粘多糖沉积病的临床表现
粘多糖病资料

黏多糖病黏多糖病(mucopolysacchrides,MPS)属于酶缺乏的溶酶体病的一种,由于酶缺陷使酸性黏多糖在体内不能降解,导致黏多糖及其降解产物在组织内沉积,引发一组多脏器异常的先天性疾病。
多以骨骼畸形为主要症状,广泛涉及神经系统、心血管系统、消化系统各组织。
【发病机制】黏多糖又称糖胺多糖(glycosaminoglycan,GAG),是骨基质及结缔组织细胞内的主要成分,由糖醛酸(uronicacid)和N-乙酰已糖胺(N-acetylhexosamine)或硫酸酯组成的二糖单位重复序列的大分子,是多阴离子多聚体(Polyanionic polymers)的糖胺多糖,是一种酸性黏多糖(acid mucopolysacchrides,MPS)。
黏多糖主要成分有:4-硫酸软骨素(chondroitin-4-sulfate,C4S)、6-硫酸软骨素(C6S)、硫酸皮肤素(dermatan sulfate,SD)、硫酸角质素(karatan sulfate,KS)、透明质酸(hyaluronicacid,HA)、硫酸肝素(heparin sulfate,HS)、硫酸乙酰肝素(hapa然sulfate),硫酸肝素与硫酸乙酰肝素统称硫酸类肝素。
这些糖胺多糖与核心蛋白连接组成蛋白多糖(Proteoglycan,PG)成为体内组织的成分。
【临床表现】MPS可以根据缺陷酶不同分为八型(见表),多数为常染色体隐性遗传,其中II型为X性联遗传。
自生后即可发现症状,有其骨骼发育异常时发现较早。
骨骼异常不明显的患儿新生儿期可以是正常的,随年龄的长大逐渐出现智力落后、肝脾肿大,长椅小儿生长发育落后、睡觉打鼾、面容丑陋就诊。
不同型别的患儿初发症状不同,除Ⅳ型智力发育正常外,其余各型均有不同程度的智力落后,甚至智力倒退。
黏多糖分型、缺陷酶、体征————————————————————————————————————类型综合名称缺陷酶沉积黏多糖成分体征————————————————————————————————————MPSⅠ-H Hurler L-艾杜糖醛酸苷酶DS,HS KS3:1 MPSⅠ-D Scheie a-L-艾杜糖醛酸苷酶(轻型)DSMPSⅠ-H/S Hurler/Scheie a-L-艾杜糖醛酸苷酶(中间型)DS,HS MPSⅡ-A Hunter艾杜糖醛酸-2-硫酸酯酶DS,HS 经典型,智力落后,肝脾肿大,面容丑陋。
粘多糖贮积症
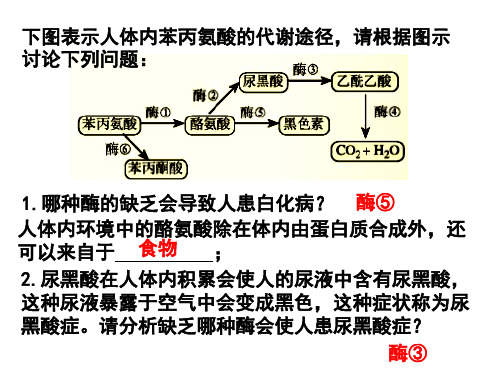
人体内苯丙氨酸的代谢途径如下图所示,人群中有若干遗传病 是由于苯丙氨酸的代谢缺陷所的。例如苯丙氨酸的代谢产物之 一苯丙酮酸在脑中积累可阻碍脑的发育,造成智力低下。
(4)粘多糖贮积症是一种高致病性、高致死性的遗传病。其致病 机理是水解粘多糖的酶缺失,导致粘多糖沉积于细胞内的_溶_酶_体 中,进而影响多种组织细胞的正常功能。 (5)为减少粘多糖贮积症的发生,该家庭第IV代中的_2_,4_,6_(填 写序号)应注意进行遗传咨询和产前诊断。进行产前诊断时可以 抽取羊水,检测胎儿脱落上皮细胞的_致_病_基_因_(或_相关_酶_的_活性_) 。
某两岁男孩(IV—5)患粘多糖贮积症,骨骼畸形,智力低下, 其父母非近亲结婚。经调查,该家族遗传系谱图如下。
(3)测定有关基因中,父亲、母亲和患者相应的碱基序列分别为:
父亲:....TGGACTTTCAGGTAT.... 母亲:....TGGA... 患者:....TGGACTTCAGGTACA....
1.在此途径中表现正常的人,其基因型有: PPEE,PPEe,PpEE,PpEe
2.导致病人既“白”(白化病)又“痴”的直接原因是: 酶2的合成受阻,没有酪氨酸和黑色素生成,同时苯丙酮酸增多 3.导致病人只“白”不“痴”的直接原因是: 酶3的合成受阻,没有黑色素生成,同时苯丙酮酸积累较少
人体内苯丙氨酸的代谢途径如下图所示,人群中有若干遗传病 是由于苯丙氨酸的代谢缺陷所的。例如苯丙氨酸的代谢产物之 一苯丙酮酸在脑中积累可阻碍脑的发育,造成智力低下。
粘多糖病(mucopolysacharidosis,MPS)

概述粘多糖病(mucopolysacharidosis,MPS)是因酸性粘多糖降解酶缺乏,使之不能完全降解,其产物在体内堆积所致。
由于各种成分在体内分布的不同,以及不同酶的缺乏,粘多糖病在临床上表现亦各异,多以骨骼病变为主,还可累及中枢神经系统、心血管系统以及肝、脾、关节、肌腱、皮肤等。
按其代谢产物和临床表现共分为8型,有的还有数种亚型。
病因和发病机制粘多糖(macopolg sacrotein MPS)是骨基质和结缔组织的主要成分之一。
粘多糖的正确命名应该是糖胺多糖(glycosaminoglycan,GAG),它是由糖醛酸与乙酰氨基糖或其硫酸酯组成的二糖单位形成的重复序列。
已知哺乳动物的GAG包括4-硫酸软骨素(chondroitin-4-sulfate,C4S),6-硫酸软骨素(C6S),硫酸皮肤素(dermatan sulfate,SD),硫酸角质(素)(Karatansulfate,KS)和肝素(haparin,HP)还有硫酸肝素或硫酸乙酰肝素(haparan,HS)总称为类肝素,及透明质酸(hyaluronic acid,HA)七种。
这些糖胺多糖与核心蛋白连接后组成蛋白多糖(proteoglycan,PG)。
蛋白多糖的旧名称粘蛋白。
糖胺多糖就是酸性粘多糖(acid mucopolysaccharide,AMPS)。
这些多糖的降解必须在溶酶体中进行,目前已知有10种溶酶体糖苷酶、硫酸酯酶和乙酰转移酶参与其降解过程,任何一种酶的缺陷都会造成氨基葡聚糖链的分解障碍而积聚体内,并自尿中排出。
病理改变粘多糖在纤维细胞内沉积,染色成为气球样细胞,称为Hurler细胞,存在于肝、脾、淋巴组织的网状细胞中,在软骨细胞和成骨细胞,中枢神经系统和周围神经节,视网膜细胞和角膜细胞中也均有类似的物质堆积。
在心内膜沉积形成斑状增厚,主动脉,肺动脉、冠状动脉和脑、肾、肝、脾和四肢的动脉壁均有沉积。
临床表现出生后发育正常,1岁前逐渐出现体征。
粘多糖

(三)分析门诊资料
1.尿粘多糖测定 通常用甲苯胺蓝法做定性试验,MPS病儿尿液呈阳性反应。
(二)临床类型
目前对引起粘多糖病的酶缺陷都已鉴定,根据酶缺陷的不同,MPS共分为六型。
1.粘多糖病I-H型(Hurler综合征)
临床表现该型是最严重的一型,常在10岁左右死亡。全身脏器如角膜、软骨、骨骼、皮肤、心肌内膜、血管结缔组织等均受累。表现为智力低下,面容丑陋,肝脾大,骨骼病变,心血管病变,角膜混浊,耳聋。末梢血白细胞、淋巴细胞可见到大小不等、形态不同的深染颗粒,有时呈空泡状。尿中排出大量酸性粘多糖(>100 mg/24小时,正常为3-25 mg/24小时)。
2.粘多糖病I-S型(Scheie综合征)
原先分类为粘多糖病V型,属中等度严重类型粘多糖病,临床症状一般在5岁后出现,智能发育正常。遗传类型和致病基因同粘多糖病I-H型。
3.粘多糖病Ⅱ型(Hunter综合征)
临床表现:临床重型与粘多糖I-H型相似,2-6岁起病。表现为智力低下,特殊面容,肝脾大,骨骼畸形,但脊椎无鸟嘴样突畸形,可并发充血性心力衰竭,无角膜混浊,有进行性耳聋。
Sanfilippo B a-N-乙酰巳糖胺酶 产物:HS
Sanfilippo C a-葡糖胺N-乙酰转移酶 产物:HS
Sanfilippo D N-乙酰葡糖胺-6-硫酸酯酶 产物:HS
Ⅳ型 Morquio综合征
Morquio A N-乙酰半乳糖胺-6-硫酸酯酶 产物:KS,CS
粘多糖贮积症基于GAG累积的检测方法

粘多糖贮积症基于GAG 累积的检测方法 苑晓舟,段晋燕,孟岩,王成彬苑晓舟,解放军医学院2014级硕士研究生,主要研究方向为遗传代谢性疾病的实验室诊断。
[摘要] 粘多糖贮积症(mucopolysaccharidosis,MPS )是由于溶酶体内水解酶失活或者活性降低,从而造成粘多糖(glycosaminoglycans ,GAGs )在各组织器官累积的一种遗传代谢性疾病。
由于GAG 累积是MPS 主要的病理原因,基于GAG 贮积的生物标志物可作为一种潜在的简单便捷的生物标志物,用来诊断并且监测疾病进展和治疗效果。
本文总结了通过检测GAG 含量,对MPS 病人进行疾病诊断、病情评估和疗效评价的各种方法,包括染料结合法、抗体免疫法、质谱法,为临床选择提供参考。
[关键词] 粘多糖贮积症;糖胺聚糖;糖类标志物;方法学[中图分类号] R725.8 [文献标志码] A [文章编号] 2095-2775(2015)04-1008-06[Abstact ] The mucopolysaccharidoses(MPS) result from attenuation or loss of enzyme activities required for lysosomal degradation of the glycosaminoglycans(GAG). GAG storage is the primary biochemical event in MPS; thus biomarkers based on GAG storage can report directly the severity of the disease. This review provides a summary of glycan biomarkers that have been used for diagnosis of patients and for monitoring disease progression and therapy. We discuss various approaches for assessing GAG accumulation in MPS patients,including dye binding methods ,antibody-based assays and total GAG analysis by mass spectrometry ,and provide choices for the physicians.[Key words ] Mucopolysaccharidoses; Glycosaminoglycans; Glycan biomarker; Methodology[作者简介] 苑晓舟,硕士研究生。
《粘多糖贮积症》课件

诊断标准
根据患者临床表现、家族史、实验室检查等进行综合评估, 确诊需依赖基因检测。
流程
收集病史→体格检查→初步实验室检查→遗传咨询与家族史 调查→基因检测→确诊。
鉴别诊断与其他相关疾病
鉴别诊断
需与戈谢病、尼曼匹克病等其他溶酶体贮积症相鉴别,主要依据临床表现、实验 室检查和基因检测结果。
相关疾病
粘多糖贮积症各型之间、以及与其他溶酶体贮积症之间的临床表现存在重叠,需 仔细鉴别。
诊断技术
目前已经开发出多种诊断粘多糖贮积症的方 法,如基因检测、生化检测和影像学检查等 ,这些方法能够较为准确地诊断该疾病。
治疗手段
针对不同类型的粘多糖贮积症,已经探索出 多种治疗手段,如酶替代治疗、基因治疗和 干细胞治疗等,这些方法在临床试验中取得
了一定的疗效。
研究热点与方向
新药研发
目前正在积极开展针对粘多糖贮积症的新药研发,旨在开发出更加安全、有效的治疗药 物。
《粘多糖贮积症》PPT课件
目录 Contents
• 粘多糖贮积症简介 • 粘多糖贮积症的诊断与鉴别诊断 • 粘多糖贮积症的治疗与护理 • 粘多糖贮积症的预防与控制 • 粘多糖贮积症的科研进展与展望
01
粘多糖贮积症简介
定义与分类
定义
粘多糖贮积症是一组罕见的遗传代谢性疾病,由于缺乏特定的酶导致体内粘多 糖代谢异常,粘多糖在组织中大量贮积,引Байду номын сангаас一系列的病理生理改变。
给药方案
根据患者的年龄、体重和病情,制定个性化的给药方案,确保药物的有效性和安全性。
手术治疗与适应症
手术治疗
对于某些类型的粘多糖贮积症,如影响到重要脏器功能的病例,可能需要手术治疗。
粘多糖贮积症的症状

粘多糖贮积症的症状
一、粘多糖贮积症的症状二、粘多糖贮积症怎么治疗三、粘多糖贮积症Ⅵ型如何鉴别
粘多糖贮积症的症状1、粘多糖贮积症的症状
经典型的患者,以Ⅰ型为例:
1.1、粗糙面容:头大,舟型头,前额突出,眉毛浓密,眼睛突出,眼睑肿胀,鼻梁低平,鼻孔上翻。
1.2.角膜混浊:随着疾病的进展,角膜混浊逐渐明显严重,可致失明。
1.3.关节僵硬:累及大关节,如肘关节,肩关节及膝关节,使这些关节的活动度受限。
1.4.身材矮小:患者脖子短,脊柱后凸,2~3岁生长几乎停止。
1.5.肝脾增大:腹部膨隆,腹腔压力大导致脐疝和腹股沟疝,手术修复后仍易复发。
1.6.智力落后:患者在1岁左右可能就表现有智力落后。
2、粘多糖贮积症是什么病
黏多糖贮积症是由于人体细胞的溶酶体内降解黏多糖的水解酶发生突变导致其活性丧失,黏多糖不能被降解代谢,最终贮积在体内而发生的疾病。
该病是溶酶体贮积病中非常重要的一类,可分为Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅵ,Ⅶ,Ⅸ型等7种型,其中Ⅲ又分为ⅢA,ⅢB,ⅢC,ⅢD四个亚型,Ⅳ型分为ⅣA和ⅣB亚型。
3、粘多糖贮积症的病因
病因:身体缺乏能分解粘多糖的酶
粘多糖症(全名为粘多糖贮积症,英:Mucopolysaccharidoses ,简称。
亨特征名词解释

亨特征名词解释
亨特氏综合征(Hunter's syndrome),也称为黏多糖贮积症II型(Mucopolysaccharidosis type II,MPS II),是一种罕见的遗传性代谢障碍疾病。
它是由于身体缺乏一种名为艾杜糖醛酸-2-硫酸酯酶(iduronate-2-sulfatase)的酶而引起的,这种酶对于分解体内的特定类型糖分子——黏多糖(glycosaminoglycans,GAGs)是必需的。
当这种酶的功能受损或缺失时,黏多糖无法被正常分解,导致其在身体的细胞和组织中积累,进而引发多种症状和并发症。
亨特氏综合征通常影响男性儿童,因为这种病通常是X染色体隐性遗传,女性通常是携带者。
亨特氏综合征的症状可能包括:
1. 粗犷的面部特征:随着黏多糖在面部组织中的积累,患者可能出现粗糙的面部外观。
2. 关节僵硬:黏多糖在关节中的积累可能导致关节僵硬和运动受限。
3. 呼吸道问题:积累的黏多糖可能影响呼吸道的正常功能,导致呼吸困难或睡眠呼吸暂停。
4. 心脏问题:心脏受累可能导致心脏瓣膜病变或其他心脏并发症。
5. 听力损失:黏多糖在耳朵中的积累可能导致听力下降甚至耳聋。
6. 发育迟缓:患者可能在身体和智力发展上出现迟缓。
亨特氏综合征的治疗目前主要是对症治疗,包括物理治疗、呼吸支持和听力辅助设备。
在某些情况下,可能会使用骨髓移植或基因疗法来尝试减缓疾病的进展。
患者还需要定期接受专业医疗团队的监测和管理,以减轻症状并提高生活质量。
粘多糖沉积病的检查项目有哪些?

粘多糖沉积病的检查项目有哪些?检查项目:尿液粘多糖检测、遗传筛查粘多糖沉积病是由于细胞溶酶体酸性水解酶先天性缺陷所致。
根据病因目前本病可分为八大类型,我国已报告200从余例,主要表现为严重的骨骼畸型、肿脾肿大,智力障碍以及其它畸形。
粘多糖沉积病产前诊断以测定培养羊水细胞内特异的酶活力最为可靠,但实验要求高,一般实验室难开展。
两种较简单的实用的方法是甲苯胺蓝定性及糖醛酸法半定量测定。
1.甲苯胺蓝定性:方法同尿液粘多糖检查。
妊娠早期羊水正常者可呈阳性,妊娠中后期为阴性如阳性提示胎儿患粘多糖沉积病。
2.糖醛酸半定量测定:羊水中酸性粘多糖与四硼酸钠硫酸溶液反应生成糖醛酸,以每毫无肌酐中糖醛酸的量反映酸性糖多糖的多少。
随着妊娠的进展,糖醛酸含量逐渐减少,妊娠16-20周的参考值为3.3~7.0mg/mgCr,如高于此值应考虑有粘多糖沉积肥病。
本法除Morguio综合征外对其它类型的粘多糖沉积病均有诊断意义。
3.常染色体隐性遗传病(autosomalrecessivedisorder):致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。
此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。
子代有1/4的概率患病,子女患病概率均等。
许多遗传代谢异常的疾病,属常染色体隐性遗传病。
按照“一个基因、一个酶”(onegeneoneenzyme)或“一个顺反子、一个多肽”(onecistrononepolypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。
常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性(Wilson病)及半乳糖血症等。
粘多糖累积病继发青光眼1例

7 0
网孔流 出的 ,网孔 大小直接 影 响房水 引流 的通畅 程度 。本 病例 眼压升高是 由于粘 多糖在细胞 外沉 积所致 , 目前 尚无根 治方法 , 小梁 切除术可 能使部 分症状缓解 , 但其预后差 。
病 , 病是 由于肝 脏 、 肉等 组织 中缺 乏分解糖 原 该 肌 的某些酶引起 的 ,是一种 以组ห้องสมุดไป่ตู้ 中糖浓度 异常 或糖分子结 构异 常为特点 的代谢性疾 病 。该 患者 症 状 、体征 检查结 果均符合 粘多糖 累积病这 一诊 断。 粘多糖包括氨 基葡聚糖和蛋 白聚糖 , 是细胞外
维普资讯
蓑刺 獭麟 藏萋 鲤; 誊 囊 曩纂 囊纂 囊 囊 ; 萋;
床并 不多见 , 一旦发 生且未得 到有效 治疗 , 可能造
局 部处 理相结 合 , 彻底 止血 , 清除 眶 隔 内积血 , 降 低 眶内压 , 有效 预防感染 。 患有血液病更 易导致 此 种并 发症 , 术前 注意询 问病 史 , 重视全 身及血液 检
杂 志 , 9 ,: 4 2 6 1 662 - 8 . 9 8
2杨群.眼袋整形 术并发症 的防治 .实用 整形美 容外科 杂 志, 9 ,: 1 66 9
2 0 2 2. 9 - 9
3 茂 昌 , 编 . 代 眼 部 整 形 美 容 学 . 1版 , 安 : 界 图 书 出 版 西 安 林 主 现 第 西 世
( 收稿 日期 :0 2 0 — 9) 20 - 3 1
多糖累积病继发性青光 眼 ( ) 人 院后 给予 降眼 双 。 压 , 一步查房角 , 压 , B超 , 进 眼 眼 确诊 后行双 眼小 梁切除术 , 手术顺利 , 术后 l , d 前房消失 , 滤过泡苍 白弥漫 , 给予 1g 0/ L阿托品散瞳 , 疗效不佳 , 急诊行 前 房成 形 术 , 术后 前 房 仍 浅 , 1 C , 续 给 予 约 / T继 2
Ⅳ型粘多糖症的X线表现

变短 变粗 ,椎体变扁 ,上 F面凸陷如凹透镜 ,扁平颅骨 ,无
颅外形 良好 ,角膜无异 常 ,鼻端 正 .牙齿发育 不全 鸡胸 、 肺部可 闻及广泛干湿罗音 ,肋骨 畸形 .心 ( 。肝脾j 常 。 一) F
腰背 部后 突 畸 形。积 下肢 呈 “ ” 型,肌 力 减 退 .五指 正 x
型粘多糖症特征改变
l 0余天”为代诉 ^院。患儿 1 岁时能站立 ,但不能行 走 , 周
后逐渐四肢肌肉及腹部脂肪 消退 .不能站立 ;2年前 始双下 肢畸形呈 “ x”腿。 曾在 市妇 幼 保 健所 诊 为佝偻 病 . 予以
4 晚发型脊柱骨骺 发育 不 良:为一 种遗传性 脊柱骨骺
发育不 良致成短躯 干型侏懦 发病 晚 .株儒 轻 ,仅 见男性 。 无严重骨关节畸形 .尿 中无 异常粘多糖 ,推体变扁只发生于 椎体前部 .而中部则呈驼峰样隆起。椎 间隙变狭窄 。可累及 髋 膝 、踝等负重关节
侏儒症 、无粘 多糖代谢紊乱 。
小结 本 文报告 粘 多糖病 Ⅳ型 1 ,着重 讨论 x线征 例
象 .提出鉴别 诊断要点 Ⅳ型的 x线特征 为椎体普遍 变扁 , 椎间隙增 宽,前缘 中部呈舌状突起 ,长骨骨骺及 干骺端有无
菌性 坏 死 。 ( 收稿 :2 0 —0 1) 0 1 7 2
。
v 治疗 ,症状无好转 。1 前突发 发热 咳喘 ,以重症 t 0天
肺炎 、骨髂畸形 收住 ^院。
体格检查 :体温 3 .℃ 神 志清 楚 ,精 神倦 怠 ,营 养 85
不 良外观 ( 重度 ) ,发育欠佳 ,侏懦 外形 .但 智力 正常 头
5 成骨不全 :骨皮 质变薄 ,易骨折 ,骨 痂形成 。四肢
维普资讯
粘多糖病病程进行性

粘多糖病病程进行性*导读:粘多糖病如同其他溶酶体累积病一样,各型MPS患儿大多在周岁左右发病,病程都是进行性的,并且累及多个系统,有着类似的临床症状。
但各型的病情轻重不一,且有各自的特征。
其中以IH型最典型,预后最差,常在lO岁以前死亡,IS型最轻。
……粘多糖病(MPS)是一组由于酶缺陷造成的酸性粘多糖分子不能降解的溶酶体累积病。
粘多糖是构成细胞间结缔组织的主要成份,也广泛存在于哺乳动物各种细胞内。
重要的粘多糖有:硫酸皮肤素(DS),硫酸类肝素(HS),硫酸角质素(HS),硫酸软骨素(CS)和透明质酸(KS)等,前3种与本组疾病关系密切。
这些多糖都是含氮的直链杂多糖,由不同的双糖单位重复连接而成;其中一个成份是N-乙酰氨基己糖,另一个则为糖醛酸或己糖,如:DS为N-乙酰氨基半乳糖(Gal N)和艾杜糖醛酸(Id UA)或葡萄糖醛酸(Glue UA);KS为N-乙酰氨基半乳糖(Gal N)和艾杜糖醛酸(或葡萄糖醛酸);l(S为N-乙酰氨基葡糖和半乳糖(Gal);CS为N-乙酰氨基半乳糖和葡萄糖醛酸。
每个氨基葡糖直链约由50~100个分子组成,许多直链同时又与一条肽链结合,即构成一个更大分子量的聚合体,结缔组织便是由这类大分子所形成。
这些多糖的降解必须在溶酶体中进行,目前已知有10种溶酶体糖苷酶、硫酸脂酶和乙酰基转移酶参与其降解过程,其中任何一个酶的缺陷都会造成氨基葡聚糖链的分解障碍而积聚在体内,并自尿中排出。
患儿缺陷酶的活性常仅及正常人的l%~10%。
【分型】粘多糖在各系统器官内的累积导致了这些器官的病理改变和临床症状。
根据临床表现和酶缺陷,MPS可以分为Ⅰ-Ⅶ等6型,其中V型已改称为IH/S型。
除Ⅱ型为性连锁隐性遗传外,其余均属常染色体隐性遗传病。
【临床表现】如同其他溶酶体累积病一样,各型MPS患儿大多在周岁左右发病,病程都是进行性的,并且累及多个系统,有着类似的临床症状。
但各型的病情轻重不一,且有各自的特征。
代谢底物堆积引起的疾病

代谢底物堆积引起的疾病
代谢底物堆积是一种与代谢过程相关的疾病,常见的疾病包括:
1. 酮症酸中毒:当体内糖原储备不足时,机体开始分解脂肪产生酮体作为能量来源。
然而,在一些代谢障碍的情况下,酮体无法正常代谢,导致酮体在体内堆积,引发酮症酸中毒。
2. 铜沉积病:铜沉积病是一组遗传性疾病,其中铜在体内不能正常排泄、处理和合成。
这导致铜在体内堆积,最常见的铜沉积病是威尔逊病,可以影响肝脏、中枢神经系统和其他器官。
3. 粘多糖病:粘多糖病是一组遗传性疾病,它们影响糖蛋白的降解酶的产生或功能。
这导致糖蛋白在细胞内无法正常降解,积累在细胞内,造成组织和器官损害。
粘多糖病有多种亚型,如猩红病、科布病等。
4. 糖原贮积病:糖原贮积病是一组遗传性疾病,影响到体内糖原的合成、降解或转化。
这些疾病导致体内糖原无法正常代谢,导致糖原在组织和器官中过度堆积。
糖原贮积病有多种类型,如麦考尔-克滕病、Andersen病等。
5. 高胆固醇/高甘油三酯血症:这是一种与脂质代谢障碍相关
的疾病,主要是由于体内胆固醇或甘油三酯代谢异常,导致胆固醇或甘油三酯在血液中过度堆积,增加心血管疾病的风险。
这些疾病的症状和严重程度各不相同,治疗方法也因疾病而异,
包括节食、药物治疗和酶替代治疗等。
及早诊断和治疗可以减轻疾病的影响。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代谢障碍,将导致产生各种类型的粘多糖沉积症(mucopo-lysaccharidoses)。
其特征是过多的寡聚糖堆积与排泄。
各型粘多糖沉积症的代谢基础相似,但遗传类型和临床表现各不相同。
1、粘多糖贮积症I型又称Hurler综合征、承雷病。
AR.为溶酶体a-L-艾杜糖醛酸酶缺乏,发病率1/10万新生儿。
主征;进行性发育迟缓,体矮,智力低下,舟状头,颈短,面容粗陋,有角膜混浊,关节僵硬,活动受限,驼背。
其他:视网膜色素沉着,耳聋,胸廊畸形,肝脾肿大,腹大,心脏瓣膜缺损和动脉硬化等。
粘多糖筛查阳性,根据酶活性测定可检出携带者。
预后差,多于儿童期死亡。
2、粘多糖贮积症II型又称Hunter综合征。
XR,无女性患者,发病率为6/10万。
为艾杜糖醛酸硫酸酯酶缺乏。
主征:与I型相似,但无角膜混浊,背不驼。
其他:多毛,进行性耳聋;肌肉痉挛,行为莽撞。
10岁后2/3患者有惊厥发作。
生化诊断如I型,主要根据临床表现相鉴别。
重型者多在青春期前死亡。