一种新型Au(Ⅲ)-吡啶酰胺配合物的合成,表征及其与5′-单磷酸鸟苷的反应研究
3_甲基吡啶合成工艺路线进展.

(Sc hool of Che m istry and Che m ical Engineeri n g , Sou t h eastUn i v ersity , Jiangsu N anji n g 211189
Abstr act The application and productive situa ti o n of 3-m ethyl p yri d i n e are i n troduced briefly . Its synthesi s
进步,该产品的新用途将不断被开发应用。
比利时的Re illy Che m ica ls公司及美国的Reill y Industries Inc公司是世界吡啶及其衍生物产量最大
的公司[1]
,欧洲是世界上最大的吡啶系列产品生产和消费地区, 2001年产量约60k, t消费量约36k, t其余产品全部出口,销往世界各地,是世界上吡啶系列产品出口量最大的地区。
以乙醛、甲醛、铵盐为原料,反应式如下
:
RolfD i n kel等
[12]
采用液相反应:在高压釜中加
入磷酸氢二铵的溶液并加热到235e ,然后在63m i n
之内连续加入乙醛与甲醛水溶液,控制反应压力3. 8~4. 0MPa。反应结束时用甲苯萃取产物。主要产物3-甲基吡啶68. 0%, 3-乙基吡啶15%。
收稿日期:2009-09-28
作者简介:姜枫(1987~,女,硕士生;通讯作者:肖国民(1967~,男,博士,教授,博导,主要从事吡啶及其衍生物的生产工艺研究,
E -ma i :l xiaogm @seu . edu . cn
P rogress of P re paration of 3-m ethyl pyr i d i ne
酰胺衍生寡聚DTPA钆配合物的合成、表征及弛豫性能

1 3 配 合物 弛豫 时间的测 试 .
按 照文献 [9 方 法测定 纵 向弛豫 时间 .称取 一定 量 的寡 聚配合 物 , 1] 加入 适量 N O a H溶 液配制 成 1 5m o L系列浓 度 的水溶液 (H= . 70 .分别 取 1 L不 同浓 度 的溶 液 和 40 LD O注入 — ml / p 65— . ) 0 0 N MR样 品管 中 , 荡 ,} 均 匀 后 封 口待 用 。在 N 振 昆合 MR波 谱 仪 上 启 动 IV E . u 自动 程 序 ,脉 宽 N RcA t 9 。=28I , 冲间隔 t 5 , 4 0MH 磁场 强度 和 3 0 ) . t 脉 ( s 为 在 0 z 5℃恒 温下 ,用反 转 恢 复法分 别 测 定水
其 中 G ¨含量 用络 合滴定 法测定 . d
H O oc
N H00C-N N / 厶 00I c O  ̄- O H C
T c 0 A2 厂 OH c 0
。
Hz N —
。。
NH 。 NH:
,
— y 面 丽I u
N。3。。 。 。。。。。。’AP 。’0。’ D。。。 。。。。‘ 。。。*。。M 。。。 。 。  ̄ 。 。。。 。。 S‘f M / C 。 。 O。’ — D
毒性亦有所降低. 但是螯合物的分子量过大会使其在体 内停 留时间过长 , 必然存在螯合物分解 , 生成 游 离 G 3的可 能 , d+ 而游 离 G 3 细胞 吸 收后会 产生 极大 的细 胞毒性 .鉴 于此 , 开潮 等 d 被 俞 曾用多胺 多羧酸与二醇化合物聚合 , 得到了分子量适中的寡聚脂类配合物 , 这类配合物作为造影剂具有毒性低
郝 志峰 吴雅红 汪朝 阳 , , , 黄卓 亮 , 余 坚 余 , 林
2一氨基一3一羟基吡啶的合成

从简到繁,由浅入深地探讨"2一氨基一3一羟基吡啶的合成"这一主题。
一、2一氨基一3一羟基吡啶的概念2一氨基一3一羟基吡啶,又称2aminopyridine3iol,是一种含氮杂环化合物,具有重要的应用价值和研究意义。
它可以作为有机合成的中间体,也可以用于药物合成等领域。
二、2一氨基一3一羟基吡啶的合成方法1. 传统方法:通过取代反应合成。
首先合成氨基吡啶,然后进行羟基化反应。
2. 新型方法:采用金属催化剂进行合成,提高产率和反应选择性。
三、2一氨基一3一羟基吡啶的应用领域1. 有机合成领域:作为重要的中间体,参与多种有机合成反应,例如偶氮染料的合成等。
2. 药物合成领域:作为药物分子的结构骨架,具有抗癌、抗病毒等药理活性,被广泛应用于药物研发。
四、2一氨基一3一羟基吡啶合成的研究进展1. 合成方法的改进:研究者不断探索新的合成路径,提高产率和反应选择性。
2. 应用领域的拓展:研究者发现了2一氨基一3一羟基吡啶在新领域的应用潜力,如光催化反应等。
五、个人观点和理解2一氨基一3一羟基吡啶作为一种重要的有机合成中间体,在化学品合成和药物合成领域具有广阔的应用前景。
随着合成方法的不断改进和应用领域的不断拓展,相信它会在更多领域发挥作用,为化学和药物领域带来新的发展机遇。
文章总结:通过对2一氨基一3一羟基吡啶的概念、合成方法、应用领域和研究进展的全面探讨,我们更加深入地了解了这一化合物的重要性和潜在价值。
我个人对其应用前景感到乐观,期待未来能够看到更多关于2一氨基一3一羟基吡啶的突破性研究成果。
以上就是对"2一氨基一3一羟基吡啶的合成"主题的深度和广度兼具的文章,希望能帮助你更好地理解和掌握这一领域的知识。
2-aminopyridine-3-ol的合成可以采用多种不同的方法和途径。
一种传统的合成方法是通过取代反应来实现。
氨基吡啶可以经过亲核取代反应,例如在邻位或对位原子上进行氢原子的替换,接着进行羟基化反应,从而得到2-aminopyridine-3-ol。
3-氨基吡啶的合成
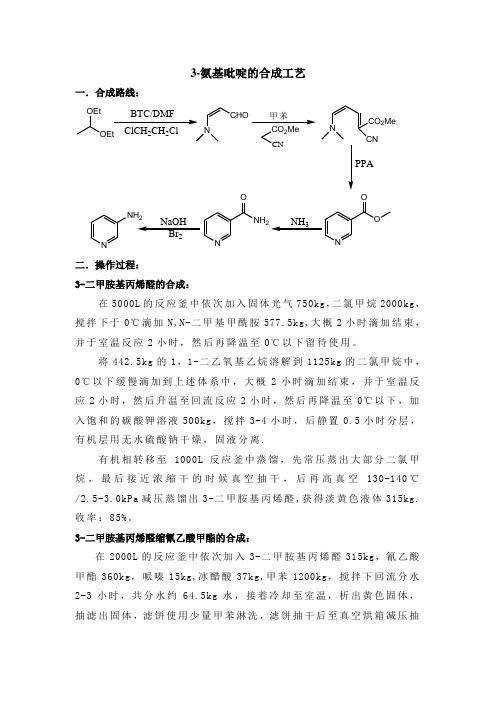
3-氨基吡啶的合成工艺一.合成路线:OEtOEt BTC/DMF22NCHO甲苯2CNNCO2MeCNPPANOONNH2ON NH2NH3 NaOHBr2二.操作过程:3-二甲胺基丙烯醛的合成:在5000L的反应釜中依次加入固体光气750kg,二氯甲烷2000kg,搅拌下于0℃滴加N,N-二甲基甲酰胺577.5kg,大概2小时滴加结束,并于室温反应2小时,然后再降温至0℃以下留待使用。
将442.5kg的1,1-二乙氧基乙烷溶解到1125kg的二氯甲烷中,0℃以下缓慢滴加到上述体系中,大概2小时滴加结束,并于室温反应2小时,然后升温至回流反应2小时,然后再降温至0℃以下,加入饱和的碳酸钾溶液500kg,搅拌3-4小时,后静置0.5小时分层,有机层用无水硫酸钠干燥,固液分离.有机相转移至1000L反应釜中蒸馏,先常压蒸出大部分二氯甲烷,最后接近浓缩干的时候真空抽干,后再高真空130-140℃/2.5-3.0kPa减压蒸馏出3-二甲胺基丙烯醛,获得淡黄色液体315kg.收率:85%。
3-二甲胺基丙烯醛缩氰乙酸甲酯的合成:在2000L的反应釜中依次加入3-二甲胺基丙烯醛315kg,氰乙酸甲酯360kg,哌嗪15kg,冰醋酸37kg,甲苯1200kg,搅拌下回流分水2-3小时,共分水约64.5kg水,接着冷却至室温,析出黄色固体,抽滤出固体,滤饼使用少量甲苯淋洗,滤饼抽干后至真空烘箱减压抽干,共计获得547kg产品3-二甲胺基丙烯醛缩氰乙酸甲酯干品,收率:90%。
母液去回收甲苯,可以反复套用。
3-甲酰胺基吡啶的合成:在2000L的反应釜中依次加入3-二甲胺基丙烯醛缩氰乙酸甲酯547kg,无水乙醇1200kg,搅拌下加入100kg的多聚磷酸PPA,搅拌回流反应10小时,后降温至室温,然后再降温至0℃以下,通氨气至PH值到9-10,过滤除去形成的磷酸铵盐,母液继续通氨气至饱和,搅拌反应24小时,后减压浓缩尽溶剂乙醇,打入1200kg的丙酮,搅拌下升温至回流,压滤除去少量不溶性固体,母液冷冻至-5-0℃析晶,降温至0℃以下后保持搅拌析晶5小时,放料滤出固体,减压烘干,得到白色粉末状固体3-甲酰胺基吡啶210kg, 熔点:128-130℃。
重组大肠杆菌生产核苷磷酸转移酶的发酵条件优化

重组大肠杆菌生产核苷磷酸转移酶的发酵条件优化沈爱萍。
何宝龙。
孙丽慧。
郑裕国(浙江工业大学生物工程研究所,浙江杭州310014)摘要:核苷磷酸转移酶能够特异地将无机焦磷酸(PPi)的磷酸根转移到核苷5’一位的羟基上,该过程并不需要A T P的参与,且具有选择性高和反应条件温和等特点。
以实验室前期构建的重组大肠杆菌E.∞f f B L21(D E3)/pE T28b—A P/P T为茵株,进行了5L发酵罐中的培养条件优化,确定了以乳糖作为诱导剂时,最佳诱导时机oD鲫为7左右,诱导剂乳糖的浓度为10g/L;确定了最适通气量为1.5L/m i n。
在该条件下,核苷磷酸转移酶酶活达221.4U/L,O D600为20.4。
关键词:重组大肠杆茵;核苷磷酸转移酶;发酵;条件优化O pt i m i zat i on of f e r m e nt a t i on condi t i ons f or t he pr oduct i on ofphosphot r ansf e r as e by E sc her i chi a col iS H E N A i—pi ng,H E B ao—l ong,SU N L i—hui,Z H EN G Y u—guo(I nst i t ut e of B i oengi neer i ng,Zhej i ang U ni vers i t y of t e chnol ogy,H angzhou310014,C hi na)A bst r ac t:P hos phot r ans f e r as e,poss ess ed hi gh r egi o s peci f i ci t y of t he nucl eosi de phosphor yl at i ng act i vi t y t o t he C-57posi t on,i s of hi gh pot ent i al f or t he pr oduct i on of i nos i ne-5'-m onophosphat e due t o m i l d r eact i on condi t i ons.I n t hi s pa pe r,r ecom bi nant E c ol iB L21(D E3)/pE T28b-A P/PT w hi ch has e xpr e ss ed ac i d phos phat a se s w as use d,and t he ef fect s of cul t ur e condi t i ons o n ce l l gr ow t h and enzy m e act i vi t y w e r e i nves t i gat ed i n5L f e r m e nt er.T he opt i m al condi t i ons of cul t ur e w e r e as f ol l o w i ng:i ndu ct i on t i m e w as at O D c劬o=7,t he l act os e concent r a t i on w as of10g/L,vent i l at i on vol um e w as of1.5L/m i n.U nder t he cul t ur e condi t i ons,t he enzym e act i vi t y and O D600c oul d r ea ch221.4U几and20.4.r es pect i vel y.K ey w or ds:r ec om bi na nt E.col i;phosphot r ansf er ase;f er m ent at i on;opt i m i zi ng condi t i ons以57一肌苷酸(5'-I M P)为代表的呈味核苷酸,是新一代食品增鲜剂,常与5’一鸟苷酸(5’一G M P)按1:1混合而成。
药物作用机制及不良反应表格总结

乙酰苯胺类
对乙酰氨基酚
阿司匹林抗凝机制:抑制血小板TXA2合成。
芳基乙酸类
吲哚美辛、双氯芬酸、奈美丁酮
芳基丙酸类
布洛芬、萘普生
1,2-苯并噻嗪类
昔康类
选择性COX2
塞来昔布、尼美舒利、依托考昔
选择性抑制COX-2。
保泰松
抑制前列腺素合成。
抗痛风药
别嘌醇、非索步坦
三唑类、咪唑类
肝毒性,AST/ALT升高。
两性霉素B
肾毒,高热。
制霉菌素
利巴韦林
肾毒,骨髓抑制。
阿昔洛韦
肾毒。
拉米夫定
肝毒,骨髓抑制。
奎宁/氯喹
金鸡纳反应。
伯氨喹,磺胺类
特异质反应(葡萄糖6磷酸缺乏者溶血性贫血)。
环磷酰胺
出血性膀胱炎。
塞替派
高尿酸。
顺铂
耳毒,肾毒。
奥沙利铂
神经毒。
博来霉素
间质性肺炎。【飞人博尔特】
贝丁酸类
消化道反应,肌毒肝毒,胆结石。
烟酸
肌毒肝毒血管毒。
硫酸亚铁糖浆
黑便,腹泻,牙齿变黑。
蔗糖铁注射液
过敏,色素沉着,局部疼痛,幼儿致死。
重组人促红素
类流感样症状,升血压,癫痫。
呋塞米(高)
低钾,耳毒,高尿酸,过敏。
氢氯噻嗪(中)
低钾,低氯,低钠,低镁,高钙,高尿酸,高血糖。
螺内酯(低)
高钾,性激素异常。
利血平
交感神经末梢抑制剂,是递质传递越来越少。
甲基多巴、可乐定
激动中枢α2受体。
硝普钠、肼屈嗪
直接舒张血管平滑肌。
哌唑嗪、特拉唑嗪
一种新型磷酰胺吗啉代寡鸟苷酸单体的合成及工艺优化

收稿日期:2023G09G15.基金项目:国家自然科学基金资助项目(21863006);江西省教育厅科学技术研究项目(G J J 208903);抚州市科技指导项目(20220906).作者简介:欧阳红霞(1983 ),女,讲师.㊀∗通信作者:李亮荣(1986 ),男,副教授.E Gm a i l :n c u r o n g@163.c o m .欧阳红霞,杨小喆,李海峰,等.一种新型磷酰胺吗啉代寡鸟苷酸单体的合成及工艺优化[J ].南昌大学学报(理科版),2024,48(1):71G76.O U Y A N G H X ,Y A N G XZ ,L IH F ,e t a l .S y n t h e s i s a n d p r o c e s s o p t i m i z a t i o n f o r an o v e l p h o s p h o r o d i a m i d a t em o r p h o l i n o gu a Gn y l i cm o n o m e r [J ].J o u r n a l o fN a n c h a n g U n i v e r s i t y(N a t u r a l S c i e n c e ),2024,48(1):71G76.一种新型磷酰胺吗啉代寡鸟苷酸单体的合成及工艺优化欧阳红霞1,3,杨小喆1,李海峰2,丁永红1,李亮荣1,3∗(1.南昌大学抚州医学院,江西抚州344000;2.江西省肿瘤医院核医学,江西南昌330029;3.抚州市A I 药物技术创新中心,江西抚州344000)㊀㊀摘要:发展了一种以鸟苷为原料的新型磷酰胺吗啉代寡鸟苷酸单体的合成路线.在对碱基上氨基和核糖5'位羟基进行保护的基础上,将核糖环转变为吗啉环,得到保护的吗啉代鸟苷.在吗啉环上进行三苯甲基化反应,并利用M i t s u n o b u 反应对嘌呤6GO 位进行修饰;对脱保护后的羟基进行N ,N G二甲基氨基磷酰化反应,合成得到目标产物.对M i t s u n o b u 反应和磷酰化反应的条件进行了优化.在优化的条件下,新合成路线的总产率为14.1%.通过L C GM S ㊁1H NM R 和31PNM R 等技术手段对中间产物和目标产物进行了结构表征.关键词:合成;鸟苷;吗啉代寡鸟苷酸;M i t s u n o b u 反应;磷酰化反应中图分类号:O 624.6㊀㊀㊀㊀文献标志码:A㊀㊀㊀㊀㊀㊀文章编号:1006G0464(2024)01G0071G06S y n t h e s i s a n d p r o c e s s o p t i m i z a t i o n f o r an o v e l p h o s ph o r o d i a m i d a t e m o r p h o l i n o g u a n yl i cm o n o m e r O U Y A N G H o n g x i a 1,3,Y A N G X i a o z h e 1,L IH a i f e n g 2,D I N G Y o n g h o n g 1,L IL i a n g r o n g1,3∗(1.F u z h o u M e d i c a l C o l l e g e o fN a n c h a n g U n i v e r s i t y ,F u z h o u J i a n gx i 344000,C h i n a ;2.D e p a r t m e n t o fN u c l e a rM e d i c i n e ,J i a n g x i C a n c e rH o s p i t a l ,N a n c h a n g 330029,C h i n a ;3.F u z h o u I n n o v a t i o nC e n t e r f o rA ID r u g ,F u z h o u M e d i c a l C o l l e g e o fN a n C h a n g U n i v e r s i t y ,F u z h o u J i a n gx i 344000,C h i n a )A b s t r a c t :As y n t h e s i s r o u t e o f a n o v e l p h o s p h o r a m i d em o r p h o l i n o l i n e Go l i g o g u a n y l a t em o n o m e r u s i n ggu a n o s i n e a s r a w m a Gt e r i a lw a s d e v e l o p e d .B a s e d o n t h e p r o t e c t i o n o f t h e a m i n o g r o u p o n t h e b a s e a n d 5'GOHi n r i b o s e ,t h e r i b o s e r i n g i s c o n v e r t e d i n Gt oam o r p h o l i n e r i n g t o o b t a i n t h e p r o t e c t e dm o r p h o l i n e g u a n o s i n e .At r i p h e n y l m e t h y l a t i o n r e a c t i o nw a s c a r r i e d o u t o n t h em o r Gp h o l i n e r i n g a n d p u r i n e 6GO p o s i t i o nw a sm o d i f i e du s i n g t h eM i t s u n o b u r e a c t i o n .N ,N Gd i m e t h y l a m i n o p h o s p h o r yl a t i o n r e a c t i o n i s p e r f o r m e do n t h e d e p r o t e c t e dh y d r o x y l g r o u p t o s y n t h e s i z e t h e t a r g e t p r o d u c t .I n t h i s s t u d y ,t h e c o n d i t i o n s o f t h eM i t s u n o b u r e Ga c t i o na n d p h o s p h o r y l a t i o n r e a c t i o nw e r e o p t i m i z e d .U n d e r o p t i m i z e dc o n d i t i o n s ,t h e t o t a l y i e l do f t h en e ws yn t h e t i c r o u t ew a s 14.1%.T h e s t r u c t u r e o f i n t e r m e d i a t e p r o d u c t s a n d t a r g e t p r o d u c t sw e r e c h a r a c t e r i z e db y LC GM S ,1H NM Ra n d 31PNM R.K e yw o r d s :s y n t h e s i s ,g u a n o s i n e ,m o r p h o l i n o Go l i g o g u a n y l a t e ,m i t s u n o b u r e a c t i o n ,p h o s p h o r y l a t i o n r e a c t i o n ㊀㊀当前,现代药物研究的焦点之一是开发能够修正致病基因的治疗技术.这些技术可以在体内靶向关闭具有缺陷或突变的基因或m R N A ,或者有效地修复基因或补充m R N A 以产生类似功能的蛋白质,从而治疗包括遗传性疾病在内的各种疾病.通过分子生物技术,合成的核酸药物可以实现对R N A 的关闭㊁补充或修改,展现出潜在的治疗潜力,这一趋势在临床应用中逐渐显现[1,2].为了改善核酸药物的药理活性,对天然寡核苷酸的磷酸二酯键㊁碱基或核糖等进行化学修饰,成为当前药物化学研究的重要领域[3-5].磷酰胺吗啉代寡核苷(p h o s p h o r o d i a m i d a t e m o r p h o l i n oo l i go n u c l e o t i d e ,P MO ,图1)是一种修饰反义寡核苷酸,具有独特的结构.它采用吗啉环第48卷第1期2024年2月㊀㊀㊀㊀㊀㊀南昌大学学报(理科版)J o u r n a l o fN a n c h a n g U n i v e r s i t y(N a t u r a l S c i e n c e )V o l .48N o .1F e b .2024㊀替代脱氧核糖或核糖,并使用磷酰二胺结构代替磷酸二酯键连接每个单体.磷酰二胺结构单元的引入减少了P MO与蛋白质之间的相互作用,因此具有较低的毒副作用.最重要的是,P MO能够与D N A 和R N A进行碱基配对,并且不容易被体内核酸酶降解,具有足够的稳定性.这使得P MO能以立体位阻的方式干扰靶向R N A的下游活动,如前信使R N A(p r eGm R N A)的剪切㊁m R N A的翻译㊁微小R N A(m i R N A)的加工或与其他分子的结合,导致相关蛋白质的合成受到阻断,从而实现在R N A水平上纠正蛋白质功能的治疗目标,或者用于研究特定基因或R N A的功能[6-7].此外,以P MO 为基础图1㊀P M O的结构F i g.1㊀t h e s t r u c t u r e o fP M O 的反义寡核苷酸在抗病毒[8-9]㊁抗菌[10]以及生物发育系统[11]等领域研究应用上也取得了一定成果. P MO的单体构建[12]和链合成方法[13-14]的不断改进是推动P MO应用研究的前提.研究表明,对碱基的化学修饰可显著改善核酸的化学性质及生物活性[15],在吗啉代寡核苷酸结构中亦是如此.引用保护基团来保护碱基上的氨基㊁羟基㊁酰胺基团,不仅对吗啉代寡核苷单体缩合成核苷酸起着至关重要的作用,还能得到功能多样的吗啉基修饰核苷单体[16],如在嘧啶碱基的C-5位引入丙炔基,可以增强其双螺旋稳定性,在嘌呤C-8位引入芳环或取代芳环可以进一步扩展P MO在分子生物学中的研究和应用.然而,对于嘌呤6GO的修饰仅在核苷及核酸结构中曾经报道过[17],而在P MO单体结构中,碱基嘌呤6GO位上的结构修饰及研究未曾见过报道.本文以鸟苷为起始原料,参照文献[12,18-19]的方法合成了中间体GN2G异丁酰基G7 GOG叔丁基二苯基硅基GNG三苯甲基吗啉代鸟苷(5).随后,通过M i tGs u n o b u反应在鸟嘌呤6GO位引入4G硝基苯乙基对其进行修饰.最后,经羟基脱保护和羟基磷酰化反应,成功合成了一种新型磷酰胺吗啉代寡鸟苷酸单体(P MOGG),并对反应条件进行了优化.该结构增强了单体分子的稳定性,降低了分子极性.因此,在聚合成磷酰胺吗啉代寡核苷酸后,将可能会增强其脂溶性并改善其生物活性.合成路线如下:图2㊀目标分子(P M OGG)的合成路线F i g.2㊀S y n t h e s i s r o u t e o f t a r g e tm o l e c u l e(P M OGG)1㊀实验部分1.1㊀仪器与试剂A v a n c e400MH z核磁共振仪(瑞士B r u c k公司);U P L CGS QGD E T E C T O R液质联用仪(美国W a t e r s公司);R E52GA型真空旋转蒸发器(上海亚荣生化仪器厂).鸟苷(99%,北京百灵威科技有限公司);N,NG 27 南昌大学学报(理科版)2024年㊀二甲基氨基磷酰二氯(S i g m aGA l d r i c h);三甲基氯硅烷(98%)㊁异丁酰氯(98%)㊁叔丁基二苯基氯硅烷(T B D P S C l,98%)㊁高碘酸钠(N a I O4,99%)(国药集团化学试剂有限公司);硼酸铵四水合物((N H4)2B2O7 4H2O,99%)㊁氰基硼氢化钠(N a B H3C N,95%)㊁三苯基氯甲烷(T r t C l,97%)㊁偶氮二甲酸二异丙酯(D I A D,95%)㊁四丁基氟化铵(T B A F,98%)㊁二氮杂二环(D B U,99%)㊁对硝基苯乙醇(98%)㊁溴化锂(L i B r,98%)㊁N,NG二甲基甲酰胺㊁四氢呋喃,二氯甲烷㊁甲醇㊁乙酸乙酯㊁乙腈㊁异丙醇(I P A)(上海阿拉丁生化科技股份有限公司).1.2㊀实验方法1.2.1㊀N2G异丁酰基鸟苷(2)的合成将鸟苷5.00g(17.7mm o l)溶于吡啶,加入23.07g三甲基氯硅烷(212.4mm o l),在室温下搅拌2h,然后冷却至0ħ.随后滴加5.65g异丁酰氯(53.1mm o l),滴加完毕后,在室温下继续搅拌3h.冷却至0ħ后,用150m L水淬灭反应,搅拌约10m i n后,加入95m L浓氨水,继续在室温下搅拌约15m i n.将反应液稀释至500m L,使用C H2C l2(300m Lˑ3)进行萃取,通过减压旋蒸除去溶剂,得到粗产物.通过柱层析(V(C H2C l2) V(M e O H)=8 1)纯化得到4.84g白色固体2,收率75%, L C M S纯度96.9%(R T=1.150m i n).E S IGM S,C14H19N5O6,实测值(理论值),m/Z:354.1(354.1)[M +H]+.1.2.2㊀N2G异丁酰基G5 GOG叔丁基二苯基硅基鸟苷(3)的合成将10.03g化合物2(28.4mm o l)溶于150m L 二甲基甲酰胺(D M F)中,并加入4.83g(71.02mm o l)咪唑.然后将反应液冷却至0ħ,并滴加含有8.59g(31.2mm o l)叔丁基二苯基氯硅烷的D M F (16m L)溶液,反应混合物在室温下搅拌反应6h.反应完成后,使用300m L H2O淬灭反应,随后用乙酸乙酯(200m Lˑ3)萃取,将有机层合并,使用饱和食盐水(150m Lˑ2次)进行洗涤,继而经无水N a2S O4干燥㊁过滤,通过减压蒸干溶剂,得到粗产品.通过用1000m L乙酸乙酯打浆,获得12.51g 化合物3,收率79%,L C M S纯度94%(R T=1.870m i n).E S IGM S,C30H37N5O6S i,实测值(理论值), m/Z:592.4(592.3)[M+H]+.1.2.3㊀N2G异丁酰基G7 GOG叔丁基二苯基硅基吗啉代鸟苷(4)的合成将4.14g(6.99mm o l)化合物3溶于50m L甲醇中,稍加热以溶解,然后加入1.64g(7.71mm o l) N a I O4水溶液(30m L),室温下搅拌反应2h.反应完成后,通过减压蒸馏将溶剂除去,得到残留物.将残留物用30m L水稀释,然后用乙酸乙酯(30m Lˑ3)进行萃取,将有机层合并,用饱和食盐水洗涤有机层,经无水N a2S O4干燥㊁过滤㊁减压蒸干溶剂得白色固体粗产物4.38g.将此粗产物溶于50m L甲醇,加入硼酸铵四水合物3.12g(16.35mm o l),在室温下搅拌反应过夜.随后加入4.38g(72.99mm o l)4A分子筛,0.89g(14.85mm o l)醋酸和0.93g (14.85mm o l)N a B H3C N,继续在室温下反应2h,然后用10m L甲醇淬灭反应,将大部分溶剂蒸干,所得油状物用50m L乙酸乙酯溶解,用水萃取(50m Lˑ3),随后经50m L饱和食盐水洗涤㊁无水N a2S O4干燥㊁过滤㊁减压蒸干溶剂等过程,得到3.54g白色固体4,此粗产品无需纯化可直接用于下步反应.E S IGM S,C30H38N6O4S i,实测值(理论值),m/ Z:575.6(575.3)[M+H]+.1.2.4㊀N2G异丁酰基G7 GOG叔丁基二苯基硅基GNG三苯甲基吗啉代鸟苷(5)的合成将2.94g(5.12mm o l)化合物4溶于30m L D M F中,然后加入1.03g(10.24mm o l)E t3N和1.57g(5.63mm o l)三苯基氯甲烷,室温搅拌2h,然后用2m L甲醇淬灭反应,用30m L水稀释反应液,用乙酸乙酯(100m Lˑ3)萃取,随后经100m L饱和食盐水洗涤㊁无水N a2S O4干燥㊁过滤,通过减压蒸干溶剂,所得粗产品用柱色谱(V(M e O H) V (C H2C l2)=1 20~1 10)梯度洗脱纯化得到2.74g化合物5,收率61%,L C M S纯度93%(R T=2.44m i n).E S IGM S,C49H52N6O4S i,实测值(理论值), m/Z:817.4(817.4)[M+H]+.1H NM R(400MH z, C D C l3,ˑ10-6),δ:11.89(s,1H),8.07(s,1H),7.57~7.38(m,12H),7.33~7.19(m,13H),5.97(d d,J=2.0,10.0H z,1H),4.30~4.24(m,1H),3.74(d d,J=4.8,10.4H z,1H),3.58(d d,J=4.8,10.4H z,1H),3.40(d,J=11.2H z,1H),3.30(d, J=12.0H z,1H),2.62(t,J=6.8H z,1H),1.76(s,1H),1.70(d,J=10.0H z,1H),1.55(t,J=11.2H z,1H),1.30(d,J=7.2H z,6H),0.95(s,9H).1.2.5㊀N2G异丁酰基GO6G对硝基苯乙基G7 GOG叔丁基二苯基硅基GNG三苯甲基吗啉代鸟苷(6)的合成将5.00g(6.13mm o l)化合物5和2.05g(12.26mm o l)对硝基苯乙醇溶于100m L四氢呋喃(T H F)中,然后加入2.09g(7.97mm o l)P P h3和1.61g37第1期㊀㊀㊀㊀㊀欧阳红霞等:一种新型磷酰胺吗啉代寡鸟苷酸单体的合成及工艺优化(7.97mm o l)偶氮二甲酸二异丙酯(D I A D),室温搅拌4h,反应完成后用150m L水稀释反应液,经乙酸乙酯萃取(150m Lˑ3),合并有机层,有机层用150m L饱和食盐水洗涤㊁无水N a2S O4干燥㊁过滤,减压蒸干溶剂,所得粗产品用柱色谱(V(石油醚) V(乙酸乙酯)=1 2)得到5.29g白色固体6,收率85.2%,L C M S纯度95%(R T=2.397m i n).E S IGM S,C57H59N7O6S i,实测值(理论值),m/Z:988.7(988.4)[M+N a]+.1.2.6㊀N2G异丁酰基GO6G对硝基苯乙基G7 G羟基GNG三苯甲基吗啉代鸟苷(7)的合成将7.40g(7.65mm o l)化合物6溶于80m L T H F中,然后加入四丁基氟化铵(T B A F)(1m o l L-1,7.16m L)的醋酸溶液,调节溶液p H至中性,室温搅拌2h,反应完成后用100m L水稀释反应液,然后用乙酸乙酯(100m Lˑ3)萃取,合并有机层,经饱和食盐水洗涤㊁无水N a2S O4干燥㊁过滤,减压蒸干溶剂,所得的粗产品用柱色谱(V(C H2C l2) V (I P A)=10 1)得到4.69g化合物7,收率86.4%, L C M S纯度96%(R T=3.913m i n).E S IGM S,C41H41N7O6,实测值(理论值),m/Z:750.4(750.3) [M+N a]+.1H NM R(400MH z,C D C l3,ˑ10-6),δ:8.17~8.10(m,3H),7.79(s,1H),7.52~7.43(m,6H),7.28(t,J=7.6H z,6H),7.26~7.08(m,3H),6.22(d d,J=2.0,10.0H z,1H),4.82~4.73(m,2H),4.34~4.24(m,1H),4.05~4.00(m,2H),3.60~3.56(m,2H),3.40(d,J=11.2H,1H),3.24~3.12(m,4H),2.08(s,1H),1.80(t,J=10.6H z,1H),1.57(t,J=11.2H z,1H),1.35~1.24(m,6H).13C NM R(100MH z,C D C l3),δ:19.9,35.0,35.6,50.5,57.8,62.6,67.1,81.2,83.5,91.2,115.3,123.8,126.7,127.0,129.4,130.6,140.3,145.3,145.8,146.2,152.3,153.2,161.2,178.5.1.2.7㊀N2G异丁酰基GO6G对硝基苯乙基G7 G(N,NG二甲基氨基)磷酰氯GNG三苯甲基吗啉代鸟苷(P MOGG)的合成将4.35g(5.97mm o l)化合物7溶于50m L C H2C l2和50m LC H3C N的混合溶剂中,加入1.03g(11.94mm o l)L i B r,搅拌2m i n,然后加入1.82g (11.94mm o l)二氮杂二环(D B U),1.45g(8.97mm o l)N,NG二甲基氨基磷酰二氯的C H2C l2(5m L)溶液,0ħ下搅拌2h,反应完成后减压蒸干溶剂,用冷却的乙酸乙酯(150m L)稀释,用预冷至0ħ~5ħ的水(100m Lˑ2)萃取,合并有机层,用无水N a2S O4干燥㊁过滤㊁减压蒸干溶剂,粗产品经柱层析(V(C H2C l2) V(I P A)=8 1)得到3.3g 油状物(P MOGG),收率61%,L C M S纯度95%(R T=4.370m i n).E S IGM S,C43H46C l N8O7,实测值(理论值),m/Z:875.3(875.3)[M+N a]+.1H NM R(400MH z,C D C l3,ˑ10-6),δ:8.18~8.04(m,2H),7.81~7.68(m,2H),7.51~7.39(m,7H),7.32~7.13(m,10H),6.20(d,J=9.7H z,1H),4.76(t,J=6.6H z,2H),4.44(d,J=5.2H z,1H),4.16~3.98(m,2H),3.44(d,J=11.2H z,1H),3.30~3.16(m,3H),2.61~2.57(m,6H),1.79~1.65(m,2H),1.56(t,J=11.2H z,1H),1.38~1.26(m,6H).13C NM R(100MH z,C D C l3),δ:19.7,35.2,35.5,36.0,50.5,58.0,67.3,69.2,77.8,84.0,91.2,115.3,123.8,126.7,127.0,129.4,130.5,140.6,145.0,145.9,146.2,152.6,153.2,161.2,177.631P NMR(162MH z,C D C l3,ˑ10-6)δ:18.45,18.12.2㊀结果与讨论㊀㊀本文使用鸟苷为起始原料,通过七个反应步骤,成功合成了一种新型的磷酰胺吗啉代寡鸟苷酸单体(P MOGG).本工作首次在碱基鸟嘌呤的6GO位上引入了对硝基苯乙基,该基团在酸碱条件下表现较为稳定.整个合成路线主要涉及到碱基氨基的保护㊁M i t s u n o b u反应㊁羟基磷酰化等多个关键工艺步骤.2.1㊀碱基氨基的保护碱基氨基的保护是寡核苷酸单体缩合成过程中必不可少的一步.常见的氨基保护方式有两种:其中一种方法是在吗啉环形成后,以三乙胺为碱,分批加入异丁酸酐,并在140ħ下加热反应,以异丁酰基对氨基进行保护,类似于S A N K HA P[12]等报道的方法;另外一种方法则是在吗啉环形成前对氨基进行保护,类似于文献[19]中采用的方法.本文尝试采用后一种方式进行氨基保护:首先,使用三甲基氯硅烷对鸟苷的羟基进行保护,然后使用异丁酰氯对氨基进行保护,最后,使用浓氨水脱三甲基硅烷基.整个保护过程在室温下进行,工艺简单㊁温和,收率高达75%.相比之下,方式一使用异丁酸酐为保护试剂,其活性较弱,需高温反应.因此,本文选择了后一种方式进行碱基氨基的保护.2.2㊀M i t s u n o b u反应三苯基膦的用量优化化合物5经M i t s u n o b u反应以对硝基苯乙醇47 南昌大学学报(理科版)2024年㊀修饰6GO 位时,三苯基膦和D I A D 在反应过程形成中间体,用于夺取质子并形成亲核试剂.然而,三苯基膦会产生三苯氧磷副产物,在柱层析过程中容易导致拖尾现象,增加纯化的难度,降低反应收率.因此,我们需要优化三苯基膦的用量,以尽量减少三苯氧磷的产生.实验结果表明,当三苯基膦和D I A D 与化合物5的物质的量之比为1.3时,反应收率达到最高值.表1㊀P P h 3用量的优化T a b .1㊀O p t i m i z a t i o no f t h e a m o u n t o fP P h 3E n t r y n (P P h 3):n (C o m p o u n d5)Y i e l d /%11.14321.25531.38541.57352.061㊀㊀注:n (对硝基苯乙醇) n (化合物5)=2 1,n (D I A D ) n (P P h 3)=1 1.2.3㊀7’GO H 磷酰化反应条件的优化在化合物7的7 GO H 位置接入N ,N G二甲基氨基磷酰二氯是合成目标化合物P MO GG 重要反应步骤.反应中需使用L i B r 做催化剂,二氮杂二环(D B U )做碱,L i B r 的作用可能是通过L i +与7 GO 和吗啉环上的N 配位形成一个六元环过渡态,从而增强7 GO H 的酸性,使之更容易被碱进攻;同时L i GB r 的另一个作用是帮助N ,N G二甲基氨基磷酰二氯中P GC l 的断裂,因此反应过程中L i B r 和D B U 的用量对反应产率至关重要.本文在参考文献[18]的基础上对D B U 和L i B r 的用量以及反应温度㊁时间等变量进行了探索和优化,其结果见表2.研究结果表明,D B U 和L i B r 与化合物7的物质的量之比为3.2时,在混合溶剂C H 3C N 和C H 2C l 2中,0ħ下反应1h ,收率达到最高,为55%;延长反应时间并不会提高产物的收率,反而会稍微降低,可能是因为随着时间的延长,会生成少量的二聚副产物.当在室温下反应1h 时,收率为43%,主要是由于终产物的磷酰氯基团在室温下容易发生水解.我们还尝试改变D B U 和L i B r 的用量,发现当D B U 和L i B r 与化合物7的物质的量之比为2.0时,在0ħ下反应2h ,收率高达61%.相比文献中的工艺条件,虽然本工艺时间略微延长,但收率显著提高,并降低了原料成本.在大规模生产中,聚合物P MO 及其活性筛选研究需要大量的单体提供物质基础,因而成本控制尤为重要.因此,本研究选择了如实验方法1.2.7中序号6的工艺条件.表2㊀磷酰化反应条件的优化T a b .2㊀O p t i m i z a t i o no fP h o s p h o r yl a t i o n r e a c t i o n c o n d i t i o n E n t r y n (D B U ):n (C o m p o u n d7)n (L i B r):n (C o m po u n d7)S o l v e n t T e m pe r a t u r e /ħt /h Y i e l do fC o m po u n d8/%13.23.2C H 3C N GC H 2C l 200.54923.23.2C H 3C N GC H 2C l 2015533.23.2C H 3C N GC H 2C l 2024743.23.2C H 3C N GC H 2C l 2r .t 14352.02.0C H 3C N GC H 2C l 2014562.02.0C H 3C N GC H 2C l 2026172.02.0C H 3C N GC H 2C l 20350㊀㊀㊀㊀㊀注:n (N ,N G二甲基氨基磷酰二氯):n (化合物7)=1.5 1,V (C H 3C N ) V (C H 2C l 2)=1 1.3㊀结论㊀㊀本研究通过7个反应步骤,并对关键步骤中M i t s u n o b u 反应㊁磷酰化反应进行了工艺优化,成功合成了一种新型磷酰胺吗啉代寡鸟苷酸单体,总收率为14.1%.作为反义核酸药物,寡核苷酸P MO57 第1期㊀㊀㊀㊀㊀欧阳红霞等:一种新型磷酰胺吗啉代寡鸟苷酸单体的合成及工艺优化具有广泛的应用领域,可用于抗病毒㊁抗菌及基因治疗等.已上市的依利替生和抗埃博拉病毒药物A V IG7537的研究证明了P MO作为药物的可行性.本研究中合成的新型单体具有稳定的结构,并且具有增强的脂溶性,这有望提高其生物活性.下一步,研究小组将目标化合物聚合成寡核苷酸,再进行生物活性研究,同时将其用于抗病毒药物的筛选工作.参考文献:[1]㊀张文洁,魏霞,何军林.吗啉代寡核苷酸的应用研究进展[J].国际药学研究杂志,2020,47(4):251G257,262.[2]周海燕.反义寡核苷酸药物在精准治疗中的应用进展及其作用机制[J].精准医学杂志,2020,35(4):283G291.[3]WA N W B,S E T H P P.T h e m e d i c i n a lc h e m i s t r y o f t h e r a p e u t i co l i g o n u c l e o t i d e s[J].J o u r n a lo f M e d i c i n a lC h e m i s t r y,2016,59(21):9645G9667.[4]S HA R MA V K,S HA R MA R K,S I N G H S K.A n t iGs e n s e o l i g o n u c l e o t i d e s:m o d i f i c a t i o n sa n dc l i n i c a l t r i a l s[J].M e d.C h e m i c a l.C o mm u n i c a t i o n.,2014,5(10):1454G1471.[5]何军林.核酸药物的研究进展[J].国际药学研究杂志,2017,44(11):1028G1051.[6]G E R E T YSS,W I L K I N S O NDG.M o r p h o l i n o a r t i f a c t s p r o v i d e p i t f a l l s a n d r e v e a l a n o v e l r o l e f o r p r oGa p o p t o tGi c g e n e s i nh i n d b r a i nb o u n d a r y d e v e l o p m e n t[J].D e v e lGo p m e n t a l B i o l o g y,2011,350(2):279G289.[7]E I S E NJ S,S M I T HJC.C o n t r o l l i n g m o r p h o l i n o e x p e rGi m e n t s:d o n t s t o p m a k i n g a n t i s e n s e[J].D e v e l o p m e n t,2008,135(10):1735G1743.[8]H E A L D AE,I V E R S E NPL,S A O U DJ B,e t a l.S a f e t ya n d p h a r m a c o k i n e t i c p r o f i l e s o f p h o s p h o r o d i a m i d a t em o r p h o l i n oo l i g o m e r sw i t ha c t i v i t y a g a i n s t e b o l av i r u sa n d m a rb u r g v i r u s:r e s u l t s o ft w o s i n g l e a sc e nd i n gd o se s t u d i e s[J].A n t i m i c r o b i a lA g e n t s a n dC h e m o t h e rGa p y[J].2014,58(11):6639G6647.[9]P O P I K W,K HA T U A A,H I L D R E T HJEK,e t a l.A lGc e nd o r p h o s p h o r o d iGa m i d a te m o r p h o l i n o t a r g e t i n g t h e5ᶄu n t r a n s l a t e dr e g i o no ft h eZ I K V R N Ai n h i b i t sv i r u sr e p l i c a t i o n[J].V i r o l o g y,2018,519(7):77G85.[10]G E L L E R BL,G R E E N B E R G D E.P e p t i d eGc o n j u g a t e d p h o s p h o r o d i a m i d a t e m o r p h o l i n o o l i g o m e r s:a n e ws t r a t e g y f o r t a c k l i n g a n t i b i o t i c r e s i s t a n c e[J].T h e r a p e uGt i cD e l i v e r y,2014,5(3):243G245.[11]刘丰,黄慧敏,王志华,等.利用吗啉代寡核苷酸技术下调早期斑马鱼胚胎l m n a基因的初步研究[J].中国比较医学杂志,2016,26(8):85G90.[12]P A T T A N A Y A K S,P A U L S,N A N D IB,e ta l.I mGp r o v e d p r o t o c o l f o r t h e s y n t h e s i so f f l e x i b l yp r o t e c t e dm o r p h o l i n o m o n o m e r sf r o m u n p r o t e c t e d r i b o n u c l e oGs i d e s[J]N u c l e o s i d e s,N u c l e o t i d e s&N u c l e i c A c i d s,2012,31(11):763G782.[13]M 卡鲁瑟斯,S 保罗.使用亚磷酰胺化学法合成主链修饰的吗啉代寡核苷酸和嵌合体:C N116804031A[P].2023G09G26.[14]O U Y A N G X H,S H E S T O P A L O VIA,S I N HA S,e ta l.V e r s a t i l e s y n t h e s i s a n d r a t i o n a ld e s i g n o fc a g e dm o r p h o l i n o s[J].J o u r n a l A m e r i c a C h e m i c a lS o c i e t y,2009,131(37):13255G13269.[15]贾磊,朱财盛,麦曦,等.2,6,9G三取代嘌呤衍生物三维定量构效关系和靶点相互作用[J].南昌大学学报(理科版),2021,45(3):273G278,306.[16]N A N D IB,P A T T A N A Y A KS,P A U LS,e t a l.S y n t h eGs i s o f n u c l e o b a s eGf u n c t i o n a l i z e d m o r p h o l i n oGm o d i f i e dn u c l e o s i d e m o n o m e r s t h r o u g h P a l l a d i u mGC a t a l y z e dC r o s sGC o u p l i n g R e a c t i o n s[J].E u r o p e a n J o u r n a lO r g a nGi cC h e m i s t r y,2013,12(7):1271G1286.[17]L A N G H,G O T T L I E B M,S C HWA R Z M,e ta l.N e w 2G(4GN i t r o p h e n y l)e t h y l(N p e)Ga n d2G(4GN i t r o p h e n y l)e t h o x y c a r b o n y l(N p e o c)Gp r o t e c t e d2 Gd e o x y r i b o n u c l e oGs i d e s a n d t h e i r3 Gp h o s p h o r a m i d i t e sGv e r s a t i l eb u i l d i n gb l oc k s f o r o l i g o n u c l e o t ide s y n t h e s i s[J].H e l v e t i c aC h i m i c aA c t a,1999,82(12):2172G2185.[18]P A T T A N A Y A K S,S I N HA S.L i t h i u m b r o m i d eGD B U m e d i a t e d s y n t h e s i s o f c h l o r o p h o s p h o r a m i d a t eGa c t i v a t e dm o r p h o l i n on u c l e o s i d es u b u n i t s[J].T e t r a h e d r o n L e tGt e r s,2012,53(49):6714G6717.[19]陈晓睿,郭永建,宋亚彬,等.磷酰胺吗啉代寡核苷酸关键中间产物的合成方法探索与工艺优化[J].军事医学,2017,41(8):662G666.(责任编辑:杜珍珍)67 南昌大学学报(理科版)2024年㊀。
无铜_催化的点击化学

! 59 !
化工时刊 2010 Vo l 24, No. 5
论文综述 )Review s∗
图 4 硫醇和烯烃的点击化学合成树状聚合物 F ig. 4 Th io l- ene click chem istry for the syn thesis o f a [ G1] - OH6 dend rmi er
F ig. 2 Pathway o f the Rad ica l Pho toadd ition o f Th io ls onto 1, 2- PBa
图 3 顺丁烯二酰亚胺和硫醇的点击反应合成 PLA F ig. 3 Synthes is o f PLA and m a lemi ide- th io l C lick reac tion
证实了此方法是一个高效的聚合物修改方法。 该方法 也 可以 运用 到聚 合物 的合 成 领域。
H aw ker和他的实验工作小组报道了通过硫醇和烯烃 的点击反应合成稳健、高效、正交树状聚合物 ( 见图 4) [ 14] 。在室温下, 无溶剂反应 1和 2在 微量的光敏 引发剂 3 的存下在, 用手持紫外灯 ( ex 365 nm ) 照 射 30 m in。微量的光敏引发剂 3起到提高反应速度 的作用, 在 1和 2之间即烯和巯基反应产生了六羟基 树枝状物质 4。高效的巯基和烯反应没有副产物的 出现, 并且 [ G 1] - OH6 用乙醚洗涤简单纯化沉淀后 获得产量为 90% 。最近, Justin W Chan, B ing Yu等 通过膦催化巯基与烯烃反应和 RAFT 制备的均聚物 N, N - 二乙基丙烯酰胺来合成三臂星形聚合物。这 也为合成星形聚合物提供了一个快速、易行、高产率 的方法 [ 15 ] 。
点击化学一般具备以下特点: ( 1) 所用原料和试剂容易获得; ( 2) 反应条件简单, 反应过程对水和氧气不敏感; ( 3) 产率高, 没有或有无害的副产品; ( 4) 立体选择性好; ( 5) 产物净化技术简单; ( 6) 产物稳定性好。 点击反应主要有 4种类型: ∃ 环加成反应, 特别
介孔碳的研究进展及应用

2018年第37卷第1期 CHEMICAL INDUSTRY AND ENGINEERING PROGRESS·149·化 工 进展介孔碳的研究进展及应用李鹏刚,王靖轩,郭飞飞,何昱轩,唐光贝,罗永明,朱文杰(昆明理工大学环境科学与工程学院,云南 昆明 650500)摘要:介孔碳是一类新型的具有巨大比表面积和孔体积的介孔材料,可以通过不同的方法合成并对其孔结构和形貌进行调节。
本文主要综述了介孔碳及介孔碳基复合材料的合成方法,对比阐述了不同方法制备的介孔碳材料所具备的孔道结构和形貌。
介绍了将不同非金属和金属元素及其氧化物掺杂在介孔碳中合成复合材料,发现制备的复合材料具有更优的性能且掺杂元素不同复合材料的形貌和孔道结构不同。
此外,简要说明了介孔碳及碳基复合材料在环境、催化、储能、电化学和生物医学等方面的应用,指出其在各个领域的应用仍存在不足。
调整介孔碳的孔结构和表面性能、采用更简便易控制的合成方法将成为制备介孔碳及碳基材料的主要研究方向。
关键词:介孔碳;掺杂;复合材料;合成中图分类号:X522 文献标志码:A 文章编号:1000–6613(2018)01–0149–10 DOI :10.16085/j.issn.1000-6613.2017-0721Recent progress in the synthesis and applications of mesoporouscarbon materialsLI Penggang ,WANG Jingxuan ,GUO Feifei ,HE Yuxuan ,TANG Guangbei ,LUO Yongming ,ZHU Wenjie(Faculty of Environmental Science and Engineering ,Kunming University of Science and Technology ,Kunming650500,Yunnan ,China )Abstract :Mesoporous carbon with specific surface area and various pore volume is a new type mesoporous material. Usually ,the pore structures and morphology of mesoporous carbons can be adjusted by using several methods. This study mainly summarizes the synthetic method of mesoporous carbons and mesoporous carbon-based composites ,and compares the pore structure and morphology of mesoporous carbon materials prepared by different methods. Doping diverse non-metal or metal and its oxide in mesoporous carbon to prepare composite materials are also introduced. It has been found that the prepared mesoporous carbon composite material have better performance ,and the composite materials containing different doping elements possess different morphologies and textures. Moreover ,this article briefly introduces their applications in environment ,biomedicine ,energy storage ,electrochemistry ,and catalysis as well as their deficiencies in application. Finally ,we believe that adjusting pore structure and surface properties of mesoporous carbons and developing simple synthetic method will be the future research directions.Key words :mesoporous carbon ;doping ;composites ;synthesis多孔碳材料指具有不同孔道结构的材料,按其孔径大小可分为:微孔碳材料(d <2nm )、介孔碳材料(2nm <d <50nm )和大孔碳材料(d >50nm )[1]。
新型Eu(Ⅲ)三元配合物的合成及表征

新型Eu(Ⅲ)三元配合物的合成及表征白羽;李文亮;崔桂花【摘要】采用Pd/C催化水合肼还原法制备5-氨基-1,10-邻菲罗啉(APhen)配体,利用Eu(Ⅲ)与此配体和α-噻吩甲酰三氟丙酮(TTA)反应,制备新型三元红色荧光配合物Eu(TTA)3APhen.运用元素分析、FT-IR(傅立叶红外光谱)、1HNMR谱、X射线光电子能谱(XPS)测试和荧光光谱技术等对合成的配合物进行表征和分析.结果表明,配合物中Eu3+离子与TTA中的O及APhen中的N原子配位,配合物具有良好的荧光性质,在579、591、613和652 nm处的发射峰分别对应于5D0→7FJ(J=0,1,2,3)的电子跃迁,且以在613 nm处Eu3+的5D0→7F2电子跃迁所发出的荧光强度最大,是EuCl3的34.5倍.%The 5-amino-1,10-phenanthroline (APhen) was synthesized using Pd/C as catalyst and hydra?zine hydrate as reduetant by reduction method. A novel ternary fluorescent complex Eu(TTA)3APhen was syn?thesized by the reaction of APhen, 2-thenoyltrifluoroacetone (TTA) and Eu(Ⅲ). The complex was characterized by elemental analysis, FT-IR, 1HNMR, X-ray photoelectron spectroscopy (XPS) and fluorescence spectrosco?py. The results indicated that the Eu3+ ions of the complexes coordinated with O atoms of the TTA and N at?oms of the APhen, and the complex had excellent fluorescence property. The emission peaks at 579, 591, 613 and 652 nm of the complexes corresponded to the electron transition of 5D0→7FJ ( J=0,1,2,3). The emis?sion intensity of Eu3+ at 613 nm corresponded to 5D0→7F2 electron transition was maximum, which was 34.5 times of EuCl3.【期刊名称】《印染助剂》【年(卷),期】2017(034)007【总页数】4页(P20-23)【关键词】5-氨基-1,10-邻菲罗啉;三元配合物;荧光性质【作者】白羽;李文亮;崔桂花【作者单位】吉林医药学院化学教研室,吉林吉林 132013;吉林医药学院化学教研室,吉林吉林 132013;吉林医药学院化学教研室,吉林吉林 132013【正文语种】中文【中图分类】TQ610.4有机电致发光器件(OLEDs)在平板显示领域展示了巨大的科研和商业价值[1],研发高效稳定的有机电致发光材料,尤其是红色材料是全色显示OLEDs技术的关键。
3-氨基吡啶连续萃取提纯装置[实用新型专利]
![3-氨基吡啶连续萃取提纯装置[实用新型专利]](https://img.taocdn.com/s3/m/85b1973b33d4b14e84246827.png)
专利名称:3-氨基吡啶连续萃取提纯装置专利类型:实用新型专利
发明人:凌广轩,彭德彪,王鹏浩
申请号:CN201721344538.X
申请日:20171018
公开号:CN207347437U
公开日:
20180511
专利内容由知识产权出版社提供
摘要:3‑氨基吡啶连续萃取提纯装置,降解液中间罐通过降解液输送泵将降解混合物送至一级萃取塔中,氯仿中间罐通过氯仿循环上料泵将氯仿送至一级萃取塔顶部,一级萃取塔的萃余液由顶部进入二级萃余液分层罐中,二级萃余液分层罐通过萃余液倒料泵将萃余液送出,一级萃取塔底部萃取液出口与萃取液中间罐相连,萃取液中间罐出口通过氯仿蒸发上料泵与氯仿蒸发罐相连,氯仿蒸发罐顶部气相出口与冷却器相连,冷却器的凝液出口与氯仿中间罐相连。
本实用新型使用氯仿对3‑氨基吡啶水溶性混合物进行萃取提纯,整个过程为自动化连续操作,具有自动化程度高、分离效果好、能源消耗少和安全稳定的优点。
申请人:沧州临港亚诺化工有限公司
地址:061000 河北省沧州市沧州临港化工园区
国籍:CN
更多信息请下载全文后查看。
用多种元素助催化的吡啶生产用的催化剂及其制备方法[发明专利]
![用多种元素助催化的吡啶生产用的催化剂及其制备方法[发明专利]](https://img.taocdn.com/s3/m/516888626529647d2628524e.png)
专利名称:用多种元素助催化的吡啶生产用的催化剂及其制备方法
专利类型:发明专利
发明人:肖国民,张进
申请号:CN200410065715.1
申请日:20041115
公开号:CN1631536A
公开日:
20050629
专利内容由知识产权出版社提供
摘要:用多种元素助催化的吡啶生产催化剂及其制备方法涉及生产吡啶使用的催化剂及其制备方法,特别是用多种元素助催化的吡啶生产催化剂及其制备方法该催化剂以ZSM-5分子筛为载体,在该载体上载有铅元素、钴元素、钯元素,其配比为:铅元素为载体ZSM-5分子筛质量的1~20%,钴元素为载体ZSM-5分子筛质量的0.5~10%,钯元素为载体ZSM-5分子筛质量的0.01~0.2%。
该催化剂在使用过程中能够提高吡啶收率,并能长时间保持稳定性和选择性。
吡啶的收率可达70%,吡啶衍生物总收率可达87%。
本发明中催化剂的再生周期可以达到24小时,最高可以达到48小时。
催化剂制备方法简单易行。
申请人:东南大学
地址:210096 江苏省南京市四牌楼2号
国籍:CN
代理机构:南京经纬专利商标代理有限公司
代理人:叶连生
更多信息请下载全文后查看。
多硝基吡啶类化合物的合成及应用研究进展

Chemical Propellants & Polymeric Materials2010年第8卷第5期· 32 ·多硝基吡啶类化合物的合成及应用研究进展王琼,李吉祯,蔚红建,付小龙,邵重斌,吴淑新,樊学忠(西安近代化学研究所,陕西西安 710065)摘 要:综述了2,6-二氨基-3,5-二硝基吡啶(ANPy)及其氧化物(ANPyO)、2,4,6-三氨基-3,5-二硝基吡啶(TANPy)及其氧化物(TANPyO)、2,4,6-三硝基吡啶(TNPy)及其氧化物(TNPyO)等多硝基吡啶类含能化合物的合成及应用研究进展。
A N P y O 的爆轰性能和安全性能与三氨基三硝基苯(TATB)接近,可作为高能钝感炸药;理论预测TANPy 比TATB 钝感;TNPy O 具有良好的热稳定性和化学稳定性。
预计这些多硝基吡啶类含能化合物在钝感炸药、低易损发射药和钝感推进剂领域中有良好的应用前景。
关键词:多硝基吡啶类含能化合物;推进剂;合成;应用中图分类号: TQ226.4 文献标识码: A 文章编号: 1672-2191(2010)05-0032-05收稿日期:2010-03-09作者简介:王琼(1985-),男,在读硕士,从事固体推进剂配方设计等研究。
电子信箱:0304140125@163.co m多数硝基吡啶化合物具有含氮量高、生成焓高和热安定性好等特点,近年来受到含能材料研究者的广泛关注[1]。
目前,国内外广泛研究的硝基吡啶类化合物主要有2,6-二氨基-3,5-二硝基吡啶(ANPy)及其氧化物(ANPyO)、2,4,6-三氨基-3,5-二硝基吡啶(TANPy)及其氧化物(TAN-P y O )、2,4,6-三硝基吡啶(T N P y )及其氧化物(TNPyO)等。
这些多硝基吡啶化合物的感度低、热安定性好且具有优异的爆轰性能,在钝感弹药、低易损发射药和钝感推进剂中有广泛的应用前景。
g-C3N5在光催化中的应用进展杜易高智白璟垚

g-C3N5在光催化中的应用进展杜易高智白璟垚发布时间:2023-06-03T07:46:55.931Z 来源:《中国科技信息》2023年6期作者:杜易高智白璟垚[导读] 宽光谱响应性的高效光催化材料体系的设计与构筑是光催化领域的研究热点和前沿。
石墨相氮化碳g-C3N5材料具有高的N/C比,较低的禁带宽度,在光催化中有着潜在的应用前景。
本文总结了近年来基于g-C3N5材料的改性及其在光催化污染物降解、裂解水和CO2还原中的应用进展。
为未来g-C3N5的光催化研究提供了基础。
西安建筑科技大学材料与工程学院摘要:宽光谱响应性的高效光催化材料体系的设计与构筑是光催化领域的研究热点和前沿。
石墨相氮化碳g-C3N5材料具有高的N/C 比,较低的禁带宽度,在光催化中有着潜在的应用前景。
本文总结了近年来基于g-C3N5材料的改性及其在光催化污染物降解、裂解水和CO2还原中的应用进展。
为未来g-C3N5的光催化研究提供了基础。
关键词:g-C3N5,改性策略,光催化1 引言在多相光催化领域中,二维(2D)材料因其良好的电学、光学及催化特性,是光催化体系中一类重要的研究对象。
石墨相氮化碳(g-C3N4)作为一种典型的光催化剂,在光催化的应用很广泛[1]。
但其带宽较宽,只能吸收500 nm以下的可见光,对太阳能的利用率不高,并且载流子极易复合,限制了大规模应用。
为了提升其光催化性能,科研工作者发现可以通过调整C/N比例,制备出N浓度偏高的氮化碳材料,调控电子结构,减低禁带宽度,进而增强光催化性能。
近年来,一种富含N的g-C3N5材料通过硬模板法被成功制备[2],其具有高的热力学稳定性,较低的禁带宽度(2.0 eV),表现出比缺N的氮化物更优异的光催化性能。
但其较低的能带位置使得氧化还原能力不足,制约了其发展。
因此科研工作者通过深入研究发现,可以通过复合其他材料的方式提高g-C3N5材料的光催化性能。
2 g-C3N5材料的改性策略g-C3N5材料的性能受制于其高的光生电荷复合率和较低的氧化还原能力。