杂质:残留溶剂的指导原则
残留溶剂的指导原则

杂质:残留溶剂的指导原则1.介绍本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。
在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。
纯度和溶解度。
因此.有时溶剂是合成中非常关键的因素。
本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。
出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。
制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。
一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。
使用低毒溶剂(第三类,表3)较为理想。
附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。
第一、二类溶剂的建议限度或溶剂的分类会随着。
新的安全性资料的获得而调整。
含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。
2. 指导原则的范围指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。
应进行残留溶剂的检验。
也只有在上述情况下,才有必要作溶剂的检查。
虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。
如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。
化学药物杂质研究的技术指导原则

化学药物杂质研究的技术指导原则一、概述任何影响药物纯度的物质统称为杂质。
杂质的研究是药品研发的一项重要内容。
它包括选择合适的分析方法,准确地分辨与测定杂质的含量并综合药学、毒理及临床研究的结果确定杂质的合理限度。
这一研究贯穿于药品研发的整个过程。
由于药品在临床使用中产生的不良反应除了与药品本身的药理活性有关外,有时与药品中存在的杂质也有很大关系。
例如,青霉素等抗生素中的多聚物等高分子杂质是引起过敏的主要原因。
所以规范地进行杂质的研究,并将其控制在一个安全、合理的限度范围之内,将直接关系到上市药品的质量及安全性。
本指导原则是在借鉴国外相关指导原则[1][2]的基础上,结合我国新药研发的实际情况制定的。
目的是为我国的药品研发提供有益的指导,从而提高药品的质量,保证人民的用药安全。
由于新药研究的探索性很强,每种药品的具体研究情况差异有可能很大,本指导原则不可能涵盖杂质研究的全部,仅提供了一个基本的研究思路和方法。
特殊情况下,研究单位可在科学、合理的基础上,对杂质进行研究,只要能用科学的数据证明药品中存在的杂质可被控制在安全、合理的范围内,就达到了杂质研究的目的。
本指导原则涵盖的范围包括新的及仿制已有国家标准的化学原料药及制剂。
发酵工艺生产的抗生素类药物一般不包括在本原则的讨论范畴,但如有可能,也建议参考本原则的有关要求。
由于我国对临床研究也实行行政审批的管理,所以,本指导原则不仅适用于上述药品的上市生产申请,也适用于临床研究的申请。
二、杂质的分类药品中的杂质按其理化性质一般分为三类:有机杂质、无机杂质及残留溶剂。
按照其来源,杂质可以分为工艺杂质(包括合成中未反应完全的反应物及试剂、中间体、副产物等)、降解产物、从反应物及试剂中混入的杂质等。
按照其毒性分类,杂质又可分为毒性杂质和普通杂质等。
杂质还可按其化学结构分类,如其它甾体、其它生物碱、几何异构体、光学异构体和聚合物等。
本指导原则主要按照杂质的理化性质分类。
USP(467)有机挥发性杂质—残留溶剂的限制

USP(467)有机挥发性杂质—残留溶剂的限制基于药典的要求,残留溶剂在此定义为有机挥发性化学物,它用于生产药物赋形剂,或药品制备过程中,通过规范的生产技术,不能将残留溶剂完全除去,在合成原料药或生产赋形剂过程中,选择适当的溶剂可提高产量或决定药物的外观,如晶型、纯度和溶解度。
因此,有机溶剂是合成过程的关键因素。
本章的指导原则不是指谨慎地用于赋形剂的溶剂,也不是溶剂化合物。
然而,在这样的产品中溶剂的含量应该被测定和判断。
因为残留溶剂不能提供药物功效,因此应去除所有的残留溶剂,更进一步的可能以符合产品规范,GMP或者基本质量要求。
原料药中残留溶剂的含量级别不能高于安全数据支持的残留溶剂级别。
已知溶剂可导致不可接受的毒性应避免在生产中使用,除非有特别的证明它的使用是合理的(一类溶剂)。
一些有较弱毒性的溶剂(二类)应限制使用,以保护病人出现潜在的不利影响。
低毒性溶剂清单不完整,其他可能使用的溶剂,以权威组织机构的批准为准,将会在清单中依次列入。
原料药,赋形剂和药品中残留溶剂的测试,应当在生产或者纯化过程中进行,它在生产或者纯化过程中出现的残留溶剂的测试才是必要的。
虽然生产商会选择性地测试药品,会使用各个成分中残留溶剂水平累积方法计算总残留溶剂水平,如果计算的结果等于或低于本章指导原则建议的溶剂水平,不用进行再测试。
如果高于规定的水平,应进行检测,以确定是否降低制备过程相关溶剂的量,使之在可接受量范围内。
如果一种残留溶剂在药品生产中出现,也应当进行检测。
一类一类残留溶剂(表1)不应该在原料药、赋形剂和药品生产中使用,因为这些溶剂是不可接受的毒性物质,且对环境有不良影响。
但是,如果使用一类溶剂,应根据表1限定它们的量,除非有独立专文支持。
表1中的三氯乙烷是环境有害物,≤1500PPM的限量是安全数据。
当在原料药、赋形剂和药品生产中使用一类溶剂,指导原则要求任何必要的地方,都要有对残留溶剂鉴别、控制和数量测试的方法的描述,除非引入适当的验证程序,这种程序应该符合在相关独立专文中的USP标准,见ICH。
ICH杂质残留溶剂的指导原则QCR

ICH杂质残留溶剂的指导原则QCRICH杂质残留溶剂的指导原则QCR(Quality Control Residual Solvents)是国际药品监管机构-国际药品注册、技术和质量控制委员会(ICH)发布的一项指南,用于规范药品制造过程中的溶剂残留物的质量控制。
溶剂残留物是指在药品生产过程中添加的溶剂,但在制剂中未完全蒸发或被移除的溶剂。
导言溶剂在药品生产过程中广泛应用,包括分离、提取、结晶和纯化等步骤。
然而,溶剂中的残留物可能对人体产生毒性或对药品质量产生影响。
因此,在药品开发和生产过程中,需要对残留溶剂进行必要的质量控制。
溶剂分类ICHQCR指导原则将溶剂分为类1、类2A和类2B三类。
类1溶剂是无毒、无致畸、无致癌和生殖毒性的溶剂,如水、乙醇、甘油等。
这些溶剂的使用一般没有限制。
类2A溶剂是有潜在毒性或限制使用的溶剂,但在限定剂量下不会对人体产生危害。
这类溶剂的最大可容许残留量取决于药物用量、剂型和给药途径。
一些常见的类2A溶剂包括氯仿、氯化甲烷、二氯甲烷等。
类2B溶剂是有潜在毒性,并且在制剂中的残留量应尽量减少的溶剂。
这类溶剂的最大可容许残留量取决于药物用量和给药途径。
一些常见的类2B溶剂包括苯、乙酸乙酯、二甲基甲醚等。
质量控制要求ICHQCR指导原则确定了三种质量控制要求来管理溶剂残留物。
1. 可容许残留量(Permitted Daily Exposure, PDE):PDE是指人体每日接触该溶剂残留量的最大可容许量。
它可以通过毒理学评估和毒性数据来确定。
对于类1溶剂,PDE通常设置为无限大。
对于类2A和类2B 溶剂,PDE被用来确定最大可容许残留量。
2.可容许残留量限度:可容许残留量限度是指药品中残留溶剂的最大可容许浓度。
它根据预期剂量、给药途径和药物性质等因素来确定。
通过使用PDE和剂量预设的数学公式,可以计算出每种溶剂的可容许残留量限度。
3.分析方法:为了确保溶剂残留物的质量控制,需要使用适当的分析方法进行检测和分析。
原料药残留溶剂试验的要求及常见问题分析-2011.4-北京周立春.pdf

举例:头孢泊肟酯
残留溶剂 照残留溶剂测定法(附录Ⅷ P)测定。 甲醇、乙腈、丙酮、二氯甲烷、异丙醇、丁酮、乙酸乙 酯、四氢呋喃、乙酸丁酯、1,2-二氯乙烷、乙酸异丙酯、 苯、四氯化碳、环己烷、二氧六环、甲基异丁基酮、吡 啶、甲苯 色谱条件与系统适用性试验 略 内标溶液的制备 取正丙醇适量,用二甲基亚砜稀释制成 每1ml中约含200μg的溶液,作为内标溶液。
顶空进样:
原理:将含有挥发性组分的样品 置于密闭系统中,在一定温 度下使样品中的挥发性组分 在气-液或气-固两相甚至气液-固三相中的分配达到平衡, 然后取凝聚相上端的气体送 入气相色谱仪进行分析 优点:干净。样品中不挥发组分 不影响GC分析。减轻污染。 要求:样品至少在顶空条件下溶 解。
K
1 p i
0 i
p0i为纯溶质的蒸气 压 γi为组分的活度系 数
顶空条件的选择
顶空温度:残留溶剂的沸点较高,顶空温度也应 相应提高;但应兼顾供试品的热分解特性,尽量 避免供试品产生的挥发性热分解产物对测定的干 扰。 顶空平衡时间:一般为30~45分钟,以保证供试 品溶液的气-液两相有足够的时间达到平衡。顶 空时间通常不宜过长,如超过60分钟,可能引起 顶空瓶的气密性变差,导致定量准确性的降低。 传输管温度:对于有传输管的顶空进样器,传输 管温度应适当,通常设定在110℃~120℃。 对照品溶液与供试品溶液必须使用相同的顶空条 件。
原料药残留溶剂试 验的要求及常见问 题分析
周立春 2011.4
主要内容
1. 2. 3. 4.
前言 2010版药典中残留溶剂的相关内容 残留溶剂的方法学研究 残留溶剂测定的常见问题
前 言
1) 2) 3)
残留溶剂定义 残留溶剂的分类 药物中残留溶剂的特点
医药中常用有机溶剂分类及残留限度

医药中常用有机溶剂分类及残留限度医药中常用有机溶剂分类及残留限度药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
化学药物残留溶剂研究的技术指导原则

化学药物残留溶剂研究的技术指导原则一、概述药物中的残留溶剂系指在原料药或辅料的生产中、以及在制剂制备过程中使用或产生而又未能完全去除的有机溶剂。
根据国际化学品安全性纲要,以及美国环境保护机构、世界卫生组织等公布的研究结果,很多有机溶剂对环境、人体都有一定的危害,因此,为保障药物的质量和用药安全,以及保护环境,需要对残留溶剂进行研究和控制。
本指导原则是在参考人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)颁布的残留溶剂研究指导原则,美国药典(the United States Pharmacopoeia,USP)、英国药典(British Pharmacopoeia, BP)、欧洲药典(European Pharmacopoeia,EP)、中国药典(Chinese Pharmacopoeia, ChP)相关内容的基础上,结合我国药物研发的特点,通过分析、研究残留溶剂问题及药物的安全性、有效性及质量可控性之间的内在关系而制定的。
本指导原则总结了对残留溶剂问题的一般认识,旨在帮助药物研发者科学合理的进行残留溶剂方面的研究,也为药物评价者提供参考。
考虑到残留溶剂研究涉及的范围比较广泛,本指导原则主要对原料药的残留溶剂问题进行讨论,并以此为基础,探讨和总结药物研究过程中对残留溶剂问题的一般性原则。
药物研发者可参考本指导原则对制剂和辅料的残留溶剂问题进行研究。
考虑到药物研究开发的阶段性,本指导原则适用于药物研发的整个过程。
二、基本内容(一)残留溶剂研究的基本原则1、确定残留溶剂的研究对象从理论上讲,药物制备过程中所使用的有机溶剂均有残留的可能,均应进行残留量的研究。
但是,药物研发者可以通过对有机溶剂的性质、药物制备工艺等进行分析,提出科学合理的依据,有选择性的对某些溶剂进行残留量研究,这样,既可以合理有效的控制产品质量,又有利于降低药物研究的成本,避免不必要的浪费。
Q3C(R6)-20191004-S-Step 4杂质:残留溶剂指导原则
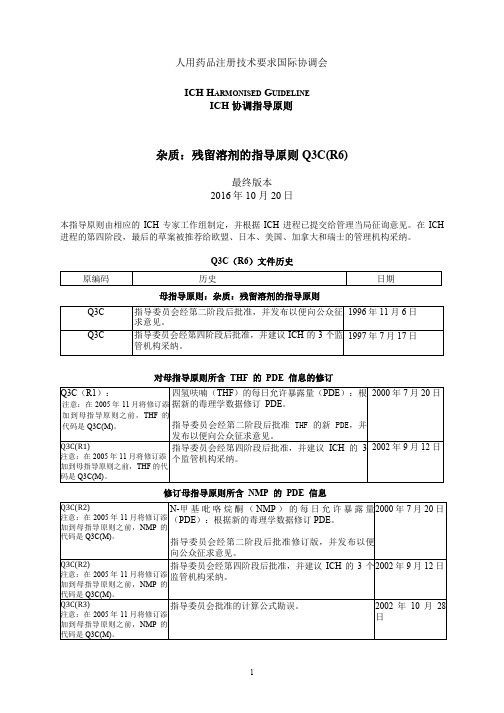
人用药品注册技术要求国际协调会ICH H ARMONISED G UIDELINEICH 协调指导原则杂质:残留溶剂的指导原则 Q3C(R6)最终版本2016年 10 月 20日本指导原则由相应的ICH 专家工作组制定,并根据ICH 进程已提交给管理当局征询意见。
在ICH 进程的第四阶段,最后的草案被推荐给欧盟、日本、美国、加拿大和瑞士的管理机构采纳。
Q3C(R6)文件历史母指导原则:杂质:残留溶剂的指导原则对母指导原则所含THF的PDE信息的修订修订母指导原则所含NMP的PDE信息母指导原则:杂质:残留溶剂的指导原则对母指导原则所含异丙基苯的PDE信息的修订修订母指导原则所含甲基异丁基酮的PDE信息,并纳入三乙胺的PDE乙二醇PDE修正I MPURITIES: G UIDELINE FOR R ESIDUAL S OLVENTS杂质:残留溶剂的指导原则TABLE OF CONTENTS目录PART I:IMPURITIES: GUIDELINE FOR RESIDUAL SOLVENTS (4)第一部分:杂质:残留溶剂的指导原则 (4)1.INTRODUCTION引言 (4)2.SCOPE OF THE GUIDELINE 指导原则的适用范围 (5)3.GENERAL PRINCIPLES通则 (6)3.1Classification of Residual Solvents by Risk Assessment基于风险评估的残留溶剂的分类 (6)3.2 Methods for Establishing Exposure Limits 建立暴露限度的方法 (6)3.3 Options for Describing Limits of Class 2 Solvents 2 类溶剂限度的表示方法 (7)3.4 Analytical Procedures分析方法 (9)3.5 Reporting levels of residual solvents残留溶剂的报告水平 (9)4.LIMITS OF RESIDUAL SOLVENTS 残留溶剂的限度 (10)4.1 Solvents to Be Avoided应避免的溶剂 (10)4.2 Solvents to Be Limited应限制的溶剂 (10)4.3 Solvents with Low Toxic Potential低潜在毒性的溶剂 (12)4.4 Solvents for which No Adequate Toxicological Data was Found 没有足够毒理学数据的溶剂 (13)GLOSSARY术语 (14)APPENDIX 1. LIST OF SOLVENTS INCLUDED IN THE GUIDELINE附录 1:指导原则中包括的溶剂列表 (15)APPENDIX 2. ADDITIONAL BACKGROUND附录 2:其他背景 (18)APPENDIX 3. METHODS FOR ESTABLISHING EXPOSURE LIMITS附录 3:建立暴露限度的方法 (19)PART II:IMPURITIES: RESIDUAL SOLVENTS (MAINTENANCE)PDE FOR TETRAHYDROFURAN第二部分:杂质:残留溶剂(修订)四氢呋喃的 PDE (23)PART III:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) PDE FOR N-METHYLPYRROLIDONE (NMP) 第三部分:杂质:残留溶剂(修订)N-甲基吡咯烷酮(NMP)的 PDE25 PART IV:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) PDE FOR CUMENEICH Harmonised Tripartite Guideline第四部分:杂质:残留溶剂(修订)异丙基苯的 PDE (27)PART V:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE)PDE FOR TRIETHYLAMINE AND PDE OF METHYLISOBUTYLKETONE第五部分:杂质:残留溶剂(修订)三乙胺的 PDE 和甲基异丁基酮的 PDE (31)PART I:IMPURITIES: GUIDELINE FOR RESIDUAL SOLVENTS第一部分:杂质:残留溶剂的指导原则Having reached Step 4 of the ICH Process at the ICH Steering Committee meeting on 17 July 1997, this Guideline is recommended for adoption to the three regulatory parties to ICH在 1997 年 7 月 17 日的 ICH 指导委员会会议上进入 ICH 进程第四阶段,并建议 ICH 的三方监管机构采纳该指导原则1.INTRODUCTION引言The objective of this guideline is to recommend acceptable amounts for residual solvents in pharmaceuticals for the safety of the patient. The guideline recommends use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则旨在建议为保证患者安全而应规定的药物中残留溶剂的可接受量。
药物中的残留溶剂测定

有机溶剂的分类
分类依据: 允许日暴露量(permitted daily exposure PDE) 定义: 是指某一有机溶剂被允许摄入而不产生毒性的日平 均最大剂量,单位为mg/天
有机溶剂的分类
类别 第一类溶剂 毒性 人体致癌物、疑为人 体致癌物或环境危害 物 有非遗传致癌毒性或 其他不可逆毒性、或 其他严重的可逆毒性 对人体低毒 没有足够毒性资料 PDE( mg/天) 0.1以下(1,1,1) 三氯乙烷除外 0.5-50
PDE : mg/天
剂量: g/天
有机溶剂的引入
根据研究对象具体情况 分析有机溶剂的引入 有机溶剂毒性
确定何种有机溶剂需要进行研究
研究方法的建立及方法学验证
研究结果的分析及质量标准的建立
有机溶剂的引入
原料药/辅料:合成过程中引入 包括: 作为合成原料或反应溶剂引入 作为反应副产物引入 由其他合成原料或其他溶剂带入 制剂 各种成份(原料药、辅料)带入 制剂制备过程中引入
有机溶剂的引入
研究集中在:原料药的第一种情况 影响因素: 有机溶剂在合成过程中使用的步骤 后续步骤中使用的有机溶剂对之前使用的溶剂的影 响 中间体的影响(中间体的纯化方法、干燥条件) 终产品精制方法和条件等等
有机溶剂的引入
制剂 控制原料药的残留溶剂,最终目的是控制制剂的残留 溶剂,使之符合规定。有时候根据制剂的一些特点, 可能对原料药残留溶剂的研究和限度要求进行特殊性 的考虑。
药物中的残留溶剂测定
浙江省药品检验所 高素英
前 言
定义:药物中的残留溶剂( Residual Solvent )系指 在原料药或辅料的生产中,以及在制剂制备过程中使 用的,但在工艺过程中未能完全去除的有机溶剂。 研究的性质:杂质研究的范畴 研究的目的:控制药物质量,保障病人用药安全。 在原料药合成中,溶剂的选择是合成中非常关键的因 素,选择适当的溶剂可提高得率或决定药物的性质,如 晶型、纯度和溶解度,从而影响疗效。同时,在某些 特定的制剂生产中,其工艺也要求使用特定的溶剂。 但由于溶剂没有疗效,故所有残留溶剂均应尽可能除 去,以使产品符合其规范、GMP或其他基本的质量要 求 。
有机溶剂残留量的测定

药物中有机溶剂的引入
▪ 研究对象:原料药/辅料、制剂 ▪ 原料药/辅料:合成过程中引入 ▪ 包括:
作为合成原料或反应溶剂引入 作为反应副产物引入 由其他合成原料或其他溶剂带入
ICH Q3c 杂质:残留溶剂的指导原则 FDA:Guidence for Industry:Q3c
Impurities:Residual Solvents
EMEA:Position Paper on Specification fro Class 1 and Class 2 Residual Solvents in Active Substances
▪ 异辛烷
Isooctane
▪ 异丙醚
Isopropyl ether
▪ 甲基异丙基酮
Methylisopropyl ketone
▪ 甲基四氢呋喃
Methyltetrahydrofuran
▪ 石油醚
Petroleum ether
▪ 三氯乙酸 Trichloroacetic acid
▪ 三氟乙酸 Trifluoroacetic acid
保障临床用药安全,控制产品质量
▪ 与其他方面研究工作密不可分
进一步验证工艺控制的合理性 为确定合理可行的质量标准提供数据支持
▪ 思考的出发点
有机溶剂的毒性 产品的合成工艺
确定何种溶剂需进行研究的原则
▪ EMEA
第一类溶剂:不宜使用。一旦作为起始原料、溶 剂,或者反应副产物,或者由其他溶剂带入时, 残留量需符合ICH相关规定。
▪ 异丙基苯 Cumene 5000
ICH 杂质 残留溶剂的指导原则 Q C R

人用药品注册技术要求国际协调会ICH协调指导原则杂质:残留溶剂的指导原则Q3C(R6)现行第四阶段版本2016年10月20日本指导原则由相应的ICH专家工作组制定,并根据ICH进程已提交给管理当局征询意见。
在ICH进程的第四阶段,最后的草案被推荐给欧盟、日本、美国、加拿大和瑞士的管理机构采纳。
Q3C(R5)文件历史母指导原则:杂质:残留溶剂的指导原则对母指导原则所含THF的PDE信息的修订修订母指导原则所含NMP的PDE信息母指导原则:杂质:残留溶剂的指导原则对母指导原则所含异丙基苯的PDE信息的修订修订母指导原则所含甲基异丁基酮的PDE信息,并纳入三乙胺的PDE杂质:残留溶剂的指导原则ICH协调指导原则目录第一部分:1. 引言 (1)2. 指导原则的适用范围 (1)3. 通则 (2)3.1 基于风险评估的残留溶剂的分类 (2)3.2 建立暴露限度的方法 (2)3.3 2类溶剂限度的表示方法 (2)3.4 分析方法 (4)3.5 残留溶剂的报告水平 (4)4. 残留溶剂的限度 (4)4.1 应避免的溶剂 (4)4.2 应限制的溶剂 (5)4.3 低潜在毒性的溶剂 (6)4.4 没有足够毒理学数据的溶剂 (7)词汇表 (8)附录1:指导原则中包括的溶剂列表 (9)附录2:其他背景 (13)A2.1 环境领域对有机挥发性溶剂的监管 (13)A2.2 药物中的残留溶剂 (13)附录3:建立暴露限度的方法 (14)第二部分:四氢呋喃的PDE (17)第三部分:N-甲基吡咯烷酮(NMP)的PDE (19)第四部分:异丙基苯的PDE (21)第五部分:三乙胺的PDE和甲基异丁基酮的PDE (24)第一部分:杂质:残留溶剂的指导原则在1997年7月17日的ICH指导委员会会议上进入ICH进程第四阶段,并建议ICH的三方监管机构采纳该指导原则1. 引言本指导原则旨在建议为保证患者安全而应规定的药物中残留溶剂的可接受量。
医药中常用有机溶剂分类及残留限度

医药中常用有机溶剂分类及残留限度医药中常用有机溶剂分类及残留限度药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。
该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。
该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂.在可能的情况下,应避免使用这类溶剂.如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
中文版VICHTopicGL18-IMPURITIESRESIDUALSOLVENTS免费哦

VICH Topic GL18(VICH的议题中指导原则8)Step 7第七阶段IMPURITIES: RESIDUAL SOL VENTS 杂质:残留溶剂IMPURITIES: RESIDUAL SOLVENTS IN NEW VETERINARY MEDICINAL PRODUCTS, ACTIVESUBSTANCES AND EXCIPIENTS杂质:新兽药、活性物质以及辅料中的残留溶剂Recommended for Implementation at Step 7 of the VICH Process on 15 June 2000 by the VICHSteering CommitteeVICH指导委员会2000年6月5日由在VICH进程的第七阶段通过并推荐THIS GUIDELINE HAS BEEN DEVELOPED BY THE APPROPRIATE VICH EXPERT WORKING GROUP ON THE BASIS OF THE ICH GUIDELINES ON THE SAME SUBJECT AND WAS SUBJECT TO CONSULTATION BY THE PARTIES, INACCORDANCE WITH THE VICH PROCESS. AT STEP 7 OF THE PROCESS THE FINAL DRAFT IS RECOMMENDED FOR ADOPTION TO THE REGULA TORY BODIES OF THE EUROPEAN UNION, JAPAN AND USA.本指南已经在ICH指南相同议题的基础上被合适的VICH专家工作小组更新,参与者会在VICH进程上就不准确的地方进一步磋商。
在VICH进程的第7阶段,这一最终草案,会被欧盟、日本和美国就调整后的文件正式通过并推荐。
1. INTRODUCTION 3 简介2. SCOPE OF THE GUIDELINE3范围3. GENERAL PRINCIPLES 4 总则3.1 Classification of Residual Solvents by Risk Assessment (4)风险评估中残留溶剂的分类3.2 Methods for Establishing Exposure Limits..........................................4建立接触限度的方法3.3 Options for Describing Limits of Class 2 Solvents..............................4第二类溶剂的相关描述3.4 Analytical Procedures ........................................................................6分析方法3.5 Reporting Levels of Residual Solvents...............................................6报道的残留溶剂的水平4. LIMITS OF RESIDUAL SOLVENTS 6 残留溶剂的限度4.1 Solvents to be Avoided ......................................................................6避免使用的溶剂4.2 Solvents to be Limited........................................................................6限制使用的熔剂4.3 Solvents with Low Toxic Potential ......................................................7具有潜在低毒的溶剂4.4 Solvents For Which No Adequate Toxicological Data Was Found.....8尚无足够的毒理数据的溶剂GLOSSARY9 术语APPENDIX 1: LIST OF SOLVENTS INCLUDED IN THE GUIDELINE 10附录1: 本指导原则中的残留溶剂APPENDIX 2: ADDITIONAL BACKGROUND 15附录2: 其他背景A2.1 Environmental Regulation of Organic Volatile Solvents (15)有机挥发性溶剂在环境方面的规定A2.2 Residual Solvents in Pharmaceuticals (15)药品中的残留溶剂APPENDIX 3: METHODS FOR ESTABLISHING EXPOSURE LIMITS 16建立接触限度的方法Table A.3.1: Values used in the calculations in this document 18表A.3.1 在这篇文档中用于计算的数值IMPURITIES: RESIDUAL SOLVENTS IN NEW VETERINARYMEDICINAL PRODUCTS, ACTIVE SUBSTANCES AND EXCIPIENTS杂质:新兽药、活性物质以及辅料中的残留溶剂1. INTRODUCTION 介绍The objective of this guideline is to recommend acceptable amounts for residual solvents in pharmaceuticals for the safety of the target animal as well as for the safety of residues in products derived from treated food producing animals. The guideline recommends use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则的目的在于推荐患病动物用含可接受的残留溶剂量的药物,保证用药动物的安全同时也适用于生产含残留物的动物食物制品的安全。
最新化学药物杂质研究的技术指导原则

化学药物杂质研究的技术指导原则化学药物杂质研究的技术指导原则一、概述任何影响药物纯度的物质统称为杂质。
杂质的研究是药品研发的一项重要内容。
它包括选择合适的分析方法,准确地分辨与测定杂质的含量并综合药学、毒理及临床研究的结果确定杂质的合理限度。
这一研究贯穿于药品研发的整个过程。
由于药品在临床使用中产生的不良反应除了与药品本身的药理活性有关外,有时与药品中存在的杂质也有很大关系。
例如,青霉素等抗生素中的多聚物等高分子杂质是引起过敏的主要原因。
所以规范地进行杂质的研究,并将其控制在一个安全、合理的限度范围之内,将直接关系到上市药品的质量及安全性。
本指导原则是在借鉴国外相关指导原则[1][2]的基础上,结合我国新药研发的实际情况制定的。
目的是为我国的药品研发提供有益的指导,从而提高药品的质量,保证人民的用药安全。
由于新药研究的探索性很强,每种药品的具体研究情况差异有可能很大,本指导原则不可能涵盖杂质研究的全部,仅提供了一个基本的研究思路和方法。
特殊情况下,研究单位可在科学、合理的基础上,对杂质进行研究,只要能用科学的数据证明药品中存在的杂质可被控制在安全、合理的范围内,就达到了杂质研究的目的。
本指导原则涵盖的范围包括新的及仿制已有国家标准的化学原料药及制剂。
发酵工艺生产的抗生素类药物一般不包括在本原则的讨论范畴,但如有可能,也建议参考本原则的有关要求。
由于我国对临床研究也实行行政审批的管理,所以,本指导原则不仅适用于上述药品的上市生产申请,也适用于临床研究的申请。
二、杂质的分类药品中的杂质按其理化性质一般分为三类:有机杂质、无机杂质及残留溶剂。
按照其来源,杂质可以分为工艺杂质(包括合成中未反应完全的反应物及试剂、中间体、副产物等)、降解产物、从反应物及试剂中混入的杂质等。
按照其毒性分类,杂质又可分为毒性杂质和普通杂质等。
杂质还可按其化学结构分类,如其它甾体、其它生物碱、几何异构体、光学异构体和聚合物等。
有机溶剂残留量的测定

Chlorobenzene 360
▪ 氯仿
Chloroform 60
▪ 环己烷 Cyclohexane 3880
▪ 1,2-二氯乙烯 1,2-Dichloroethene
1870
▪ 二氯甲烷 Dichloromethane 600
▪ 1,2-二甲氧基乙烷 1,2-Dimethoxyethane 100
第二类溶剂应该限制使用
▪ N,N-二甲氧基乙酰胺N,N-Dimethylacetamide 1090
▪ N,N-二甲氧基甲酰胺N,N-Dimethylformamide 880
▪ 1,4-二氧六环 1,4-Dioxane 380
▪ 2-乙氧基乙醇 2-Ethoxyethanol 160
▪ 乙二醇 Ethyleneglycol 62
顶空进样法的原理
▪ 顶空分析是通过样品基质上方的气体成 分来测定这些组分在原样品中的含量.这是一 种间接的分析方法,其基本理论是在一定条件 下气相和凝聚相液相或固相之间存在着分配 平衡.所以气相的组成能反映凝聚相的组成.
▪ 当样品的蒸气压相当低时,色谱峰面积Ai与挥 发性组分i的蒸气压Pi成正比 Ai=CiPi Ci与物质种类及检测器有关的特定常数.
▪
▪ 1,1-二乙氧基丙烷 1,1-Diethoxypropane
▪ 1,1-二甲氧基甲烷 1,1-Dimethoxymethane
▪ 2,2-二甲氧基丙烷 2,2-Dimethoxypropane
▪ 异辛烷
Isooctane
▪ 异丙醚
Isopropyl ether
▪ 甲基异丙基酮
Methylisopropyl ketone
天给药量计算,给出了一个比较大的安全系数. ▪ 目前情况下,采用浓度限度比较简便,而且只要
残留溶剂的指导原则

残留溶剂的指导原则残留溶剂是指在溶剂使用过程中不能完全挥发掉的溶剂残余物。
在许多行业中,残留溶剂是一种常见的环境和健康危害物质,对人体健康和环境造成潜在风险。
因此,制定和遵循残留溶剂的指导原则是非常重要的。
1.最小化使用残留溶剂的量要减少残留溶剂的问题,首先应尽量减少其使用量。
使用先进的工艺和技术,使用更低挥发性的溶剂,或者使用替代品来替代高挥发性的溶剂,能有效地降低残留溶剂的生成。
2.优先选择可再生溶剂优先选择可再生的溶剂,以最大程度减少溶剂的浪费和排放。
可再生溶剂具有更高的回收利用率和再利用价值,不仅可减少资源消耗,还能降低环境风险。
3.加强溶剂回收和再利用加强溶剂的回收和再利用,是实现降低残留溶剂的有效手段之一、通过使用适当的技术和设备,如蒸馏、吸附、膜分离等,可以将残留溶剂回收再利用,降低环境风险。
4.合理控制溶剂的使用条件在使用溶剂时,应严格遵守使用条件和操作规程,合理控制溶剂的使用量和使用方式。
遵循溶剂的最佳使用温度、浓度、搅拌速度等条件,可最大限度减少残留溶剂的生成。
5.采用防止挥发的封闭式操作在工业生产过程中,应尽可能采用封闭式设备和操作工艺,以防止溶剂的挥发和扩散。
通过密封设备、负压操作、局部排风等措施,可有效减少残留溶剂对环境和人体的危害。
6.严格遵守相关法律法规和标准制定和遵循残留溶剂管理的指导原则,既要在国家和地方层面上遵守相关的法律法规和标准,也要在企业内部建立相应的规章制度和管理体系。
通过加强监督和管理,确保残留溶剂的处理和处置符合法律法规和环保要求。
7.开展溶剂的危险性评估和监测对使用的溶剂进行危险性评估和监测,了解其挥发性、毒性、可燃性等性质和特点,及时采取相应的控制措施和防护措施。
通过定期检测和监测,及时发现和处理残留溶剂的问题,减少环境和健康风险。
总之,制定和遵循残留溶剂的指导原则是保护环境和人体健康的重要措施。
通过最小化使用残留溶剂的量、优先选择可再生溶剂、加强回收和再利用、控制使用条件、采用封闭式操作、遵守法律法规和标准、进行危险性评估和监测等措施,可以有效减少残留溶剂对环境和健康的危害。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
杂质:残留溶剂的指导原则1.介绍本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。
在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。
纯度和溶解度。
因此.有时溶剂是合成中非常关键的因素。
本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。
出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。
制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。
一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。
使用低毒溶剂(第三类,表3)较为理想。
附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。
第一、二类溶剂的建议限度或溶剂的分类会随着。
新的安全性资料的获得而调整。
含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。
2. 指导原则的范围指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。
应进行残留溶剂的检验。
也只有在上述情况下,才有必要作溶剂的检查。
虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。
如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。
果制剂生产中用到某种溶剂,也应进行测定。
本指导原则不适用于临床研究阶段的准新原料药、准赋形剂和准制剂。
也不适用于已上市的药品。
本指导原则适用于所有剂型和给药途径。
短期(如30天或更短)使用或局部使用时,允许存在的残留溶剂水平可以较高。
应根据不同的情况评判这些溶剂水平。
有关残留溶剂的背景附加说明见附录2。
3.通则3.1 根据危害程度对残留溶剂分类“可耐受的日摄人量”(TDI)是国际化学品安全纲要(IPCS)用于描述毒性化合物接触限度的术语。
“可接受的日摄人量”(ADI)是WHO及一些国家和国际卫生组织所用的术语。
新术语“允许的日接触量”(PDE)是本指导原则中用于定义药物中可接受的有机溶剂摄人量,以避免与同一物质的ADI混淆。
本原则中残留溶剂的评价以通用名和结构列于附录1,根据它们对人体可能造成的危害分为以下三类;(1)第一类溶剂:应避免的溶剂为人体致癌物、疑为人体致癌物或环境危害物。
(2)第二类溶剂。
应限制的溶剂非遗传毒性动物致癌或可能导致其他不可逆毒性测神经毒性或致畸性)的试剂。
可能具其他严重的但可逆毒性的溶剂。
(3)第三类溶剂:低毒性溶剂对人体低毒的溶剂,无须制定接触限度;第三类溶剂的PDE为每天50mg或50mg以上。
3.2 建立接触限度的方法用于建立残留溶剂的PDE方法见附录3。
用于建立限度的毒理数据的总结见Pharmeuropa,V ol . 9,No . l,Suplement,April 1997.3.3 第二类溶剂限度的选择方法制定第二类溶剂的限度时有两种选择。
方法1: 使用表2中以ppm为单位的浓度限度,假定日给药量为10g,以方程(1)计算。
方程(1) C(ppm)=PDE:mg/天剂量:g/天这些限度对所有原料药、赋形剂和制剂均适用。
因此,这一方法可用于日剂量未知或未定的情况、只要在处方中所有的赋形剂和原料药都符合方法1给定的限度,就可以以任何比例用于制剂。
只要日剂量不超过10g,就无须进一步计算。
服用剂量超过10g/天,应考虑用方法2。
方法2:制剂中的每一种成分不必符合方法1的限度。
药物中允许的残留溶剂限度水平,可根据表2中PDE mg/天及已知最大日剂量,用方程(1)来计算。
只要证明已降低至实际最低水平,便可以认为这种限度是可接受的、该限度能说明分析方法的精度、生产能力和生产工艺的合理变异,并能反映当前生产的标准水平。
应用方法2时可将药物制剂的每种成分中残留溶剂叠加起来,每天的总溶剂量应低于PDE给定的值。
下面举例说明如何用方法l和2来考虑制剂中的乙睛限度。
乙睛的允许日接触量是4.1 mg/天,因此由方法1算出限度是410PPm;如现在日最大给药量是5.0g,制剂中含两种赋形剂,制剂中的成分和计算得到的最大残留乙睛量见下表:成分处方量乙睛量日(摄人)量原料药0.3g800ppm0.24mg辅料一0.9g 400ppm 0.36mg辅料二 3.8g 800PPm 3.04mg药物制剂 5.09 728ppm 3.64mg辅料1符合方法1限度,但原料、辅料2和药物制剂不符合方法1限度,而制剂符合方法2规定的4.1mg/天,故符合本指导原则的建议值。
乙睛作为残留溶剂的另一例子,曰最大给药量5刀g,制剂中含两种赋形剂,各组分及计算得到的最大残留的乙睛最见下表:成分处方量乙睛量日(摄人)量原料药0.3g 800ppm 0.24mg辅料1 0.9g 2000ppm 1.80mg辅料 3.8g 800ppm 3.04mg药物制剂 5.0g 1016ppm 5.08mg此例制剂中乙睛限度总量既不符合方法1也不符合方法2。
生产厂可先测定制剂,以确定在处方工艺中能否降低已睛水平,如果不能将乙腈水平降至允许范围,生产厂应采取措施降低制剂中的乙腈量;若所有措施均不能降低残留溶剂的水平,厂方应提供其尝试降低残留溶剂以符合指导原则所做工作的总结报告,并以利弊分析报告证明允许该制剂存在的较高水平的残留溶剂。
3.4 分析方法残留溶剂通常用色谱技术,如用GC法测定,如可能,对药典上规定要检测的残留溶剂,应采用统一了的测定方法。
生产厂也可选用更合适的、经论证的方法来测定。
若仅存在第三类溶剂;可用非专属性的方法如干燥失重来检查。
残留溶剂的方法论证应遵循ICH指导原则:“分析方法论证:定义和术语”及“分析方法论证:方法学”。
3.5 残留溶剂的报告水平制剂生产商需要了解有关赋形剂或原料药中残留溶剂量的信息,以符合本指导原则的标准。
以下阐述了赋形剂或原料药供应商应提供给制剂牛产商的信息的~些例子。
供应商应选择以下一项:·仅可能存在第三类溶剂,干燥失重小于0.5%。
·仅可能存在第M类溶剂,X、Y……全部应低于方法1的限度。
(这里供应商应将第二类溶剂用X、Y……来表示)·仅可能存在第二类溶剂X、Y……和第三类溶剂,残留的第三类溶剂低于方法1的限度,残留的第三类溶剂低于0.5%。
如果可能存在第一类溶剂,应进行鉴定并定量。
“可能存在”系指用于工艺最后一步的溶剂和用于较前几步工艺的溶剂经论证不能全部除尽。
如果第二类溶剂高于方法1的限度或第三类溶剂高于0.5%,应鉴定并定量。
4. 残留溶剂的限度4.1应避免的溶剂因其具有不可接受的毒性或对环境造成公害,第一类溶剂在原料药、赋形剂及制剂生产中不应该使用。
但是,为了生产一种有特殊疗效的药品而不得不使用时,除非经过其他论证,否则应按表1控制,1,1,1-三氯乙烷因会造成环境公害列人表1,其限度1500ppm是基于安全性数据而定的。
表1 药物制剂中含第一类溶剂的限度(应避免使用)溶剂浓度限度(ppm)备注苯 2 致癌物四氯化碳 4 毒性及环境公害1,2-二氯乙烷5毒性1,1-二氯乙烷8毒性1,1,1-三氯乙烷1500环境公害4.2 应限制的溶剂列于表2的溶剂,由于其具毒性,在制剂中应予限制,规定PDE约0.1mg/天,浓度约10ppm。
所列值不能反映测定所必需的分析精度,精度应为方法论证的一部分。
表2 药品中第二类溶剂溶剂PDE(mg/天)浓度限度(ppm)乙腈 4.1 410氯苯 3.6 360氯仿0.6 60环氧乙烷38.8 38801,2-二氯乙烯18.7 1870二氯甲烷 6.0 6001,2-二甲亚砜 1.0 100N,N-二甲乙酰胺10.9 1090N,N-二甲基甲酰胺8.8 8801,4-二恶烷 3.8 3802-乙氧基乙醇 1.6 160乙二醇 6.2 620甲酰胺 2.2 220正己烷 2.9 290甲醇30.0 30002-甲氧基乙醇0.5 50甲基丁酮0.5 50甲基环己烷11.8 1180N-甲基吡咯烷酮48.4 4840硝基甲烷0.5 50吡啶 2.0 200二氧噻吩烷 1.6 160四氢萘 1.0 100甲苯8.9 8901,1,2-三氯乙烯0.8 80二甲苯* 21.7 2170 *通常为60% m-二甲苯,14% p-二甲苯,9% o-二甲苯和17%乙基苯。
4.3低毒溶剂第三类溶剂(见表3)可能低毒,对人体危害很小。
第三类溶剂包括人们认为在药物中以一般量存在时对人体无害的溶剂,但该类溶剂中许多尚未进行长期毒性或致癌研究。
急性毒性或短期毒性试验表明这类溶剂几乎无毒、无遗传毒性。
每日50mg或更少量无须论证即可接受(用方法1计算。
即5000ppm或0.5%)。
如果能够反映生产能力和GMP的实际情况,更大的量也可接受。
表3在GMP或其他质量要求中应限制的第三类溶剂醋酸乙醇甲乙酮丙酮醋酸乙酯甲基异丁酮苯甲醚乙醚2-甲基-1-丙醇1-丁醇甲酸乙酯戊烷2-丁醇甲酸正丙醇醋酸丁酯正庚烷正戊醇叔丁基甲基醚醋酸异丙酯醋酸异丁酯醋酸甲酯2-丙醇异丙基苯3-甲基-1-丁醇醋酸丙酯二甲亚砜四氢呋喃4.4 没有足够毒性资料的溶剂以下溶剂(表4)在赋形剂、原料药和制剂生产中也许会被生产商采用,但尚无足够的毒理学数据,故无PDE值,生产厂在使用时应提供这些溶剂在制剂中残留水平的合理性论证报告。
表4无足够毒理学数据的溶剂1,1-二乙氧基丙烷甲基异丙酮1,1-二甲基甲烷甲基四氢呋喃2,2-二甲丙烷石油醚异辛烷三氯乙酸异丙醚三氟乙酸术语遗传毒性致癌指通过影响基因或染色体而致癌。
LOEL:lowest-observed effect level的缩写。
能观察到反应的最低量(lowest-obserued effect leuel)是在研究人体或动物接触某种物质时产生任何反应的频率或严重性在生物学上显著增加的最低剂量。
修正因子是由毒理学家评定的、由生物测定的结果转换成与人体安全性相关的系数。
神经毒性某种物质引起神经系统不良反应的能力。
NOEL:no-observed effect level的缩写。