catenin knockdown attenuated KA ‐induced aberrant neurogenesis by blockade of the Wnt signallingTo test whether aberrant neurogenesis was mediated by Wnt3a/β‐catenin signal pathway in kainic acid ‐induced epilepsy,we suppressed the activity of Wnt3a/β‐catenin pathway byknockingFIGURE 1Neuronal changes in the CA1hippocampal region of rats after intracerebral injection of kainic acids.The haematoxylin and eosin (HE)staining of hippocampus of sham operation group (A and B),14days after kainic acid injection (KA ‐14,C and D)or 35days after kainic acid injection (KA ‐35,E and F).The images in B,D,F were taken under higher magnification.The neurons in CA1region and the dentate gyrus (DG)organized closely in the sham operation group,while slightly derange,degeneration,necrosis,and karyopyknosis (C and D)were observed in the CA3region of kainite treated rats 14days after injection and necrotic neurons were observed in CA3region (E and F)of kainite ‐treated rats 35days after injectiongroupFIGURE 2Immunohistochemistry staining showing the expression and localization of β‐catenin,cyclin D1and wnt3a.Left:Representativeimaging showing the expression change after 7days (KA ‐7),14days (KA ‐14),28days (KA ‐28)and 35days (KA ‐35)after kainic acid injection comparing with sham group.Right:Integrated optical density (IOD)quantification of β‐catenin,cyclin D1and wnt3a expression.*P <0.05vs sham groupdown the expression of β‐catenin by small RNA interference.Western blot result showed that in vivo injection of β‐catenin siRNA oligos dramatically suppressed the up ‐regulation of β‐catenin induced by kainic acid (Figure 4A,B).Moreover,expression levels of both cyclin D1and DCX,a marker for neurogenesis,were also rescued to sham level upon β‐catenin knockdown (Figure 4A,B).BrdU incorporation assay showed that the epilepsy ‐related aberrant neurogenesis induced by kainic acid was significantly suppressed by knocking down β‐catenin (Figure 4C,D),indicating that suppression the activation of Wnt3a/β‐catenin signal pathway can effectively block aberrant neurogenesis in kainic acid ‐induced epilepsy.More importantly,inthe behavioural assay,the β‐catenin knockdown group showed less activity and lower alert level.4|DISCUSSIONIn the present study,using kainic acid ‐induced temporal lobe epilepsy model,we found that Wnt3a/β‐catenin signal pathway was activated by up ‐regulating the expression levels of its key regulators,Wnt3a,cyclin D1,and β‐catenin.Aberrant neurogenesis was also observed in this model,which may lead to the epileptogenesis in temporallobeFIGURE 4BrdU incorporating staining showing neuronal cell proliferation.A and B,western blot showing the expression change of β‐catenin,DCX and cyclin D1after knocking down β‐catenin.*P <0.05and **P <0.01vs sham group.C and D,aberrant neuronal cell proliferation was observed after 14days kainic acid injection,but effectively diminished by knocking down β‐catenin.Scale bar:100μm.#P <0.05vs sham group.P <0.05vs KAgroupFIGURE 3Western blot showing theexpression change of β‐catenin,Wnt3a and cyclin D1after kainic acid injection.A,representative imaging of western blot results.B,collective data.*P <0.05and **P <0.01vs sham groupepilepsy.Blockade of the activation of Wnt3a/β‐catenin signal pathway by knocking downβ‐catenin effectively eliminated the aberrant neurogenesis in kainic acid‐induced epilepsy and alleviated the spontaneous seizures,which may provide potential therapeutic treatment against temporal lobe epilepsy.It has been well established that intracerebral injections of kainic acids induced epilepsy with significant phenotype of neuronal loss that has been observed in patient with temporal lobe epilepsy15.The kainite acids mainly damage the hippocampal formation,especially the pyramidal neurons of the CA3region.In our experiment,rat epilepsy model was successfully established by intracerebral injection of kainic acids.Classic neuronal cells absence in the hippocampus CA3region was observed by HE staining and TIMM staining(Figure1),which is con-sistent with the classical epileptic behaviours.In this epilepsy model, several key molecular regulators in the canonical Wnt3a/β‐catenin signal pathway,such as Wnt3a,cyclin D1andβ‐catenin,were up‐regulated(Figures2and3),indicating that this signal pathway involved in the process of kainic acids induced epilepsy.The roles of Wnt3a/β‐catenin signalling pathway involved in neurogenesis during development and other physiological process were well understood 11-14.However,the exact roles of canonical Wnt3a/β‐catenin signalling in epileptogenesis are still partially understood.Accumulating evidences showed that aberrant neurogenesis was found to be involved in the epileptogenesis process of temporal lobe epilepsy7-10.Here,BrdU incorporating staining showed that aberrant neuronal cell proliferation was observed in kainic acids induced epilepsy,which may be due to the activation of Wnt3a/β‐catenin signal pathway.Blockade of the activation of this pathway by knocking downβ‐catenin effectively eliminated the aberrant neurogenesis(Figure4),indicating that Wnt3a/β‐catenin signalling pathway is involved in the epileptogenesis process by mediating neuronal cell proliferation.In the present study, we discovered that Wnt3a/β‐catenin signalling pathway serves as a link between aberrant neurogenesis and underlying remodelling in the hippocampus,leading to temporal lobe epilepsy.ACKNOWLEDGEMENTSThe authors have no support or funding to report.CONFLICT OF INTERESTThe authors declare no potential conflicts of interest.ORCIDYulanZhu /0000-0002-0472-0473REFERENCES1.Engel J.Mesial temporal lobe epilepsy:what have we learned?Neuroscientist.2001;7:340‐352.2.Sander JWAS,Shorvon SD.Epidemiology of the epilepsies.J NeurolNeurosurg Psychiatry.1996;61:433‐443.3.Bartolomei F,Chauvel P,Wendling F.Epileptogenicity of brainstructures in human temporal lobe epilepsy:a quantified study from intracerebral EEG.Brain.2008;131:1818‐1830.4.Houser CR.Granule cell dispersion in the dentate gyrus of humans withtemporal‐lobe epilepsy.Brain Res.1990;535:195‐204.5.Harvey AS,Berkovic SF,Wrennall JA,Hopkins IJ.Temporal lobe epi-lepsy in childhood:clinical,EEG,and neuroimaging findings and syndrome classification in a cohort with new‐onset seizures.Neurology.1997;49:960‐968.6.SerranoCastro P,SanchezAlvarez JC,GarciaGomez T.Mesial temporalsclerosis(I):histological data,physio‐pathological hypothesis and aetiological factors.Rev Neurol.1997;25:584‐589.7.Parent JM,Yu TW,Leibowitz RT,Geschwind DH,Sloviter RS,Lowenstein DH.Dentate granule cell neurogenesis is increased by sei-zures and contributes to aberrant network reorganization in the adult rat hippocampus.J Neurosci.1997;17:3727‐3738.8.Scharfman HE.Epilepsy as an example of neural plasticity.Neuroscientist.2002;8:154‐173.9.Pun RYK,Rolle IJ,LaSarge CL,et al.Excessive activation of mTOR inpostnatally generated granule cells is sufficient to cause epilepsy.Neuron.2012;75:1022‐1034.10.Cho KO,Lybrand ZR,Ito N,et al.Aberrant hippocampal neurogenesiscontributes to epilepsy and associated cognitive decline.Nat Commun.2015;6:11.van Amerongen R,Nusse R.Towards an integrated view of Wntsignaling in development.Development.2009;136:3205‐3214.12.Gordon MD,Nusse R.Wnt signaling:multiple pathways,multiplereceptors,and multiple transcription factors.J Biol Chem.2006;281:22429‐22433.13.Cadigan KM,Liu YI.Wnt signaling:complexity at the surface.J Cell Sci.2006;119:395‐402.14.Huang C,Fu XH,Zhou D,Li JM.The role of Wnt/beta‐catenin signalingpathway in disrupted hippocampal neurogenesis of temporal lobe-epilepsy:a potential therapeutic target?Neurochem Res.2015;40:1319‐1332.15.Levesque M,Avoli M.The kainic acid model of temporal lobe epilepsy.Neurosci Biobehav Rev.2013;37:2887‐2899.16.Wang CJ,Zhou ZG,Holmqvist A,et al.Survivin expression quantifiedby image pro‐plus compared with visual assessment.Appl Immunohistochem Mol Morphol.2009;17:530‐535.17.Casarejos MJ,Perucho J,Lopez‐Sendon JL,et al.Trehalose improveshuman fibroblast deficits in a new CHIP‐mutation related ataxia.Plos One.2014;9:How to cite this article:Qu Z,Su F,Qi X,et al.Wnt/β‐cateninsignalling pathway mediated aberrant hippocampal neurogenesis in kainic acid‐induced epilepsy.Cell Biochem Funct.2017;1‐5.https:///10.1002/cbf.3306。
动物体内转染(Entranster)技术用来干什么?

简单地说,如果你希望影响动物某部位基因的功能,增强、减弱,以及引入外源基因。
就可以用动物体内转染技术。
主要特点优点:比较以前的病毒感染、基因敲除、转基因动物等方法,要简单得多,用合成的小RNA与体内转染试剂Entranster-in vivo混合注射动物,就可以进行类似基因敲除的实验。
同样,用质粒DNA与体内转染试剂混合注射动物就可以进行类似转基因动物的实验。
非常方便,注射几天后就可以观察结果,而且可以随时更换核酸,完成更多基因,更深入的实验。
而且,省时间和经费。
病毒感染需要动辄上万的经费和数月包装病毒的时间。
基因敲除老鼠和转基因动物也很麻烦。
vivo(动物体内转染试剂)使用手册

vivo(动物体内转染试剂)使用手册Entranster TM-in vivo(动物体内转染试剂)使用手册Cat. No. 18668-11 Size:1ml常温运输,储存于4℃。
一产品介绍英格恩生物公司(Engreen Biosystem Co, Ltd.)是专业的转染试剂研发生产厂商。
Entranster TM是英格恩生物公司研发合成的纳米聚合物转染试剂,该试剂采用纳米技术合成,是最新一代非病毒转染试剂。
由于纳米技术的应用,Entranster TM-in vivo在细胞转染过程中,表现了卓越的低毒、高效的性能。
本品可用于转染DNA,也可以转染RNA(如siRNA、miRNA、mimic和inhibitor),可用于如下研究:●基因治疗研究●RNA干扰研究●蛋白功能研究本品显著的特点是方法简单、快捷,价格低廉,对动物没有明显的炎性反应,对操作者安全。
二使用前注意一般情况下,核酸(ug)和Entranster TM-in vivo (ul)按照1:2的比例使用。
也可调整比例从1:1.5到1:4自行优化使用。
核酸的具体用量和注射用量须根据靶器官大小、动物大小、给药途径决定,具体可参考下表(核酸的用量换算成μg计算)。
表1 给药途径与核酸用量给药途径建议的核酸用量最大给药体积尾静脉50μg 200μl-400μl脑室 2.5-1μg 5μl成年小鼠腹膜100μg 0.6ml-1ml皮下肿瘤 10-50μg 100μl脑室 2-5μg 20μl成年大鼠静脉 150-300μg 1-1.5ml 一些器官比如皮下肿瘤的用量,也可以通过先确定最终的注射量,然后根据表2推算出核酸的用量和转染试剂的用量。
比如最终的注射量是100ul,那么核酸用量一般为12.5ug,Entranster TM-in vivo的用量为25ul。
表2 100ul 转染复合物的组成25μl 纯水25μl 核酸溶液12.5μg 核酸核酸稀释液25μl 的10%葡萄糖溶液25μl 转染试剂100μl 转染复合物转染试剂稀释液25μl 的10%葡萄糖溶液三操作步骤下面以50μg 的核酸与100μl 转染试剂,总注射体积400ul ,成年小鼠尾静脉注射为例说明。
DNA转染试剂-Entranster
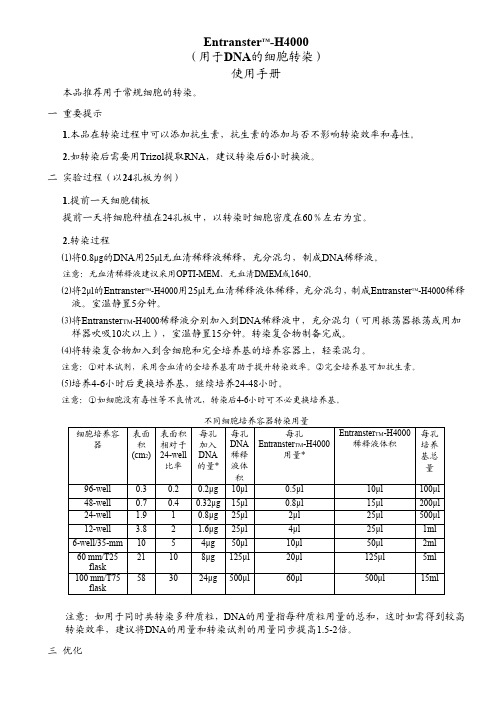
Entranster TM-H4000(用于DNA的细胞转染)使用手册本品推荐用于常规细胞的转染。
一重要提示1.本品在转染过程中可以添加抗生素,抗生素的添加与否不影响转染效率和毒性。
2.如转染后需要用Trizol提取RNA,建议转染后6小时换液。
二实验过程(以24孔板为例)1.提前一天细胞铺板提前一天将细胞种植在24孔板中,以转染时细胞密度在60%左右为宜。
2.转染过程⑴将0.8μg的DNA用25μl无血清稀释液稀释,充分混匀,制成DNA稀释液。
注意:无血清稀释液建议采用OPTI-MEM、无血清DMEM或1640。
⑵将2μl的Entranster TM-H4000用25μl无血清稀释液体稀释,充分混匀,制成Entranster TM-H4000稀释液。
室温静置5分钟。
⑶将Entranster TM-H4000稀释液分别加入到DNA稀释液中,充分混匀(可用振荡器振荡或用加样器吹吸10次以上),室温静置15分钟。
转染复合物制备完成。
⑷将转染复合物加入到含细胞和完全培养基的培养容器上,轻柔混匀。
注意:①对本试剂,采用含血清的全培养基有助于提升转染效率。
②完全培养基可加抗生素。
⑸培养4-6小时后更换培养基,继续培养24-48小时。
注意:①如细胞没有毒性等不良情况,转染后4-6小时可不必更换培养基。
注意:如用于同时共转染多种质粒,DNA的用量指每种质粒用量的总和,这时如需得到较高转染效率,建议将DNA的用量和转染试剂的用量同步提高1.5-2倍。
三优化由于DNA和转染试剂的用量比值是决定转染效率的重要因素,同时由于各实验室质粒的定量误差,质粒纯化程度不同以及细胞状态不同,造成不同细胞和实验室的最优实验条件的差异,为取得最高的转染效率,初次应用时,建议先进行优化。
最优条件确定后,实验的结果将非常稳定。
下表列出了在6-well的优化方案,供参考。
根据预实验的最优化条件,按培养器皿表面积比例应用到其他培养容器。
四常见问题与解决方案。
RNA转染研究病理性色素沉着(entranster),转染效率高

A novel P53/POMC/G a s/SASH1autoregulatory feedback loopactivates mutated SASH1to cause pathologichyperpigmentationDing’an Zhou a,b *,Zhiyun Wei c ,Zhongshu Kuang a ,Huangchao Luo a ,Jiangshu Ma a ,Xing Zeng a ,Ke Wang a ,Beizhong Liu a ,Fang Gong a ,Jing Wang a ,Shanchuan Lei a ,Dongsheng Wang d ,Jiawei Zeng e ,Teng Wang b ,Yong He a ,Yongqiang Yuan a ,Hongying Dai a ,Lin He b,c ,Qinghe Xing b,*aDepartment of Laboratory Medicine,Yongchuan Hospital,Chongqing Medical University,Chongqing,ChinabChildren’s Hospital and Institutes of Biomedical Sciences,Fudan University,Shanghai,ChinacBio-X Institute,Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education),Shanghai Jiao Tong University,Shanghai,ChinadDepartment of Laboratory Medicine,The Affiliated Hospital of North Sichuan Medical College,Nanchong,ChinaeDujiangyan People’s Hospital,Cheng du,Sichuan,ChinaReceived:August 4,2016;Accepted:September 28,2016Abstractp53-Transcriptional-regulated proteins interact with a large number of other signal transduction pathways in the cell,and a number of positive and negative autoregulatory feedback loops act upon the p53response.P53directly controls the POMC/a -MSH productions induced by ultra-violet (UV)and is associated with UV-independent pathological pigmentation.When identifying the causative gene of dyschromatosis univer-salis hereditaria (DUH),we found three mutations encoding amino acid substitutions in the gene SAM and SH3domain containing 1(SASH1),and SASH1was associated with guanine nucleotide-binding protein subunit-alpha isoforms short (G a s).However,the pathological gene and pathological mechanism of DUH remain unknown for about 90years.We demonstrate that SASH1is physiologically induced by p53upon UV stimulation and SASH and p53is reciprocally induced at physiological and pathophysiological conditions.SASH1is regulated by a novel p53/POMC/a -MSH/G a s/SASH1cascade to mediate melanogenesis.A novel p53/POMC/G a s/SASH1autoregulatory positive feedback loop is regu-lated by SASH1mutations to induce pathological hyperpigmentation phenotype.Our study demonstrates that a novel p53/POMC/G a s/SASH1autoregulatory positive feedback loop is regulated by SASH1mutations to induce pathological hyperpigmentation phenotype.Keywords:SASH1 p53 DUH hyperpigmentationIntroductionThe skin pigmentation is originated from the synthesis of melanin in the melanocytes,followed by distribution and transport of pig-ment granules to neighbouring keratinocytes [1].Variations in the coding region of the melanocortin-1-receptor (MC1R)are impor-tant for tanning and pigmentation in human beings.MC1R is a G protein-coupled receptor (GPCR)that is preferentially expressed in epidermal melanocytes [2]and is activated by its ligand a -melano-cyte-stimulating hormone (a -MSH),a propigmentation hormone which is produced and secreted by both keratinocytes andmelanocytes in the skin following UV.a -melanocyte-stimulating hormone and other bioactive peptides are cleavage products of pro-opiomelanocortin (POMC),a multi-component precursor for a -MSH (melanotropic),ACTH (adrenocorticotropic)and the opioid peptide b -endorphin.Normal synthesis of a -MSH and ACTH is an important determinant of constitutive human pigmentation and the cutaneous response to UV [2].Within melanocytes,MC1R regulates the amount and type of pig-ment production and is a major determinant of skin phototype,*Correspondence to:Prof.Qinghe XING E-mail:qhxing@ Dr.Ding’an ZHOUE-mail:081023094@ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley &Sons Ltd and Foundation for Cellular and Molecular Medicine.This is an open access article under the terms of the Creative Commons Attribution License,which permits use,distribution and reproduction in any medium,provided the original work is properly cited.doi:10.1111/jcmm.13022J.Cell.Mol.Med.Vol 21,No 4,2017pp.802-815sensitivity to UV radiation-induced damage and skin cancer risk[3]. Upon ligand binding,GPCRs impart a signal to heterotrimeric G proteins, which are composed of a-,b-and c-subunits,resulting in the detach-ment of the a-subunit from the G bc subunit of G proteins.G proteins of the G a s class directly catalyse the transformation of ATP to cAMP. cAMP is responsible for melanogenic actions of such ligands as a-MSH,including the activation of tyrosinase in melanin biosynthesis[4].The tumour-suppressor protein p53,a transcriptional factor,has been documented to directly activate transcription of numerous genes such as those that control cell-cycle,apoptosis and others.P53 directly mediates UV induction of POMC/MSH in skin and stimulates the POMC promoter in response to UV and is involved in UV-indepen-dent pathologic pigmentation and could mimic the tanning response [1].In the skin,p53function is critical for the retention of tissue integrity following UV irradiation[1].UV can exclusively induce dipyrimidine C to T substitutions that include CC to TT frameshift mutations in the p53gene,which were found in the skin of UV-irra-diated mice months before tumour development[5].In addition to the above activities,p53has been shown to be essential for the for-mation of‘sunburn cells’,which are a hallmark of sunburns[5].DUH is a clinically heterogeneous disorder that is characterized by generalized mottled pigmentation.DUH was initially described by Ichi-kawa and Hiraga in two generations of two families in1933[6].We discovered similar Chinese DUH pedigrees with dyschromatosis sym-metrica hereditaria(DSH)in2003with autosomal-dominant DUH[7] and diagnosed as DUH rather than DSH subsequently.However,the pathological gene and pathological mechanism of DUH have not been further characterized since itsfirst report in1933.SASH1was originally described as a candidate tumour-suppressor gene in the carcinomas of breast and colon and belongs to the previ-ously described novel family of putative adapter and scaffold proteins that transfer signals from the ligand to the receptor[8–10].Our previ-ousfindings indicate that SASH1binds to G a s,the downstream mole-cule of a-MSH/MC1R signalling cascade[11].Our previous study also showed that,in several DUH several affected individuals,hyperpig-mented macules were showed to become more pronounced after strong UV exposure especially in summer[7],but no further mechanism was identified the reasons of photosensitivity[12].The importance of expression of p53/POMC/a-MSH in UV-photopigmentation response and UV-independent hyperpigmentation has been elucidated[1].More-over,less observations were reported to demonstrate that the variations in SASH1gene are associated with hyperpigmentation and how these variations cause hyperpigmentation.Taken above,we hypothesise that a novel p53/POMC/a-MSH/G a s that SASH1is involved in,to mediate UV-photopigmentation response and pathological hyperpigmentation.Materials and methodsPCR,sequencing and mutation analysisTwo Chinese families from the Henan and Yunnan provinces of China and one American family with typical features of DUH were recruited for this study.Three pedigrees with DUH showed an autosomal-domi-nant inheritance pattern and were ascertained by experienced clinical dermatologists.The American family is a smaller pedigree,which could only provide three peripheral blood samples from affected indi-viduals for study.This research was approved by the ethical review committees from the appropriate institutions.Genotyping was per-formed,and the two-point LOD score was calculated as previously described[7].In total,50family members and500normal individuals (controls)participated in the study after providing informed consent. Samples of peripheral blood DNA were taken from all available family members.PCR and sequencing were performed as previously described [7].The sequencing was performed with an ABI BigDye Terminator Cycle Sequencing Kit(Applied Biosystems Inc,Foster City,CA,USA)on an ABI PRISM3130DNA Analyzer(Applied Biosystems),and data were analysed using sequence analysis software,version3.4.1(Applied Biosystems). Sequence data were compared with the SASH1reference sequence(Gen-Bank NM_015278.30)using Sequencher4.10.1(Gene Codes Corp,Ann Arbor,Michigan,USA).Nucleotide numbering reflects complementary DNA (cDNA)numbering,with+1corresponding to the A of the ATG translation initiation codon in the reference sequence[7].Construction of SASH1,G a s,POMC and p53 expression vectorsThe construction protocol of recombined vector of wt and mutant SASH1-PEGFP-C3and wt and mutant SASH1-PBABE-Flag-puro was mainly referred to our previous study[11].To construct HA-Pcna3.0-p53,myc-Pcdna3.0-POMC and GFP-G a s-Pegfp-C3vectors,PCRs of bacteria (obtained from Han jiahuai Lab,Xiamen University,Xiamen,China)contain-ing the vector of full-length CDS sequences of G a s,p53and POMC were performed with Phusion Hot Start High Fidelity Polymerase(New England Biolabs,Inc.,Ipswich,Massachusetts,USA)or GXL Polymerase(Takara, Shimogyo-ku,Kyoto,Japan),and the following primers were used:G a s primers50-ACGCGTCGACATGGGCTGCCTCGGGAAC-30(forward,Sal I site included)and50-CCGCTCGAG TTAGAGCAGCTCGTACTGACG-30(reverse, Xho I site included);p53primers50-CGCGGATCCGCCACCACCATGGAGG AGCCGCAGTCAGATCCTA-30(forward,BamH I site included)and50-CCG CTCGAGTCAGTCTGAGTCAGGCCCTTCTGT(reverse,Xho I site included); POMC primers50-CGCGGATCC ATGCCGAGATCGTGCTGC-30(forward, BamH I site included)and50-CCCAAGCTTT CACTCGCCCTTCTTGTA GGCGTTCTTGAT-30(reverse,Xho I site included).Mammalian expression vectors(Invitrogen,Carlsbad,California,USA)via the relative restriction sites were sequenced.Cell culture and transfectionA375cells,SK-MEL-28cells and HEK-293T cells were maintained as previously described[13].Normal human epithelial melanocytes (NHEMs,C-12402;PromoCell,Germany)were cultured in M2medium. A375,SK-MEL-28and HEK-293T cells were transfected using Lipofec-tamine2000(11668-027;Invitrogen)as previously described[13,14] or Entranster-D(18668-01;Engreen Biosystem Co.,Ltd,New Zealand) or polyethyleneimine(PEI)prepared by ourselves.The transfected A375 and SK-MEL-28cells were cultured in1.5l g/ml puromycin or2.0l g/ ml G418to select stable cell lines.HEK-293T cells were transiently transfected with wild-type and mutant SASH1-pEGFP-C3or co-trans-fected with wild-type SASH1-Pbabe-Flag-puro and G a s-Pegfp-C3ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley&Sons Ltd and Foundation for Cellular and Molecular Medicine.803J.Cell.Mol.Med.Vol21,No4,2017vectors for immunoprecipitation experiments.NHEMs and HEK-293or HEK-293T cells were transiently transfected with Pcdna3.0-HA-p53, Pcdna3.0-myc-POMC,Pegfp-C3-G a s and wild-type SASH1-pEGFP-C3 according to pairwise combination to analyse the expression of exoge-nous p53,POMC,G a s and SASH1using PEI prepared by ourselves or PromoFectin(PK-CT-2000-MAC-1;PromoCell,Heidelberg,Germany).HEK-293T cells were transfected with G a s-GFP,HA-p53,myc-POMC and GFP-SASH1vector and subsequently silenced by G a s-and POMC-specific siRNAs that were synthesized by Shanghai GenePharma Co., Ltd(Shanghai,China)using Entranster TM-R Transfection Reagent (18668-06;Engreen Biosystem Co.,Ltd).The sense and antisense strands of each siRNA for G a s,POMC,GAPDH and the negative control are shown in Table S3.Pull-down assay and nano-flow LC-MS/MS and bioinformatic analysisThe protocols for the pull-down assay,nano-flow LC-MS/MS,database search and bioinformatic analysis for functional classification are mainly referred to our previous report[11]. Immunoprecipitation and immunoblottingHEK-293T or HEK-293transfected cells and NHEMs were gently washed in PBS three times and then lysed ing IP-Western blot lysis buffer(P0013;Beyond Time BioScience and Tech-nology company,Jiangshu,China)in the presence of a complete protease inhibitor cocktail,1l M sodium orthovanadate and1mM sodiumfluoride per10-cm dish on ice.Cell lysates were transferred into 1.5-ml microcentrifuge tubes.Extracts were centrifuged for 10min.at13400g.at4°C.Then,600l l of supernatants was pre-cleaned with20l l of Protein A/G PLUS-Agarose(sc-2003;Santa Cruz Biotechnology,Inc,California,USA)for1hr,immunoprecipitated using6l l of GFP-Tag(7G9)mouse mAb(M20004,Shanghai Abmart,Inc.,Shanghai,China)or6l l of DYKDDDDK-Flag-Tag mouse mAb(M20008;Shanghai Abmart,Inc.)or6l l of HA-Tag mouse mAb(SG4110-25;Shanghai Genomics,Shanghai,China)at4°C for 10hr and mixed with20l l of Protein A/G PLUS-Agarose(sc-2003, Santa Cruz Biotechnology,Inc.)at4°C for4hr and assayed using co-immunoprecipitation or immunoprecipitation.The immunoprecipi-tates were washed with PBS three times and subjected to SDS-PAGE and Western blotting.The primary antibodies used in the Western blot analysis were GFP-Tag mouse Ab(M20004,Shanghai Abmart,Inc.), Flag-tag mouse mAb(M20008;Shanghai Genomics),anti-G a s rabbit polyclonal Ab(G7X105877;Gene Tex,Inc.,Irvine,CA,USA),myc-tag mAb(SG411-30,Shanghai Genomics)and HA-tag mouse mAb(SG4110-25,Shanghai Genomics),SASH1Rabbit mAb(A302-265A-1,Bethyl Laboratories,Inc.,Montgomery,Texas,USA),DYKDDDDK-Flag-Tag mouse mAb(M20008;Shanghai Abmart,Inc.),TYRP1(TA99)mouse mAb(Ab3312;Abcam,Cambridge,UK),Rab27a mouse mAb (H0005873-M01;Abnova,Taipei City,Taiwan),melanoma gp100Rabbit mAb(ab137062;Abcam,Cambridge,UK),GAPDH mouse mAb(M20005; Shanghai Abmart,Inc.)and anti-b-tubulin mouse mAb(M20005M; Shanghai Abmart,Inc.).Immunoblotting was performed as previously described[15].Immunohistochemical and immunofluorescence staining,and melanin staining Immunohistochemical stainingWritten informed consent regarding tissue and data use for scientific purposes was obtained from all participating patients.Epithelial tissues from affected individuals with the Y551D SASH1mutation from pedi-gree family I werefixed in10%formalin at4°C for24hr and then embedded in paraffin.Paraffin sections(5l m)were incubated at56°C overnight and then deparaffinized and rehydrated using xylene and an ethanol gradient.The sections were incubated with the SASH1mono-clonal antibody(A302-265A-1;Bethyl Laboratories,Inc.),Rabbit Anti-ACTH(7-23)antibody(bs-004R;biosynthesis biotechnology Co.,Ltd, Beijing,China),Mitf polyclonal antibody(BS1550;Bioworld Technology, Inc,Louis Park,MN,USA),HMB45monoclonal antibody(sc59305; Santa Cruz Biotechnology,Inc.),TYRP1(TA99)mouse mAb(Ab3312; Abcam),Rab27a mouse mAb(H0005873-M01;Abnova)and p53mon-oclonal antibody(kit-0010-2;biosynthesis biotechnology Co.,Ltd)as well as horseradish peroxidase-linked anti-rabbit and antimouse univer-sal secondary antibodies or FITC.Finally,sections were counterstained with haematoxylin and photographed under the positive position micro-scope BX51.Immunofluorescence(IF)and confocal microscopyWild-type or mutant SASH1-A375stable cells were plated in6-well chamber slides and incubated at37°C for at least48hr.Indirect immunofluorescence analysis was performed on A375cells expressing wild-type and mutant SASH1(s)in6-well chamber slides to assess SASH1localization.IF was performed as described previously using the following antibodies:SASH1Rabbit mAb(A302-265A-1;Bethyl Labora-tories,Inc.)and DYKDDDDK-Flag mouse mAb(M20008;Shanghai Genomics)[11].Melanin stainingParaffin sections(5l m)from epithelial tissues were incubated in an 80°C baking oven for30min and then kept at room temperature for 15min.Melanin staining was performed according to the manufac-turer’s protocol(GMS80023.3;GENMED SCIENTIFICS INC.,Shanghai, China)and observed under a light microscope.Quantitative real-time RT-PCRThe total RNA from the different groups of SK-MEL-28cells was iso-lated using TRIzol Reagent(Invitrogen).Reverse transcription was car-ried out according to the manufacturer’s protocol for the PrimeScript TM RT Reagent Kit(DRR037A;Takara)or PrimeScript RT reagent using the gDNA Eraser Kit(DRR047A;Takara)for qRT-PCR.The sense and anti-sense primer sequences for SASH1,TYRP1,Pmel17,Rab27a,G a s, POMC and GAPDH are presented in Table S3.The PCR products were confirmed by agarose gel electrophoresis.Real-time PCR was per-formed using the Applied Biosystems7500System with SYBR Premix Ex Taq TM(DRR041A;Takara).The quantity of each mRNA was normal-ized to that of GAPDH mRNA.804ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley&Sons Ltd and Foundation for Cellular and Molecular Medicine.UV exposureThe human foreskin tissues from a14-year-old boy were exposed for enough time under an UV phototherapy instrument(NBUVB SS-05; Sigma-Aldrich,St.Louis,Missouri,USA)to reach the expected UV intensity,thenfixed in10%formalin and embedded in paraffin for immunohistochemistry analyses.We conformed to the guidelines of the World Medical Assembly(Declaration of Helsinki)to acquire the human foreskin tissues.In the case of in vitro UV experiments which mainly referred to the protocol of our institute[16],HEK-293T cells and NHEMs transiently transfected with myc-POMC were cultured to approximately70–80% confluence in6-cm-diameter dishes and were irradiated with100mJ/ cm2UVC delivered via a HL-2000HybriLinker with a254-nm wave-length(Upvon)and followed by the indicated recovery time.Finally, cells were harvested to detect proteins’levels using immunoblot. Electrophoretic mobility shift assayThree probes binding with/without biotin,which targeted SASH1pro-moter,were synthesized.The sequence of probes was as follows:probe 1#50-GCCCAAGCTT TCACACTTGTTT-30,probe2#50-CCAAGACTTGCTA-GAAGGAACGAGTCG-30,probe3#50-CGTGGCCACCTAGACCCGAGGTG-30. Electrophoretic mobility shift assay was performed as described as the protocol provided with LightShiftâChemiluminescent EMSA Kit(20148; Thermo Scientific,Pierce Biotechnology,Rockford,USA). Statistical analysisThe data are presented as meanÆstandard error of the mean (S.E.M.)s.These data werefirst analysed using the homogeneity of variance test and followed by the change of variable test.Statistical significance was determined by a one-factor analysis of variance (ANOVA)with LSD correction on SPSS version16.0(IBM(International Business Machine))to generate the required P-values.Cartograms were plotted using GRAPHPAD PRISM5(GraphPad Software, Jolla,CA,USA)5.ResultsMutations in SASH1in DUH-affected individuals result in the up-regulation of SASH1in vitro and in vivoWe have located the gene that is responsible for DUH is local-ized to chromosome6q24.2-q25.2[7].The10.2-Mb region on chromosome6(6q24.2-q25.2)that isflanked by the markers D6S1703and D6S1708contained more than50candidate genes. We screened selected genes in this region for possible pathologi-cal mutations by directly sequencing the PCR products of exons that were amplified from genomic DNA of affected,unaffected and control individuals.We sequenced50candidate genes and found three heterozygous mutations encoding amino acid substi-tutions in SAM and SH3domain containing I(SASH1)in the probands in each of the two non-consanguineous Chinese DUH-affected families(families I and II)and in one non-consangui-neous American DUH-affected family(family III).SASH1point mutations were found in the three pedigrees.These mutations were as follows:a T?G substitution at nucleotide2126in exon 14in family I,a T?C substitution at nucleotide2019in exon 13in family II and a G?A substitution at position2000in exon 13in family III.These three nucleotide changes cause non-con-servative missense mutations in the SASH1gene,resulting in the following amino acid substitutions:Tyr to Asp at codon551 (TAC?GAC),designated as Y551D;Leu to Pro at codon515 (CTC?CCC),designated as L515P;and Glu to Lys at codon509 (GAA?AAA),designated as E509K(Fig.1A).These sequence changes were confirmed in all of the affected family members but were not observed in unaffected family members,correlating the presence of the mutations with the presence of the pheno-type.The mutations were not observed in any of the500normal controls or in any of the current databases,including the Hap-Map database.Therefore,these mutations are unlikely to be common single nucleotide polymorphisms(SNPs)[7].When SASH1mutants were stably expressed in A375cells,they significantly up-regulated SASH1(Fig.1B).Immunoblotting demon-strated that SASH1was up-regulated in A375cells stably express-ing either wild-type(WT-A375cells)or mutant SASH1(mutant-A375cells,including E509K-A375cells,L515P-A375cells and Y551D-A375cells),compared to the expression of endogenous SASH1in A375cells expressing the pBABE-puro empty vector (VECTOR-A375cells)or A375cells without any transfected vector (BLANK-A375cells)(Fig.1B).To verify the stability of SASH1pro-teins,HEK-293T cells stably expressing wild-type or mutant SASH1 were treated with20l g/ml of the protein synthesis inhibitor cyclo-heximide(CHX)for the indicated times to assess the half-life of SASH1.The protein levels of SASH1decreased in a time-course-dependent manner in response to CHX treatment.Wild-type SASH1 levels decreased with a half-life of approximately4hr.However,with CHX treatment for6hr or longer,CHX began to degrade mutant SASH1proteins.Therefore,the three mutant SASH1proteins were more stable than the wild-type,supporting the above observation that SASH1mutants are expressed at higher levels than the wild-type (Fig.S1A and B).Endogenous SASH1was an unstable protein with a half-life of approximately3hr(Fig.S1C).We characterized the subcellular localization of SASH1in A375cells and skin epithelial layers.The endogenous SASH1 protein in VECTOR-A375cells and the skin epithelial layers from normal controls demonstrated a homogeneous pattern of expres-sion(Fig.1C and Fig.S2-a4).However,in WT-A375cells and mutant-A375cells,activated SASH1(through either the overex-pression or mutation of SASH1)showed a pattern of heteroge-neous expression(Fig.S2-b4to Fig.S2-e4).The heterogeneous pattern of SASH1in vitro was also observed in vivo(Fig.1C).In addition,most of the SASH1-positive cells were melanocytes that were nucleic positive for Mitf,a melanocyte marker,andª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley&Sons Ltd and Foundation for Cellular and Molecular Medicine.805J.Cell.Mol.Med.Vol21,No4,2017Fig.1Mutations in SASH1increase SASH1expression in vitro and in vivo.(A)Mutation sites in the SASH1gene in three families with DUH.(B) Western blotting demonstrated the differential and increased expression of mutant SASH1proteins compared to that of wild-type SASH1in different A375cells.(C)HE staining,SASH1and Mitf immunohistochemical analysis of the epidermal tissues from the Y551D-mutation DUH-affected individ-uals and normal controls.Heterogeneous expression of the SASH1protein was observed in all of the epithelial layers in the epidermal tissues from the Y551D-mutation DUH-affected individuals as compared with that of normal controls(NC).Heterogeneous distribution of melanocytes was detected in the epithelial layers of DUH-affected individuals using the melanocyte marker Mitf as compared with that of normal controls.409magni-fication.Scale bar=20l m.Red arrows denote the representative positive cells of SASH1and Mitf.806ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley&Sons Ltd and Foundation for Cellular and Molecular Medicine.demonstrated a heterogeneous distribution of melanocytes in the epithelial tissues of DUH-affected individuals as compared with those of unaffected individuals.Some cytoplasm-positive staining of Mitf is false positive (Fig.1C).The phenomenon thatmelanocytes or SASH1-positive epithelial cells localized not only to the basal layers but also to the suprabasal layers of the affected epidermal tissue is coincide with our previous conclusion that SASH1mutations promote melanocyte migration[11].Fig.2G a s interacts with SASH1and is a pivotal downstream of p53/POMC cascade.(A )The associations between GFP-SASH1and endogenous G a s were identified by immunoprecipitate-Western blot (IP-WB)analysis in HEK-293T cells.HEK-293T cells were transfected with the pEGFP-C3-SASH1vectors.At 24hr post-transfection,GFP-SASH1was immunoprecipitated (IP),and the associated GFP-SASH1was detected by Western blot analysis using an anti-GFP antibody.Different sizes of G a s bands were observed,at 28,46,68and 111kD,which may be caused by post-transla-tional modifications (PTMs).(B )GFP-G a s is associated with Flag-SASH1.HEK-293T cells were co-transfected with the pEGFP-C3-G a s and pBABE-puro-Flag-SASH1vectors.At 36hr post-transfection,Flag-SASH1was immunoprecipitated,and the associated GFP-G a s was detected by Western blot analysis using an anti-GFP antibody.(C )and (D )P53,POMC and SASH1is necessary for the activation of G a s.HEK-293cells and NHEMs were transfected with HA-p53,myc-POMC and GFP-SASH1,respectively,according to different manners of combination.After 36hr after transfection,two normal cells were lysed and subjected to immunoblotting with GAPDH as loading control.(E )Exogenous G a s is induced by p53.HA-p53and GFP-G a s were introduced into HEK-293cells.After 36hr after transfection,cells were lysed and subjected to immunoblotting.Exogenous G a s was activated by gradually increased amounts of exogenous p53(HA-p53).(F )Exogenous G a s is induced by SASH1.GFP-G a s and GFP-SASH1were introduced into HEK-293T cells.Exogenous G a s was induced by gradually increased doses of exogenous SASH1.(G )and (H )Exogenous p53(HA-p53)overexpression induces exogenous POMC(myc-POMC)expression in a dose-dependent manner in HEK-293T cells and NHEMs.Different dose of HA-p53vector and a certain amounts of myc-POMC vector were transfected into HEK-293T cell for expression.Exogenous POMC RNA levels were measured by quantitative RT-PCR and normalized to GAPDH.Results of RNA levels are expressed as the mean of the experiment carried out in triplicate Æthe S.D.The expression of HA-p53and myc –POMC was analysed by Western blot as GAPDH as loading control.ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley &Sons Ltd and Foundation for Cellular and Molecular Medicine.807J.Cell.Mol.Med.Vol 21,No 4,2017SASH1is associated with G a s and induced by the canonical p53/POMC/G a s cascadeThe functional domains of SASH1(SAM and SH3)suggest that this protein plays a role in a signalling pathway as a signalling molecule adapter or as an associated scaffolding protein[8,9].Therefore,we performed a pull-down assay and a mass spectrometry analysis to investigate which signalling pathways are activated by SASH1.The pull-down assay and LC-MS/MS analysis demonstrated that SASH1 interacts with G a s and CALM,both of which are important in melano-genesis process(Table S1)in WT-A375cells.G a s is a key element of the classical signal transduction pathway linking receptor-ligand inter-actions with the activation of adenylyl cyclase and a variety of cellular responses[17].To investigate the associations between SASH1and G a s,HEK-293T cells were co-transfected with Flag-SASH1and GFP-G a s.Exogenous SASH1was immunoprecipitated with both exoge-nous G a s(GFP-G a s)and endogenous G a s.Exogenous SASH1 immunoprecipitates had different observed band sizes of G a s (Fig.2A and C).G a s mediates cAMP production in melanocytes which is stimu-lated by a-MSH and melanocortins[18],and our study here shows that G a s is associated with SASH1.Hence,we examine whether G a s is required for the induction of SASH1and how G a s mediates SASH1 expression,we introduced exogenous p53,POMC,G a s and SASH1 gene into HEK-293T and NHEMs to assess the effects of p53and POMC on G a s.Exogenous G a s was induced in the co-existence of exogenous p53and POMC(Fig.2C lane5and Fig.2D lane5),and both inducements of exogenous G a s and exogenous SASH1were observed in the co-existence of exogenous p53and POMC in two types of normal cells(Fig.2C lane6and Fig.2D lane6).Mean-while,in the presence of GFP-SASH1,GFP-G a s was also induced (Fig.2C lane4and Fig.2D lane4),which indicated that SASH1is necessary for the activation of GFP-G a s.And immunoblot showed that G a s was identified to be induced by exogenous p53and SASH1(Fig.2E and F).Our results also demonstrated that POMC was mediated by p53in HEK-293T and melanocytes were consis-tent with previous conclusions[1](Fig.2G and H).Conversely, endogenous SASH1and exogenous SASH1were induced by G a s (Fig.3A and B).To confirm the fact that POMC,p53and G a s are necessary for the inducement of SASH1,exogenous POMC,p53,G a s and SASH1were transfected into HEK-293T cells and followed by silence of G a s and POMC by two specific pairs of siRNA,respectively.As identified in HEK-293cells,knockdown of G a s gene directly induced signifi-cant reduction in SASH1(Fig.3C and D).Silencing of POMC resulted in the down-regulation of G a s and SASH1(Fig.3E and F).Taken above,it is believed that G a s serves as a pivotal downstream of p53/ POMC cascade and SASH1is regulated by a novel p53/POMC/G a s cascade.SASH1is physiologically induced by p53upon UV stimulationTo verify that SASH1is induced physiologically by p53,discarded normal human foreskin specimens were exposed to gradually increased dose of UV and stained for the histological analyses of p53, ACTH/POMC and SASH1.Immunohistochemical(IHC)analyses revealed p53is rapidly induced in basal layers at the0.5J/cm2dose of UV irradiation.The rapid induction of SASH1and POMC/ACTH at 1.0J/cm2dose of UV irradiation in melanocytes is followed by p53 up-regulation(Fig.4A).Previous reports had suggested that the up-regulation of POMC gene is induced at both protein and mRNA levels following UV irradiation of skin[19,20].Followed the previous descriptions[1],a100J/m2UVB dose was administered in this experiment.This dose is equivalent to the standard erythema dose (SED),which is commonly used as a measure of sunlight[21].So HEK-293T cells and NHEMs were transfected with exogenous POMC and followed by UV irradiation,both endogenous p53and SASH1pro-tein levels were assessed by immunoblot.UV markedly induced expression of exogenous POMC and endogenous SASH1by6hr,and p53induction was already maximal by3hr,which is consistent with its known stabilization by UV in NHEMs.At24hr,the levels of POMC, p53and SASH1protein were maximally induced by UV in NHEMs (Fig.4B).Similar inducement of exogenous POMC and endogenous p53and SASH1by UV irradiation was observed in HEK-293T cells (Fig.4C).Hence,we believe that not only POMC but also SASH1acts as a novel downstream partner which is responsive to the induction of p53by UV irradiation.Reciprocal induction between p53and SASH1is induced in normal cellsTo examine whether p53is required for the induction of SASH1, we introduced exogenous p53and POMC gene into HEK-293T and NHEMs to assess the induction of p53and POMC to SASH1. Exogenous SASH1was induced by p53in the presence of POMC (myc-POMC)in NHEMs and HEK-293T cells(Fig.S3).Exogenous SASH1was induced by increasing amounts of exogenous p53in two normal cells(Fig.5A and B).Conversely,exogenous p53was promoted by increasing amounts of exogenous SASH1(Fig.5CFig.3A novel p53/POMC/G a s/SASH1cascade regulates the expression of SASH1.(A)and(B)Endogenous or exogenous SASH1is induced by G a s.Gradually increasing amounts of exogenous G a s(GFP-G a s)and exogenous SASH1(GFP-SASH1)or only different doses of exogenous G a s were transfected into HEK-293T cells.The expression of endogenous or exogenous SASH1was analysed by immunoblotting along with GAPDH as loading control.(C)and(D)G a s is necessary for the inducement of SASH1.After transfection with GFP-G a s,myc-POMC and GFP-SASH1as well as increasing doses of HA-p53according to different combinations,two groups of HEK-293cells were subsequently introduced with two pairs of effective G a s siRNAs and negative control(NC)siRNA.Protein levels were detected by immunoblot.(E)and(F)POMC is necessary for the induce-ment of SASH1and G a s.After introduction into GFP-G a s,myc-POMC and GFP-SASH1as well as increasing dose of HA-p53according to different manner of combinations,two groups of HEK-293cells were subsequently silenced with two pairs of effective POMC siRNAs and NC siRNA.808ª2016The Authors.Journal of Cellular and Molecular Medicine published by John Wiley&Sons Ltd and Foundation for Cellular and Molecular Medicine.。
动物细胞转染基础知识大全(共5篇)

动物细胞转染基础知识大全(共5篇)第一篇:动物细胞转染基础知识大全转染转染(transfection)指真核细胞由于外源DNA掺入而获得新的遗传标志的过程。
转染方法的分类对外源基因转染方法的要求包括:转移效率高,不影响细胞正常生理活动,低毒性,容易使用,重复性好,易获得稳定转化子.根据转染的机制不同可划分为化学转染法和物理转染法两大类.一、化学转染法1.DEAE-葡聚糖法DEAE葡聚是最早应用哺乳动物细胞转染试剂之一,DEAE-葡聚糖是阳离子多聚物,它与带负电的核酸结合后接近细胞膜而被摄取,用DEAE-葡聚糖转染成功地用用于瞬时表达的研究,但用于稳定转染却不是十分可靠。
2.磷酸钙法磷酸钙法是磷酸钙共沉淀转染法,因为试剂易取得,价格便宜而被广泛用于瞬时转染和稳定转染的研究,先将DNA和氯化钙混合,然后加入到PBS中慢慢形成DNA磷酸钙沉淀,最后把含有沉淀的混悬液加到培养的细胞上,通过细胞胞膜的内吞作用摄入DNA。
磷酸钙似乎还通过抑制血清中和细胞内的核酸酶活性而保护外源DNA 免受降解.3.人工脂质体法人工脂质体法采用阳离子脂质体,具有较高的转染效率,不但可以转染其他化学方法不易转染的细胞系,而且还能转染从寡核苷酸到人工酵母染色体不同长度的DNA,以及RNA和蛋白质。
此外,脂质体体外转染同时适用于瞬时表达和稳定表达,与以往不同的是脂质体还可以介导DNA和RNA 转入动物和人的体内用于基因治疗。
人工合成的阳离子脂质体和带负电荷的核酸结合后形成复合物,当复合物接近细胞膜时被内吞成为内体进入细胞质,随后DNA 复合物被释放进入细胞核内,至于DNA 是如何穿过核膜的,其机理目前还不十分清楚。
二、物理方法1.显微注射显微注射虽然费力,但是非常有效的将核酸导入细胞或细胞核的方法。
2.电穿孔这种方法常用来制备转基因动物,但却不适用于需要大量转染细胞的研究。
电穿孔法常用来转染如植物园生智体这样的常规方法不容易转染的细胞。
Entranster-E:电转染过程中的影响因素

一电转的简介电穿孔转染,作为物理方法的一种,不仅能将DNA、RNA,还能将抗体、酶及其他生物活性分子转入细菌、酵母、动物细胞和植物细胞。
它是一种高效。
简便的基因转移系统,具有其他转移方法无法比拟的优越性,如操作简便、快捷,可重复性强,转染率高,适用广谱等,尤其对目前一般认为难转的悬浮培养细胞也能获得较高的转染。
二电转原理电穿孔法是通过电场作用于细胞几微秒到几毫秒之后,在细胞膜上暂时形成小孔或开口,把大分子如DNA等导入细胞并最终进入细胞核的技术。
其过程简述如下:首先在电击过程中,细胞膜上出现穿孔,质粒在电泳力的作用下与细胞膜接触,并在细胞膜上电穿孔的区域形成一种可转移的复合物。
再次电击后,质粒脱离复合物并扩散至细胞质内,开始瞬转;同时小部分质粒进入核内与染色体整合,开始稳转。
一旦DNA扩散至细胞,膜上小孔会关闭。
三电穿孔转染条件的选择1 电场参数电场是电转染的重要因素,细胞在电场的作用下,膜通透性增加或是形成小孔,以完成转染过程。
因此电场强度是应该被优化的主要参数。
电场强度不能过高,过高会增加细胞的死亡率;也不能过低,过低不能增加膜的通透性或在膜上形成小孔。
因此,一个适宜的电场强度至关重要。
不同细胞系具有不同的最佳场强值,其确定方法除了实验直接测定比较不同场强下转染率的高低外,还可以采取较为简便的间接法。
文献显示存活率在50%左右的电场参数为理想参数,故可间接测定存活率来确定最佳场强值。
2脉冲过程1)脉冲波形脉冲波形主要分为两种:1、方波脉冲;2、指数递减波脉冲。
方波脉冲是指:电压瞬间升至预设电压,保持电压放电,然后瞬间终止放电。
一般哺乳动物细胞电转染时选择方波脉冲,有较高的转染效率和细胞存活率。
指数递减波脉冲是指:先对电容充电,然后让电容完全放电,其电压变化呈指数递减。
一般这种波形的脉冲电转染适用于细菌、酵母菌、昆虫细胞。
2)脉冲时间脉冲时间的选定主要取决与脉冲波形。
在方波脉冲中,脉冲时间可直接设定。
转染资料整理终结版

转染转染——让克隆的核酸进入真核细胞中,已经成为研究和控制真核细胞基因表达的重要手段。
比如表达纯化特定的蛋白;鉴定一个基因的生物学特性;突变分析;研究基因表达对细胞生长的影响,研究基因表达的调控机制等等,等等。
如今广义的转染不单包括了DNA、RNA,还有蛋白质等生物大分子。
生物通在前面已经为大家集中介绍了8个不同的品牌的转染试剂——不是选美,没有包医百病的万灵丹,只是希望有助于你了解每种产品的特性。
这里我们最后“唐僧”一下,转染中一些值得注意的事项。
很多因素会影响转染效率,细胞株本身啦,细胞培养环境啦,转染的DNA、RNA 或者蛋白的质量和特性啦,转染方法啦,等等。
每个转染高手也必然有自己的独门心经,只可惜大家都为实验或者生活而疲于奔命,没有几个人愿意静下心情来仔细总结经验汇集成文字——那些曾经用无数失败的痛苦烦恼换回来的宝贵经验,那些曾经以为会永远铭刻于脑海的教训,随时间的流逝而终于渐渐褪色,到模糊。
即使偶尔有珠玉掩埋于浩瀚的口水中,也难以汇串成珠。
试剂盒的盛行,让实验更快,更简单,也更机械化,相信更多人更愿意先抓起Protocol 123地往下做,直到碰壁再苦思原因。
其实等碰得遍体鳞伤回头一看就会醒悟,很多坑,只要事前注意了就不会陷进去的。
幸好还有一些资料可寻,生物通姑且在这里抛砖引玉,罗列一些吧。
这一part很闷的。
DNA转染后,转入基因的表达可以在1-4天内检测到——仅有一部分转入细胞的DNA被转运到细胞核内进行转录并最终输出mRNA到细胞质进行蛋白合成。
几天内,大部分外源DNA会被核酸酶降解或随细胞分裂而稀释;一周后就检测不到其存在了。
因此转染也可以分为瞬时转染和稳定转染。
瞬时转染(transient transfection)指的是转染的核酸不整合到染色体上,结果是短暂的高水平表达,可在24—96小时内检测表达效果,表达水平与位置无关,不会受到周围染色体元件的影响。
瞬时表达分析所需的人力和时间比稳定表达少,但因为DNA摄入效率和表达水平在不同实验中差异较大,不长久也不稳定。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
动物体内转染实验的重复性/可靠性如何?
运用体内转染试剂Entranster进行动物体内实验时,实验的重复性或可靠性和整体的实验分不开,如果基础实验(比如细胞实验)充分,而且又有其他的方法进行印证,那么实验的可靠性就很高,这时动物体内转染就不需要太多动物,几十只动物,能说明问题就可以。
但如果其他的实验较少,单纯用一种方法进行实验,就需要排除实验过程的客观因素的影响,比如动物本身的差异,比如实验过程的差异,比如检测方法的误差等。
这时就需要动物数量足够,以达到统计学上的误差要求。