美国药典残留溶剂限度
ICH常用有机溶剂分类及残留限度

ICH常用有机溶剂分类及残留限度2009-12-04 11:50残留溶剂无防治作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
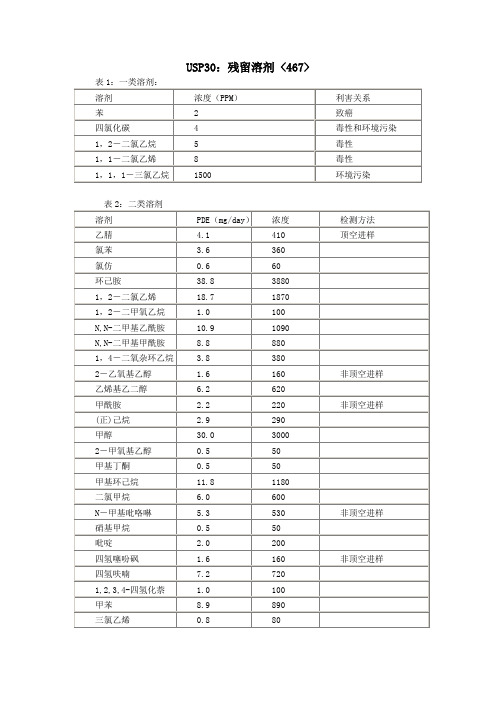
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
美国药典通则467残留溶剂的检测

美國藥典通則<467>殘留溶劑的檢測<467> RESIDUAL SOLVENTSSpectrum 质检部斯百全化学(上海)有限公司800.720.5720 通則適用範圍•所有藥品原料,輔料,製劑中可能存在的溶劑通則目標•為病人的安全,規定藥品中可接受的殘留溶劑量殘留溶劑分類•一類(class 1): 已知會產生不可接受毒性的一些溶劑(Solvents that are know to cause unacceptabletoxicities)•二類(class 2):與並不嚴重毒性相關的一些溶劑(Solvents associated with less severe toxicity)•三類(class 3):較少毒性的溶劑(Less toxic solvents )•其他殘留溶劑:無足夠毒理學數據(Other residual solvents)未列在本通則的溶劑怎麼處理?•當工廠使用經法定監管機構批准使用的未在本通則中羅列的新溶劑時,工廠有責任告知USP 在該品種標準中這一溶劑的鑒定、限度、測試方法,USP將會在個論中加上這一項。
•ICH 認可的新溶劑,USP將直接加在該通則中。
測試範圍•使用過的溶劑•生產或純化過程中產生的溶劑測試方式•直接測最終產品•或者各成份累加:如果各成分累加的值大于本通则的限度,则需测最终产品。
Colchicine秋水仙堿•Colchicine•C22H25NO6 399.44•Acetamide, N-(5,6,7,9-tetrahydro-1,2,3,10-tetramethoxy-9-oxobenzo[a]heptalen-7-yl)-, (S)-.Colchicine[64-86-8].•»Colchicine is an alkaloid contained in various species of Colchicum and in other genera. It contains not less than 94.0 percent and not more than 101.0 percent of C22H25NO6, calculated on the anhydrous, solvent-free basis.•Caution—Colchicine is extremely poisonous.•Packaging and storage—Preserve in tight, light-resistant containers.•USP Reference standards 11—USP Colchicine RS.•Identification, Infrared Absorption 197K—[note—Disregard any peak occurring at 1735 cm1.]•Specific rotation 781S: between 240 and 250, calculated on the anhydrous, solvent-free basis. Test solution: 10 mg per mL, in alcohol. •Water, Method I 921: not more than 2.0%.•Limit of colchiceine—To 5 mL of a solution (1 in 100) add 2 drops of ferric chloride TS: no definite green color is produced. •Limit of ethyl acetate—Internal standard solution—Dilute 0.5 mL of n-propyl alcohol with water to 100.0 mL. •Standard solution—Pipet1 mL of ethyl acetate and 0.5 mL of n-propyl alcohol into a 1000-mL volumetric flask, add water to volume, and mix. Each mL of Standard solution contains 0.90 mg of ethyl acetate.•Test solution—Place about 250 mg of Colchicine, accurately weighed, in a 10-mL volumetric flask, dissolve in about 8 mL of water, and add 1.0 mL of Internal standard solution. Add water to volume, and mix.•Procedure—Determine appropriate sensitivity settings on a gas chromatograph (see Chromatography 621) fitted with a 4-mm ×1.5-m column packed with 20% (w/v) phase G14 on support S1, maintaining the column temperature at75, using nitrogen as the carrier gas, and using a flame-ionization detector. Inject the Standard solution and the Test solution, determine the peak height for ethyl acetate relative to the peak height for n-propyl alcohol, and calculate the percentage, by weight, of ethyl acetate in the portion of Colchicine taken: not more than 8.0% is found.•Chromatographic purity—The sum of the responses of any peaks other than that due to colchicine, eluting within 1.5 times the retention time for colchicine, is not more than 5.0% of the sum of all responses, obtained as directed in the Assay.•Residual solvents 467: meets the requirements, except that the limit of chloroform is 100 ppm.二類溶劑計算選項•選項1:Dose ≤10g/dayConcentration(ppm)=(1000ug/mg ×PDE)/dose•選項2:根據藥品實際情況直接計算PDE是否符合要求,若超出限度則需測試。
467 残留溶剂 usp

<467> RESIDUAL SOLVENTS残留溶剂(Chapter under this new title—to become official July 1, 2008)(这个新名称下的通则----2008年7月1日起生效)(Current chapter title is <467> Organic Volatile Impurities)(当前通则名称是<467>有机挥发性杂质)INTRODUCTION介绍This general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本通则适用于现有原料药、辅助剂、成药。
所有原料药和成药均需对可能存在其中的溶剂进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs because the solvents employed may vary from one manufacturer to another.在所适用的限度与下面所述相符的情况下,残留溶剂检测一般不在具体各论中提及,因为每个生产商所使用的溶剂可能都不相同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The general chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本通则的目的是为了保障病人的安全,提供在药物中残留溶剂的可接受数量。
USP-467 溶剂残留

[467]有机挥发性杂质残留溶剂限度根据药典要求,药品中残留溶剂定义:在药物或赋形剂制造或使用过程中,或药物制剂生产过程中残存的有机挥发性物质。
目前制药技术不能完全去除残留溶剂。
药物或赋形剂合成中选择适当的溶剂可提高得率,或获得某些特性如晶型、纯度和溶解性。
因此,合成中所用溶剂有时可能是危险因素。
本章节不包括涉及用于组分的溶剂或溶剂化物。
尽管如此,以上产品仍应标明所含溶剂含量并证明对人体安全。
由于残留溶剂不用于治疗用途,因此应尽可能除去,以符合药物、赋形剂和产品规格要求、GMP或其他质量标准。
药物制剂中所含溶剂不得高于安全性评价规定限度。
众所周知,药物、赋形剂、药物制剂不应含能引起不可逆毒副作用(Ⅰ级,见表1)的残留溶剂,除非证明有很好的风险-效益比。
应对引起次级严重毒副作用的残留溶剂(Ⅱ级,见表2)规定限度,以防止病人发生潜在的不良反应。
理想状态下,应尽可能使用低毒性溶剂(Ⅲ级,见表3)。
本章节所有溶剂列表见附录1。
下述表格及限度并不代表全部。
当医药工业发展需使用其他溶剂情况下,应将新溶剂添加到列表中。
当药物、赋形剂、药物制剂生产和纯化过程中残留已知有机溶剂时,应依法检查溶剂限度。
本限度检查仅检查用于制造和纯化加工所用的溶剂。
虽然制造商可能选择测试药物,我们可采用累积法从制造工艺水平计算产品中残留溶剂。
如残留溶剂计算结果等于或低于本章节规定限度,可不进行残留溶剂检查。
如残留溶剂计算结果高于本章节规定限度,仍需检查残留溶剂限度以确定是否精加工降低了有关溶剂水平至可接受量。
如残留溶剂为制造所用溶剂,必须检查药物制剂残留溶剂限度。
附录2为有关残留溶剂的相关背景资料。
根据安全评估残留溶剂分类国际化学安全机构用术语“可忍受日摄食量”(TDI)描述有毒化学物质残留限度。
世界卫生组织和其他国家和国际卫生机构用术语“可接受日摄食量”(ADI)来描述残留限度。
术语“允许日接触量”(PDE)定义为根据药效学残留溶剂可接受摄入量,避免与ADIs混淆。
美国食品中农药残留限量标准

美国食品中农药残留限量标准美国农药残留容许量标准主要由美国环保局(EPA)负责制定,在美国联邦法规汇编(CFR)第40篇―环境保护‖第180节―化学农药在食品中的残留容许量与残留容许量豁免‖公布。
该节包括5个分节,即A分节―定义和解释性法规‖、B分节―程序性规定‖、C分节―具体容许量‖、D分节―容许量豁免‖及E分节―不需要制订限量的农用化学物‖。
C分节,D分节及E分节部分数据可在数据库中查询。
A分节,B分节,D分节及E分节部分内容见附件1。
同时,美国食品药品管理局(FDA)对食品和饲料中的不可避免的农药残留制定了行动水平(Action level),在FDA符合性政策指南(CPG Sec. 575.100)公布(附件2)。
此外,美国联邦法典(US Code)第21篇―食品和药品‖第9章346a部分(附件3)还对农药残留容许量以及残留容许量豁免的原则性问题进行了规定,如规定含有无残留容许量标准或者残留容许量豁免农药的食品不安全等。
附件1美国联邦法规汇编(CFR)第40篇“环境保护”第180节化学农药在食品中的残留容许量与残留容许量豁免A分节定义和解释性法规§ 180.1 定义和解释。
(a) 管理者,如无限定条件,则指环境保护局的管理者。
(b) 机构,如无限定条件,则指环境保护局。
(c) FFDCA是指《联邦食品,药物和化妆品法案》,经21 U.S.C. 301–392修订。
(d)初级农产品包括:新鲜水果,不管是否已清洗和着色或已经进行了其它处理,只要还保持它们带皮的自然形式;在未加工和自然状态下的蔬菜,无论它们是否已剥掉了边叶,封装或制成新鲜绿色沙拉等;以及谷物、坚果、鸡蛋、生牛奶、肉类以及其他类似的农产品。
不包括已经加工的食品以及通过烹饪、冷冻、脱水、磨碎等方式加工的产品。
(e) 根据FFDCA 402节和408(a) 节的规定,如果原料中所含化学农药属于豁免物质,或其残留水平不超过FFDCA 法408节制定的残留容许量标准,只要符合下列条件,则用其制成的加工品被认为是安全的,即使未针对该化学农药在该加工品的残留制定专门容许量或未规定容许量豁免时也是如此:(1) 化学农药已在初级农产品上使用,并符合本节的残留容许量要求;(2) 化学农药残留已通过良好生产规范尽可能去除;并且(3)加工食品的残留水平不高于相应的初级农产品原料的残留容许量。
医药中常用有机溶剂分类及残留限度

医药中常用有机溶剂分类及残留限度医药中常用有机溶剂分类及残留限度药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
USP(467)有机挥发性杂质—残留溶剂的限制

USP(467)有机挥发性杂质—残留溶剂的限制基于药典的要求,残留溶剂在此定义为有机挥发性化学物,它用于生产药物赋形剂,或药品制备过程中,通过规范的生产技术,不能将残留溶剂完全除去,在合成原料药或生产赋形剂过程中,选择适当的溶剂可提高产量或决定药物的外观,如晶型、纯度和溶解度。
因此,有机溶剂是合成过程的关键因素。
本章的指导原则不是指谨慎地用于赋形剂的溶剂,也不是溶剂化合物。
然而,在这样的产品中溶剂的含量应该被测定和判断。
因为残留溶剂不能提供药物功效,因此应去除所有的残留溶剂,更进一步的可能以符合产品规范,GMP或者基本质量要求。
原料药中残留溶剂的含量级别不能高于安全数据支持的残留溶剂级别。
已知溶剂可导致不可接受的毒性应避免在生产中使用,除非有特别的证明它的使用是合理的(一类溶剂)。
一些有较弱毒性的溶剂(二类)应限制使用,以保护病人出现潜在的不利影响。
低毒性溶剂清单不完整,其他可能使用的溶剂,以权威组织机构的批准为准,将会在清单中依次列入。
原料药,赋形剂和药品中残留溶剂的测试,应当在生产或者纯化过程中进行,它在生产或者纯化过程中出现的残留溶剂的测试才是必要的。
虽然生产商会选择性地测试药品,会使用各个成分中残留溶剂水平累积方法计算总残留溶剂水平,如果计算的结果等于或低于本章指导原则建议的溶剂水平,不用进行再测试。
如果高于规定的水平,应进行检测,以确定是否降低制备过程相关溶剂的量,使之在可接受量范围内。
如果一种残留溶剂在药品生产中出现,也应当进行检测。
一类一类残留溶剂(表1)不应该在原料药、赋形剂和药品生产中使用,因为这些溶剂是不可接受的毒性物质,且对环境有不良影响。
但是,如果使用一类溶剂,应根据表1限定它们的量,除非有独立专文支持。
表1中的三氯乙烷是环境有害物,≤1500PPM的限量是安全数据。
当在原料药、赋形剂和药品生产中使用一类溶剂,指导原则要求任何必要的地方,都要有对残留溶剂鉴别、控制和数量测试的方法的描述,除非引入适当的验证程序,这种程序应该符合在相关独立专文中的USP标准,见ICH。
USP-467 溶剂残留
[467]有机挥发性杂质残留溶剂限度根据药典要求,药品中残留溶剂定义:在药物或赋形剂制造或使用过程中,或药物制剂生产过程中残存的有机挥发性物质。
目前制药技术不能完全去除残留溶剂。
药物或赋形剂合成中选择适当的溶剂可提高得率,或获得某些特性如晶型、纯度和溶解性。
因此,合成中所用溶剂有时可能是危险因素。
本章节不包括涉及用于组分的溶剂或溶剂化物。
尽管如此,以上产品仍应标明所含溶剂含量并证明对人体安全。
由于残留溶剂不用于治疗用途,因此应尽可能除去,以符合药物、赋形剂和产品规格要求、GMP或其他质量标准。
药物制剂中所含溶剂不得高于安全性评价规定限度。
众所周知,药物、赋形剂、药物制剂不应含能引起不可逆毒副作用(Ⅰ级,见表1)的残留溶剂,除非证明有很好的风险-效益比。
应对引起次级严重毒副作用的残留溶剂(Ⅱ级,见表2)规定限度,以防止病人发生潜在的不良反应。
理想状态下,应尽可能使用低毒性溶剂(Ⅲ级,见表3)。
本章节所有溶剂列表见附录1。
下述表格及限度并不代表全部。
当医药工业发展需使用其他溶剂情况下,应将新溶剂添加到列表中。
当药物、赋形剂、药物制剂生产和纯化过程中残留已知有机溶剂时,应依法检查溶剂限度。
本限度检查仅检查用于制造和纯化加工所用的溶剂。
虽然制造商可能选择测试药物,我们可采用累积法从制造工艺水平计算产品中残留溶剂。
如残留溶剂计算结果等于或低于本章节规定限度,可不进行残留溶剂检查。
如残留溶剂计算结果高于本章节规定限度,仍需检查残留溶剂限度以确定是否精加工降低了有关溶剂水平至可接受量。
如残留溶剂为制造所用溶剂,必须检查药物制剂残留溶剂限度。
附录2为有关残留溶剂的相关背景资料。
根据安全评估残留溶剂分类国际化学安全机构用术语“可忍受日摄食量”(TDI)描述有毒化学物质残留限度。
世界卫生组织和其他国家和国际卫生机构用术语“可接受日摄食量”(ADI)来描述残留限度。
术语“允许日接触量”(PDE)定义为根据药效学残留溶剂可接受摄入量,避免与ADIs混淆。
医药中常用有机溶剂分类及残留限度

医药中常用有机溶剂分类及残留限度医药中常用有机溶剂分类及残留限度药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
ICH常用有机溶剂分类及残留限度

ICH常用有机溶剂分类及残留限度2009-12-0411:50??????残留溶剂无防治作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
????药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
????各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
???按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
USP30残留溶剂
USP30:残留溶剂 <467>一类和二类溶剂(水溶性的物品)程序A:一类残留溶剂标准贮备液:取1.0ml的USP一类残留溶剂混合物RS到100ml的容量瓶中,加入9ml的二甲基亚砜,用水稀释到刻度,混匀。
取1.0ml该溶液到100ml的容量瓶中,用水稀释到刻度,混匀。
取1.0ml该溶液到10ml的容量瓶中,加水稀释到刻度,混匀。
一类残留溶剂标准液:移取1.0ml的一类残留溶剂标准贮备液到合适顶空安瓿瓶中,加5.0ml的水,加塞,加盖,混匀。
二类残留溶剂标准贮备液:取1.0ml的USP二类残留溶剂混合物A(RS)到100ml的容量瓶中,加水稀释到刻度,混匀,制得二类残留溶剂标准储备液A。
取1.0ml的USP二类残留溶剂混合物B(RS)到100ml 的容量瓶中,加水稀释到刻度,混匀,制得二类残留溶剂标准储备液B。
二类残留溶剂A标准液:移取1.0ml的二类残留溶剂标准贮备液A到合适顶空安瓿瓶中,加5.0ml的水,加塞,加盖,混匀。
二类残留溶剂B标准液:移取5.0ml的二类残留溶剂标准贮备液B到合适顶空安瓿瓶中,加1.0ml的水,加塞,加盖,混匀。
测试贮备液:取约250mg的测试品,准确称量,加入25ml的容量瓶中,加水溶解并稀释到刻度,混匀。
测试液:取测试贮备液5.0ml到合适顶空安瓿瓶中,加1.0ml的水,加塞,加盖,混匀。
一类系统适用性溶液:取1.0ml的一类残留溶剂标准贮备液到合适顶空安瓿瓶中,加入5.0ml的测试贮备液,加塞加盖,混匀。
色谱系统<见色谱621>:气相色谱,配制:FID检测器,0.32mm×30mm的炭化硅胶柱,涂布1.8µm厚的G43或0.53mm×30m的广口柱,涂布3µm厚的G43。
载气为氮气或氢气,线速约为35cm/秒,分流比为1:5。
柱温:40℃保持20分钟,然后以10℃/分钟的速度升温至240℃,保持20分钟;进样口和检测器的温度分别为140℃和250℃。
USP美国药典70种农残检测限度

26
硫丹
Endosulfan (sum of isomers and endosulfan sulfate)
3
27
异狄氏剂
Endrin
0.05
28
乙硫磷
Ethion
2
பைடு நூலகம்29
乙嘧硫磷
Etrimphos
0.05
30
皮蝇硫磷
Fenchlorophos (sum of fenchlorophos and fenchlorophos-oxon)
1.5
36
氟氰戊菊酯
Flucytrinate
0.05
37
氟胺氰菊酯
τ-Fluvalinate(Tau-Fluvalinate (F) )
0.05
38
地虫硫磷
Fonophos
0.05
39
七氯
Heptachlor (sum of heptachlor, cis-heptachlorepoxide and trans-heptachlorepoxide)
3
除虫菊酯
Pyrethrins
65
喹硫磷
Quinalphos
0.05
66
五氯硝基苯
Quintozene (sum of quintozene, pentachloraniline and methyl penthachlorphenyl sulfide)
1
67
八氯二丙醚
S-421
0.02
68
四氯硝基苯
Bromide, inorganic (calculated as bromide ion)
50
7
乙基溴硫磷
usp36 467溶剂残留

<467>溶剂残留简介:INTRODUCTIONThis general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本章节适用于现有的原料药,辅料和制剂。
应对原料药或制剂产品中可能存在溶剂的所有原料及制剂产品进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs, because the solvents employed may vary from one manufacturer to another.当限值与下面提供的数值相符合,残留溶剂的测试方法一般不会在专论中特别,因为不同制造商所使用的溶剂不同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
USP32残留溶剂完整版

USP32:残留溶剂<467> 完整翻译概述该通那么适用于现有的原料药,辅料和成品。
所有原料药和成药均需对可能存在其中的溶剂进行操纵。
凡例中包括了对残留溶剂操纵的总的要求,通那么<467>将对溶剂残留所利用的检测方式与限度做出详细的规定,由于各药品厂家利用的溶剂种类截然不同,因此各论中再也不涉及。
本通那么的成立目的是保障病人的服药平安,提供残留溶剂的可同意限度。
USP建议尽可能利用低毒性的溶剂,并描述了关于某些残留溶剂而言其毒性可同意的限度水平。
药物中的残留溶剂系指在原料药或辅料的生产中,和在制剂制备进程中利用或产生的,但在工艺进程中未能完全去除的有机溶剂。
在原料药的合成和赋形剂的生产进程中,适当的溶剂可提高产量、阻碍药物的外观与形状(如晶型、纯度、溶解度等)。
因此,有机溶剂是合成进程的关键因素。
本通那么尽管没有对作为辅料直接利用的溶剂和溶剂化物形态的溶剂进行单独的强调,只是此类产品中的溶剂含量一样应该进行评估与论证。
药物中残留的溶剂不能产生药效,应尽最大可能去除,以知足GMP或其它质量标准的要求。
药品中含有的残留溶剂应该不超过已有平安数据所支持水平。
一类溶剂(见表1)为幸免利用的溶剂,有不可同意的毒性,除非它们的利用有风险-收益评估结果的强力支持,应幸免在生产中利用。
二类溶剂(见表2)为限制利用的溶剂,有较弱毒性的溶剂,应限制利用以避免显现潜在的不良阻碍。
三类溶剂(见表3)为低毒溶剂,对人体及环境的危害较小,应尽可能利用。
注:本通那么涉及的所有溶剂见附件1,这些表格中的溶剂及限度并非全数,随着医药工业的进展,尔后会将新的溶剂添加到列表中。
表1:一类溶剂(应幸免利用)表2:二类溶剂(应限制利用)表3:三类溶剂(GMP或其他质量要求限制利用,对人体低毒的溶剂)本版药典为了保障人们的用药平安,特做出以下规定:当生产商通过法规部门的批准,被授权许诺利用不在本版USP药典清单上的溶剂或许诺利用溶剂的残留量高于规定限度时,生产商有责任将相关信息(溶剂的辨别方式,残留溶剂许诺限度,采纳的检测方式)报送给USP,USP会及时的在相应的各论中引入该事项,更新完善药典章程。
美国药典标准

盐酸缬更昔洛韦C14H22N6O5·HCl 390.82L-缬氨酸,9-[[2-羟基-1-(羟甲基)乙氧基]甲基]鸟嘌呤酯,盐酸盐L-缬氨酸,2-[(2-氨基-1,6-二氢-6-氧代-9H-嘌呤-9-yl)甲氧基]-3-羟丙基酯,盐酸盐[175868-59-5]按干燥品计算,本品含C14H22N6O5·HCl应为97.0%~102.0%。
包装与贮存——密封包装,25℃保存,允许温度浮动范围15℃~30℃。
鉴别A:红外吸收(197K)B:紫外吸收(197U)溶液:10μg/ml溶剂:甲醇C:本品的水溶液(1→20)显氯化物的鉴别反应。
(191)水分,方法一(921):取本品100mg,依法检查,含水分不得过8.0%。
炽灼残渣(281):取本品1g,依法检查,遗留残渣不得过0.10%。
重金属,方法一(231):不得过0.002%。
异丙醇内标溶液——取100μL的1,4-二氧六环到100mL容量瓶,用二甲基甲酰胺定容。
标准储备液——分别取1.0mL异丙醇与0.1mL甲苯到同一100mL容量瓶,用二甲基甲酰胺定容。
(注:甲苯用来调整系统适用性)标准溶液——取2.0mL内标溶液置于反应瓶中,再加入100μL标准储备液,混匀,待用。
系统适用性溶液——使用标准溶液供试品溶液——精密称取90~100mg的盐酸缬更昔洛韦,置于反应瓶中,精密量取2.0mL的内标溶液加入其中,混匀待用。
色谱系统(见色谱法(621))——配有氢火焰离子化检测器的气相色谱仪,色谱柱为涂布3.0-µm G43固定相的0.53mm× 30m毛细管柱,载气为氦气,流速为10.5mL/min,分流比为1:15。
色谱程序设定如下:初始柱温为40℃,保持10min,后在25℃/min下逐渐升温至240℃。
(注:每次进样后调节柱温至240℃,并保持15min左右)进样口温度保持在250℃,检测器温度保持在300℃。
美国药典USP

美国药典USP美国药典USP浓度重量克分子浓度、容量克分子浓度和当量浓度用于本药典内大部分化学含量测定和检测方法中(亦见容量溶液于试剂、指示剂和溶液篇章中)重量克分子浓度用m表示,前面有一个数目字即为该溶质的克分子数(1公斤的所标明的溶液中)容量克分子浓度用M表示,前面的数目字表示在制备1立升溶液中所含的该溶质的克分子数当量浓度用N表示,前面的数目字表示制备1立升溶液中该溶质的克当量数。
百分比计量—百分比浓度如下表示:重量于重量百分比—(w/w)表示一个组分在100克溶液或混合物中的克数重量于容量百分比—(w/v)表示一个组分在100毫升溶液中的毫升数(不管溶剂是水还是其他液体)容量于容量百分比—(v/v)表示一个组分在100毫升溶液中的毫升数用百分比这个名词没有限定意义,对固体和半固体用w/w;对溶液或固体在溶液中的悬浮液用w/v;对液体在液体溶液用v/v;气体在液体内用w/v,例:一个1%的溶液是由溶解1克固体或半固体或1毫升溶液于足够的溶剂使成100毫升溶液。
由于室温的差异,微小的容量计量差异可忽略。
有效数据和允许偏差此处表示的数限是上限和下限并包括此二值以及其间的所有数字,但在限度以外的数值不在内。
在供试品的专篇内,检测中不管数字是以百分比还是绝对数字来表示都是表示最末的数字。
在容量滴定法中相当的叙述——容量滴定法故采用包括相当于标化滴定液的每毫升相当于供试品的重量,在这样的相当陈述,滴定液的浓度中的有效数字被认为相当于供试品的重量有效数字,所有的容量滴定应做空白校正。
允许偏差—药典所述的供试品的专篇内所规定的那些限度的建立是把供试品作为药物来使用或作为营养剂、饮食补充剂使用,除非另有其他指定。
药物的活性成分用分子式来计其强度标明了化学本质,如同已给的供试品的化学全名其绝对纯度为100%。
中略。
一个可写在检测报告上的值通常是几个单独测定值的合计,此结果是按规定所做的一个完整的测定所得而用于可接受的标示值进行比较,当需舍入时,5以下舍去,5(包含5)以上进1,如:标准含量为≥98.0% 实测值舍入前97.95% 舍入后98.0% 判断合格舍入前97.94% 舍入后97.9% 判断不合格标准限度≤0.02% 实测值舍入前0.025% 舍入后0.03% 判断不合格舍入前0.015% 舍入后0.02% 判断合格舍入前0.024% 舍入后0.02% 判断合格检测和含量测定设施——在检测或含量测定中所用的容器或设施的大小型式不过是一种推荐。