欧盟医疗器械指令的基本要求及协调标准
欧盟ivdd协调标准

欧盟ivdd协调标准全文共四篇示例,供读者参考第一篇示例:欧盟IVDD协调标准,即欧盟体外诊断医疗器械指令协调标准,是欧洲联盟为了保障体外诊断医疗器械产品的质量和安全性而制定的一系列标准。
随着医疗技术的不断进步和诊断需求的增加,体外诊断医疗器械在医疗领域中扮演着越来越重要的角色。
为了确保这些产品的质量和安全性,欧盟采取了一系列措施,其中包括制定IVDD协调标准。
IVDD协调标准主要包括产品设计、生产和销售等各个环节的要求和规范。
产品设计方面,IVDD协调标准要求产品必须符合相关的技术规范和性能要求,且必须具有明确的诊断功能。
生产方面,IVDD协调标准要求生产商必须符合ISO9001质量管理体系的要求,并进行相关的风险评估和控制。
销售方面,IVDD协调标准要求销售商必须向用户提供相关的产品信息和使用说明,且必须确保产品的追溯性和可溯源性。
IVDD协调标准的制定和执行对保障体外诊断医疗器械产品的质量和安全性具有重要意义。
IVDD协调标准可以统一欧盟各成员国对体外诊断医疗器械产品的要求和标准,减少市场准入的障碍,促进产品的流通和交易。
IVDD协调标准可以加强对生产商和销售商的监管,确保他们遵守相关的法规和规范,提高产品的质量和安全性。
IVDD协调标准可以增加消费者和医护人员对体外诊断医疗器械产品的信任度,增强市场竞争力,促进医疗器械行业的发展。
在执行IVDD协调标准的过程中,欧盟通过设立专门的机构和部门来负责协调和监督各个环节的工作。
欧盟委员会负责制定IVDD协调标准的具体内容和要求,并不断更新和调整标准以适应市场和技术的变化。
欧盟成员国的相关部门和机构则负责监督和检查生产商和销售商的生产和销售行为,对违法违规行为进行处罚和惩罚。
在未来,随着医疗技术的不断发展和体外诊断医疗器械产品的需求不断增加,IVDD协调标准将会继续发挥重要作用。
欧盟将继续加强对体外诊断医疗器械产品的监管和管理,完善IVDD协调标准,使其更加符合市场的需求和产品的特点,促进医疗器械行业的健康发展。
欧洲医疗器械领域标准

欧洲医疗器械领域标准一、医疗器械通用要求医疗器械通用要求是欧洲医疗器械法规中的基础性要求,主要包括以下几个方面:1. 医疗器械的设计、生产、检验、使用和销毁应符合安全和性能要求。
2. 医疗器械应配备有使用说明、维护和保养指南以及合格证明文件。
3. 医疗器械应当能够满足预期用途,并能够根据医疗需求进行调整和修改。
二、医疗器械标签和说明书医疗器械标签和说明书是向医护人员和患者提供有关医疗器械的重要信息。
它们应清晰、易于理解,并包含以下内容:1. 医疗器械的名称、型号和序列号。
2. 生产商名称、地址和联系方式。
3. 使用说明、操作方法和注意事项。
4. 医疗器械的性能参数和有效性评估结果。
5. 医疗器械的安全性数据和使用风险。
6. 医疗器械的保质期和使用寿命。
7. 医疗器械的包装和标识要求。
三、医疗器械安全性测试医疗器械安全性测试是确保医疗器械在使用过程中安全可靠的重要手段。
测试应包括以下方面:1. 物理性能测试,如尺寸、重量、形状等。
2. 化学性能测试,如成分分析、稳定性等。
3. 生物学性能测试,如细胞毒性、致敏性等。
4. 电磁兼容性测试,以确保医疗器械在使用过程中不会对其他设备产生干扰。
5. 环境适应性测试,以评估医疗器械在不同环境条件下的性能表现。
6. 灭菌过程验证,以确保医疗器械经过正确的灭菌程序后能够达到预期的消毒效果。
7. 安全性评估报告,详细记录测试结果和分析结论,为医疗器械的安全性提供有力支持。
8. 安全性监测,对已上市的医疗器械进行跟踪监测,及时发现和处理潜在的安全风险。
9. 不良事件报告制度,要求生产商、销售商和医疗机构及时报告医疗器械的不良事件,为改进产品质量和提高安全性提供依据。
中文版欧盟mdr法规

中文版欧盟mdr法规欧盟医疗器械法规(Medical Device Regulation, MDR)是欧洲联盟针对医疗器械领域制定的一份重要法规。
它于2017年5月发布,并于2020年5月正式生效。
本文将根据各项规定,为您详细介绍中文版欧盟MDR法规的内容。
第一部分:导言这一部分主要介绍了欧盟MDR法规的背景和目的,以及适用范围和基本定义。
MDR法规的目标是确保欧盟市场上的医疗器械的安全性、有效性和可靠性,以保障公众的健康和安全。
第二部分:一般要求1. 医疗器械分类和命名MDR法规对医疗器械进行了新的分类,以便更好地管理和监督不同风险等级的产品。
同时,对医疗器械的命名也有明确规定,以避免混淆和误导。
2. 表演的规定MDR法规要求制造商对其产品进行临床评价,并根据风险等级制定相应的技术文件和技术规格。
此外,MDR还提出了对制造商质量管理体系的要求,以确保其产品符合法规的要求。
3. CE标志的规定MDR法规明确了CE标志的使用规定,要求制造商在产品上正确使用并保持其可见性。
这是制造商将其产品引入欧盟市场的必要步骤。
第三部分:具体要求1. 临床评价和临床试验MDR法规对医疗器械的临床评价和临床试验进行了详细规定。
制造商需要对产品进行充分的临床评估,以确保其安全性和有效性符合要求。
2. 医疗器械监督和报告MDR法规明确了各方在市场监督和产品报告方面的责任。
制造商需要建立有效的市场监督和报告机制,并及时识别、评估和报告任何可能危害人类健康的事件。
3. 医疗器械技术文献和标签MDR法规要求制造商提供充分的技术文献和产品标签,以便医疗专业人员和最终用户正确使用和了解产品。
第四部分:认证和授权1. 医疗器械的市场准入MDR法规规定了医疗器械的市场准入程序,制造商需要满足一系列要求,包括提交技术文件、进行质量控制审核等。
只有通过认证的产品才能进入欧盟市场。
2. 第三方评估机构MDR法规对第三方评估机构的审核和授权进行了规定,确保其独立性和专业性。
医疗器械定义在欧盟市场中的法规要求及其市场准入流程

THANKS感谢观看05欧盟市场准入挑战与对策
BIG DATA EMPOWERS TO CREATE A NEW
ERA
技术壁垒与标准差异
技术标准不统一
01
欧盟各成员国之间存在技术标准差异,对医疗器械的性能、安
全性和有效性要求各不相同。
认证程序繁琐
02
医疗器械需要在欧盟各成员国分别进行认证,程序繁琐且耗时
较长。
语言和文化差异
全过程符合相关法规和标准。
02
建立完善的质量管理体系
企业应建立完善的质量管理体系,确保产品质量的一致性和可靠性,提
高产品的市场竞争力。
03
积极寻求专业支持
企业可以积极寻求专业的法规咨询、技术支持和市场推广服务,以更好
地应对市场挑战和抓住发展机遇。
未来发展趋势及影响
法规要求的不断更新和完善
随着医疗技术的不断进步和市场需求的变化,欧盟医疗器 械法规要求将不断更新和完善,对企业提出更高的要求。
建立合作伙伴关系
与供应商、销售商等合作伙伴建立良好的合作关系,共同应对法 规挑战,提高市场竞争力。
07
总结与展望
BIG DATA EMPOWERS TO CREATE A NEW
ERA
欧盟医疗器械法规要求的重要性
保障公共安全
欧盟医疗器械法规要求确保医疗器械的安全性和有效性,保护患者 和公众免受潜在风险,维护公共健康和安全。
定期组织医疗器械法规培训
确保企业内部相关人员对欧盟医疗器械法规有全面、深入的了解 。
提高法规意识
强化员工对法规的遵守和执行意识,确保企业日常运营符合法规要 求。
关注法规动态
及时关注欧盟医疗器械法规的更新和变化,以便调整企业策略和措 施。
欧盟,医疗器械标准

欧洲共同体理事会关于医疗器械的93/42/EEC指令1993年6月14日欧洲共同体理事会考虑到建立欧洲经济共同体的条约,特别是其第100a条;考虑到欧洲共同体委员会提交的议案;考虑到与欧洲议会合作;考虑到经济与社会委员会的意见;鉴于应在欧洲共同体内部市场范围内正式通过必要的措施;鉴于欧洲共同体内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于各成员国有关医疗器械的安全、健康保护和工作特性的法律、法规和行政条款的内容和范围是不同的;鉴于各成员国之间对这类器械的认证和检验程序存在差异;鉴于这种差异在欧洲共同体内形成贸易壁垒;鉴于各国针对医疗器械的使用所制定的有关患者、使用者及其他人员的安全和健康保护的条款应予以协调,以保证此类器械在欧洲共同体内部市场自由流通;鉴于协调条款必须与成员国为管理直接或间接与这类器械有关的公共健康和医疗保险计划的资金筹措而采取的措施相区别;鉴于因而只要遵守欧洲共同体法律,这些条款并不影响各成员国实施上述措施的能力;鉴于医疗器械应向患者、使用者及第三方提供高水平的保护并达到制造商赋予其的性能水平;鉴于因此保持和提高各成员国已达到的保护水平是本指令的基本目的之一;鉴于在1965年1月26日欧洲共同体理事会关于使有关特许专卖药品的法律、法规或行政措施趋于一致的65/65/EEC指令中,某些医疗器械是用于施药的;鉴于在这种情况下,医疗器械投放市场通常由本指令管理,而药品投放市场由65/65/EEC指令管理;鉴于如果这种器械投放市场的方式使器械与药品构成一种规定只供组合使用且不能再次使用的整体,则这个单一整体产品应由65/65/EEC指令管理;鉴于必须将上述器械与包含某种物质,特别是当其单独使用时,按65/65/EEC指令可视为药物的医疗器械相区别;鉴于在这种情况下,若这种物质能配合医疗器械对人体产生辅助作用,则这类器械投放市场由本指令管理;鉴于这类物质的安全性、质量和有效性必须比照欧洲共同体理事会1975年5月20日关于使成员国有关分析标准、药物毒理学标准和临床标准及特许专卖药品试验协议的法律趋于一致的75/318/EEC指令规定的适当方法加以验证;鉴于本指令附录中的基本要求和其他要求,包括对“减小”或“降低”危险的任何引用,在解释和实施时必须考虑设计时的技术现状和实际做法及高水平的健康和安全相适应的技术与经济条件;鉴于按照1985年5月7日欧洲共同体理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和生产的规定必须限于满足基本要求所必须的条款;鉴于因为这些要求是基本的,因此它们应取代各国相应的条款;鉴于实施基本要求应审慎考虑设计时的技术水平和与高水平的健康及安全保护相适应的技术、经济条件;鉴于1990年6月20日欧洲共同体理事会关于使成员国有关有源植入式医疗器械的法律趋于一致的90/385/EEC指令是新方法指令在医疗器械领域中的首次应用案例;鉴于为了使欧洲共同体的统一规定适用于所有医疗器械,本指令基本上是以90/385/EEC指令的条款为依据的;鉴于为此必须修订90/385/EEC指令,以便放入本指令规定的一般性条款;鉴于电磁兼容性问题是医疗器械安全的一个组成部分;鉴于就1989年5月3日欧洲共同体理事会关于使成员国有关电磁兼容的法律趋于一致的89/336/EEC指令而言,应包含这方面的专门规定;鉴于本指令应包括有关发射电离辐射的器械在设计和制造方面的要求;鉴于本指令既不影响1980年7月15日欧洲共同体理事会80/836/Euratom指令对有关保护公众和工人免受电离辐射危险的基本安全标准的指令进行修订所要求的授权,也不影响欧洲共同体理事会1984年9月3日对接受医疗检查和治疗的人员的辐射防护规定了基本措施的84/466/Euratom指令的实施;鉴于欧洲共同体理事会1989年6月12日关于采取措施鼓励改善工人工作中的安全和健康的89/391/EEC指令以及有关这一问题的专门指令应继续予以实施;鉴于为了证实符合基本要求并使这种符合得到验证,需要制定欧洲协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方机构制定的,应保持其非强制性的地位;鉴于为此欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)按照1984年11月13日欧洲共同体委员会与这两个机构之间签署的合作总指导原则,被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲共同体委员会委托,由上述两机构之一,或两个机构共同根据欧洲共同体理事会1983年3月18日关于在技术标准和法规领域提供信息程序的83/189/EEC指令,依照上述总指导原则而批准的技术规范(欧洲标准或协调文件);鉴于对协调标准进行修订,欧洲共同体委员会应得到根据83/189/EEC指令建立的常设委员会的帮助;鉴于应采取的措施必须按欧洲共同体理事会87/373/EEC决定中规定的程序Ⅰ而规定;鉴于在特定领域,欧洲药典专著这类现有的形式应包括在本指令的范围内;鉴于因此有几部欧洲药典专著可视为等同于上述协调标准。
欧盟对医疗器械的法规要求及分类标准

D
02 欧盟医疗器械法规概述
医疗器械指令(MDD)
01
MDD是欧盟针对医疗器械的基本法规,规定了医疗器 械的定义、分类、基本要求、评估程序和市场监管等内 容。
02
MDD要求医疗器械必须符合安全性和性能的基本要求 ,并通过相应的符合性评估程序获得CE标志,才能在欧 盟市场销售和使用。
03
MDD还规定了医疗器械制造商、进口商和经销商的责 任和义务,包括建立质量管理体系、进行临床评估、报 告不良事件等。
体外诊断医疗器械法规(IVDR)
IVDR对体外诊断医疗器械的分类更加详细,要求制 造商提供更多的临床数据和性能评估报告,证明产品 的准确性和可靠性。
IVDR是欧盟针对体外诊断医疗器械的法规,于2017 年发布,2022年5月起强制执行。
IVDR还要求制造商建立严格的质量管理体系和生产 过程控制,确保产品的稳定性和一致性。同时, IVDR还规定了更加严格的市场监管措施,包括加强 不良事件报告和召回制度等。
技术文档应包括医疗器械的设计 、制造、性能、安全性、有效性 等方面的详细信息,以及相关的
试验、验证和评估结果。
技术文档应随时可供欧盟监管机 构检查,并应随着医疗器械的更
新和改进而不断更新。
上市后监管义务
制造商应对其已上市的医疗器械进行持 续的监管,以确保其在使用过程中的安
全性和有效性。
制造商应建立上市后监管计划,包括定 期收集和分析医疗器械的使用数据、不 良事件报告等信息,以及采取必要的纠
临床试验要求
对于高风险医疗器械,欧盟要求 进行临床试验以验证其安全性和
有效性。
临床试验必须在欧盟境内进行, 并符合欧盟相关法规和标准的要
求。
申请临床试验需要提交试验方案 、研究者资质、伦理委员会批准 等文件,并接受监管机构的审核
欧洲共同体理事会医疗器械的EEC指令

欧洲共同体理事会医疗器械的EEC指令欧洲共同体理事会医疗器械的EEC指令,是欧洲联盟(EU)颁布的医疗器械指令,旨在保证医疗器械具有高度的质量和安全性能。
该指令的意义在于,促进欧洲范围内医疗器械的自由流通,并提高市场竞争和医疗保健的效率。
欧洲共同体理事会医疗器械的EEC指令自1985年起开始实施,在近40年的时间里,其规定逐步演化和完善,内部体系逐渐健全。
目前,该指令已被重新修订,于2017年逐步推行至2020年。
医疗器械指令是欧盟的一项重要法规,在欧洲范围内对所有医疗器械的设计、制造、销售等进行严格监管。
医疗器械是指所有用户带入体内的物品,如手术工具、假肢、血压计等等。
这些设备和工具如不合适制造,或使用不当,很可能对患者造成不可恢复的损伤和伤害。
因此,自1985年起,欧洲共同体理事会医疗器械的EEC指令就要求所有生产商和销售商遵守该指令,以确保所有投入市场的医疗器械都满足高质量和安全性的标准。
该指令的要求主要包括以下四个方面:1、医疗器械的安全性:该指令规定,所有医疗器械必须满足高度的安全性标准。
生产商需要证明其产品对患者的安全性,避免不必要的风险,如感染、毒素、过敏反应等。
2、设备的医疗功能:该指令要求所有医疗器械必须标明其设备的医疗功能,以便医生和患者更好地了解其用途和功能。
3、采用质量管理体系:该指令强调生产商必须采用质量管理体系,以确保其设备符合规定的标准。
体系内包括内部的质量管理制度、产品质量检验、反馈机制等。
4、审核机构的认证:该指令要求医疗器械生产商必须得到批准,并通过审核机构的考察认证。
审核机构负责审核生产商的质量管理体系、设备制造过程、产品检验等整个生产和销售过程。
值得一提的是,该指令还划分了三类设备,分别是I类、II类、III类。
不同的设备类别对应的监管要求也不同,按照严格程度依次递增。
根据类别划分,市场监管部门能够更好地识别,对违规设备实施更有针对性的惩罚措施。
欧洲共同体理事会医疗器械的EEC指令的重要性在于,其为医疗器械及相关行业制定了清晰的标准,建立了开放的市场竞争,以改善医疗保健质量和效率。
医疗器械欧盟协调标准2012-8-30
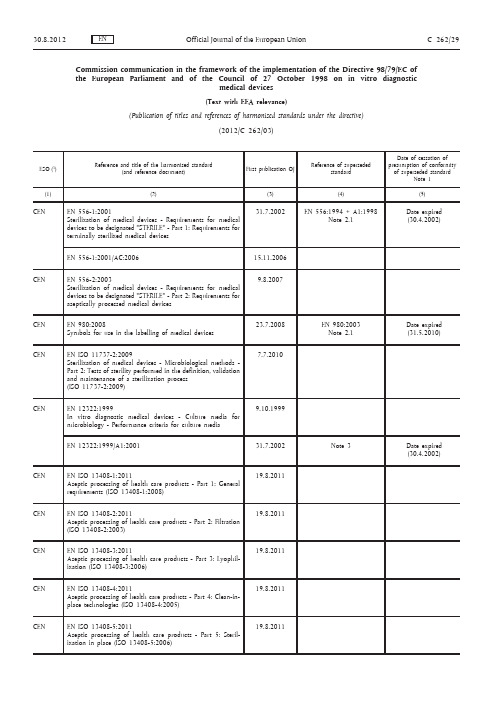
Commission communication in the framework of the implementation of the Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnosticmedical devices(Text with EEA relevance)(Publication of titles and references of harmonised standards under the directive)(2012/C 262/03)(1) ESO: European Standards Organisation:— CEN: Avenue Marnix 17, 1000 Bruxelles/Brussel, BELGIQUE/BELGIË, Tel. +32 25500811; fax +32 25500819 (http://www.cen.eu)— Cenelec: Avenue Marnix 17, 1000 Bruxelles/Brussel, BELGIQUE/BELGIË, Tel. +32 25196871; fax +32 25196919 (http://www.cenelec.eu) — ETSI: 650 route des Lucioles, 06921 Sophia Antipolis, FRANCE, Tel. +33 492944200; fax +33 493654716, (http://www.etsi.eu)Note 1: Generally the date of cessation of presumption of conformity will be the date of withdrawal (“dow”), set by the European Standardisation Organisation, but attention of users of thesestandards is drawn to the fact that in certain exceptional cases this can be otherwise.Note 2.1: The new (or amended) standard has the same scope as the superseded standard. On the date stated, the superseded standard ceases to give presumption of conformity with the essentialrequirements of the directive.Note 2.2: The new standard has a broader scope than the superseded standard. On the date stated the superseded standard ceases to give presumption of conformity with the essential requirements ofthe directive.Note 2.3: The new standard has a narrower scope than the superseded standard. On the date stated the (partially) superseded standard ceases to give presumption of conformity with the essentialrequirements of the directive for those products that fall within the scope of the newstandard. Presumption of conformity with the essential requirements of the directive forproducts that still fall within the scope of the (partially) superseded standard, but that do notfall within the scope of the new standard, is unaffected.Note 3: In case of amendments, the referenced standard is EN CCCCC:YYYY, its previous amendments, if any, and the new, quoted amendment. The superseded standard therefore consists of ENCCCCC:YYYY and its previous amendments, if any, but without the new quoted amendment.On the date stated, the superseded standard ceases to give presumption of conformity with theessential requirements of the directive.NOTE:— Any information concerning the availability of the standards can be obtained either from the European Standardisation Organisations or from the national standardisation bodies of which the list is annexed tothe Directive 98/34/EC of the European Parliament and Council (1) amended by the Directive98/48/EC (2).— Harmonised standards are adopted by the European Standardisation Organisations in English (CEN and Cenelec also publish in French and German). Subsequently, the titles of the harmonised standards aretranslated into all other required official languages of the European Union by the National StandardsBodies. The European Commission is not responsible for the correctness of the titles which have beenpresented for publication in the Official Journal.(1) OJ L 204, 21.7.1998, p. 37.(2) OJ L 217, 5.8.1998, p. 18.— Publication of the references in the Official Journal of the European Union does not imply that the standards are available in all the Community languages.— This list replaces all the previous lists published in the Official Journal of the European Union. The Commission ensures the updating of this list.— More information about harmonised standards on the Internet athttp://ec.europa.eu/enterprise/policies/european-standards/harmonised-standards/i n dex_en.htm。
医疗器械进入欧盟医疗器械场要求

WOR/式可以任意编辑入欧盟的相关知识一、欧盟国家(共31家)1、2022年5月1日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典;2、2022年5月1日参加:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他;3、欧洲自由贸易协会EFTA挪威、列支敦士登、冰岛、瑞士;4、新入欧盟:罗马利亚、保加利亚;5、瑞士不要求产品携带CE标志.二、医疗器械CE认证的通用要求1、根本要求(总要求)(1)平安性(任何风险与器械提供的益处相比拟,必须在可以接受的范围内,故亦称风险分析);(2)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);(3)性能符合性(产品的根本要求);(4)器械性能和平安的效期(器械的平安和性能必须在器械的使用寿命内得到保证.);(5)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响).2、根本要求的具体包括如下14条:(1)器械设计和生产必须保证:根据其预定和条件使用,器械不会损害医疗环境、患者平安、操作者或其他人员的平安和健康;使用时的潜在危险与患者受益相比拟可以为人们所接受,但应具有高水平的防护方法.(2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守平安准那么、生产者应:首先应尽可能降低甚至预防危险.其次,对无法预防的危险采取适当的防护举措,包括安装报警装置;最后,告知用户所提供防护举措的弱点及其可能带来的危险.(3)器械必须取得生产者期望获得的功能.器械设计制造和包装应有利于第一条(2)(A) D多规定的各项功能的发挥.(4)在生产线者确定的器械使用寿命期内,在正常使用可能由现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康.(5)器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变.(6)副作用的大小同器械的使用性能相比拟可以为人们所接受.(7)化学、物理和生物性能(8)感染和微生物污染.(9)组装和环境因素(10)检测器械(11)辐射防护(12)带有能源或与其他能源相连接的器械(13)生产者提供的操作信息(14)如果需要根据医疗数据确定器械是否满足根本要求,如第六款的情形,有关数据必须根据附录X专业资料整理分享的规定取得三、申请CE 认证(一)一般流程(所有的产品)1、确认由口国家.2、确认产品类别及欧盟相关产品指令.(1)假设产品属于这里所列22类中的任何一个,一般地讲,那么需要进行CE 认证.假设一个产品同时属于一个以上的类别,那么必须满足所有类别相对应的产品指令中所列由的要求.(2)某些产品指令中有时会列由一些排除在指令外的产品.3、指定“欧盟授权代理〞(AuthorizedRepresentati .ve)(1)为了能保证前CE 标志(CEMarking)认证实施过程中4项要求得以满足,欧盟法律要求位于述的28个EEA 盟国境外的制造商必须在欧盟境内指定一家欧盟授权代(AuthorizedRepresentativ .e)4项要求:保证产品投放到欧洲市场后,在流通过程及使用期间产品“平安〞的一贯性;技术文件File 必须存放于欧盟境内供监督机构随时检查;对被市场监督机构发现的不CE 要求的产品、 s) 合或者使用过程中由现事故但是已加贴CE 标签的产品,必须采取补救举措.(比方从货架上暂时拿掉,或从市场中永久地撤除);已加贴CE 标签之产品型号在投放到欧洲市场后,假设遇到欧盟有关的法律更改或变化,其后续生产的同型号产品也必须相应地加以更改或修正,以便符合欧盟新的法律要求.4、确认认证所需的模式(Module).5、采用“自我声明〞模式还是“必须通过第三方认证机构〞的选择.(1)允许风险水(RiskLeve 较低(MinimalRisk)欧盟的产品指令制造商选择模A: “内部生产平l)式控(自我声明)〞的方式进CE 认证.制 行(2)风险水平较高的产品必须通过第三方认证机构NB(NotifiedBody) .(3)介入对于风险水平较高的产品,具制造商必须选择模式A 以外的其它模式,或者模式A 外加其它模式来到达CE 认证.也就是说,必须通过第三方认证机构NB(Notified Body) 介入.模式 A 以外的其它模式的认证过程中,通常均需要至少一家欧盟认可的认证机构NB 参于认证过程中的一局部或全部.根据不同的模式,NB 那么可能分别以:来样检测,抽样检测,工厂审查,年检,不同的质量体系审核,等等方式介入认证过 程,并由具相应的检测报告,证书等.(4)目前,已经有1200多家认证机构获得欧盟认可,这些认证机构中的绝大多数位于欧盟盟国境内.WOR 幅式可以任意编辑(2) (Technical通常情况下,一家NB仅被欧盟授权可针对某一类或几类产品进行某一或几种模式下的认证.换言之,一家欧盟授权的认证机构并不可能针对所有的产品种类进行认证,即使对其被授权的产品种类,通常情况下也并非被授权所有的模式.对于每一个欧盟的产品指令,通常都有一个针对该产品指令的授权认证机构NB名录.6、建立技术文件(TechnicalFiles)及其维护与更新.(1)欧盟法律要求,加贴了CE标签的产品投放到欧洲市场后,其技术文件(Technical Files)必须存放于欧盟境内供监督机构随时检查.技术文件中所包涵的内容假设有变化,技术文件也应及时地更新.(2)欧盟对技术文件内容有要求.(二)CE认证的一般程序(医疗器械)1、分析器械及特点,确定它是否在欧盟的3个医疗器械指令的范围内;2、确认适用的根本要求;专业资料整理分享WOR幅式可以任意编专业资料整理分享辑指令规定,任何医疗器械必须满足相关指令中所规定的预期用途,所以对制造商来说,首先要做的而且是最重要的事情就是确认所有的适用于其产品的根本条件.WOR外式可以任意编辑3、确认任何有关的欧洲协调标准协调标准是由欧洲标淮委员会(CEN和欧洲电气技术委员会(CENELEC利定的公布在欧盟官方杂志上的标准,对于某种医疗器械来说,可能有多种协调标准适用于它,因此在确认哪些协调标准适用于某种产品对应十分仔细.4、产品分类根据指令附录IX的分类规那么,医疗器械分成4类,即I、IIa、IIb和III类,不同类型的产品,具获得CE标志的途径(符合性评价程序)不同,因此对制造商来说,如何准确地确定其产品的类型,是十分关键的.5、保证产品满足根本要求或协调标准的要求并且使证据文件化(技术文档的整理)制造商应能提由充分的证据(如,由认证机构或其他检测机构依据协调标准进行的检测等)来证实产品符合根本要求.6、确定相应的符合性评价程序如附图I所示,对于IIa、IIb和III类医疗器械的制造商来说,存在着如何选择符合性评价程序途径的问题.主要的区别是选择型式试验的方式,还是选择质量体系的方式,这两种途径各有其特点,制造商应根据自己的实际情况选择最为适合的途径.7、选择认证机构对于IIa、IIb和III类医疗器械,以及无菌的或具有测量功能的I类医疗器械,应选择一个认证机构并进行符合性评价程序.在欧盟官方杂志上公布的认证机构名单上,对每个认证机构可以从事的医疗器械认证范围以及可进行的符合性评价程序途径都有严格的规定,制造商在选择认证机构时,必须非常谨慎,预防造成不必要的损失.8、起草符合性声明并加贴"CE’证标志认可以说符合性声明是最重要的文件.每一种器械必须包括医疗器械指令的附录中所描述的符合性声明.(三)咨询公司一流程1、咨询(1)可以以、、电子邮件等任一方式,提由初步的申请意向(如:哪几类产品、申请何种认证、具体的型号及规格、产品样本及描述等).(2)根据您所提供的大致情况,建议最正确的认证方案,提由测试前的文件及图纸等资料要求,并初步估算相关的费用.同时,我们会向您提供?申请书?、?代理授权通知书?等文件以备填写.2、申请(1)请您提交申请资料按要求准备相关的资料;(2)在收到上述的文件及资料后,正式受理申请并确立工程号,同时拟定?认证代理委托协议?(双方缔结的认证业务条约,以明确相互的责任及义务,一式两份).3、签约(1)请您将签字盖章的上述?协议?返回我司 ,并按?协议?条款支付相关费用;(2)在收到签字盖章的?协议?和有关付款凭证后,指定工程工程师负责此工程,并选择与适宜的实验室和工程师联系.4、技术支持(可选择)(1)应您的要求,我们的工程工程师会向您讲解与您的产品相关的标准及平安要求;安排摸底测试和结构预检;将中文资料译成英文等.(2)上述技术支持的费用水平将根据具体的工作量来确定.专业资料整理分享WOR/式可以任意编辑5、送检准备(1)我们的工程工程师会及时反响同认证机构的联系的进展情况,并通知您测试或重复测试的样品要求和确切费用,以及认证机构要求签署的一系列文件(如认证机构的申请表、结构参数表、跟踪效劳协议等),(2)请您按要求准备好样品、文件资料、测试费用,并送交我们的工程工程师.6、送检(1)我们的工程工程师会将您提供的样品、资料、费用一并提交到相应的认证机构或实验室,并及时地跟踪认证工程的进展情况、反响测试信息,直至该工程结束.7、重复测试(1)如果测试由现不合格项,您可以进行样品整改,再次送样,重复测试;也可以取消工程,在成熟时再提由申请.(2)我们的工程师将给由整改意见,并协助您进行样品整改.8、首次工厂检查或发证前检验(1)工程开始后,会有认证机构或其指定的检验机构来您工厂进行首次工厂检查或发证前检验(CE认证除外),以考察工厂的测试和生产过程是否符合要求.通过该项检查检查,也是您取得授权或证书的必要条件.(2)我们的工程工程师可以协助您进行有关的准备工作.8、申请人签署CE保证自我声明,并在产品上贴附CE标示.9、采用“自我声明〞模式还是“必须通过第三方认证机构认证NB(NotifiedBody) 〞10、风险水(RiskLeve 较低(MinimalRisk)欧盟的产品指令允许某些类别中风险水(RiskLevel)平l)平较(MinimalRis的产品之制造商选择以模A: “内部生产限制(自我声明)〞的方式进CE认证.低k)式行11、风险水平较高的产品必须通过第三方认证机构介入,对于风险水平较高的产品,具制造商必须选择模式A以外的其它模式,或者模式A外加其它模式来到达CE认证.也就是说,必须通过第三方认证机构NB(NotifiedBody) 介入.模式A以外的其它模式的认证过程中,通常均需要至少一家欧盟认可的认证机构NB参于认证过程中的一局部或全部.根据不同的模式,NB那么可能分别以:来样检测,抽样检测,工厂审查,年检,不同的质量体系审核等等方式介入认证过程,并由具相应的检测报告、证书等.12、后续效劳(可选择)(1)在认证结束以后,我们可以根据您的不同的要求提供一系列的年度效劳,如:购置标签及黄卡、来往函件及资料的译、代付费用、更换修改页、申请变更等.(具体可参见?年度效劳操作方法?).专业资料整理分享认证机构初审通知, 确定产品平安分类按认证机构要求编 写并提供TCF 文件发放CE 证书〔四〕APRAGA 流程1〕企业向认证机构提由认证申请,并填写认证询价单交认证机构;2〕认证机构向申请认证企业提由报价单,企业签字确认即完成合约;3〕企业向认证机构提交ISO9000+ISO13485质量体系文件即质量手册和程序文件,供认证机构进行体系文件审核;质量体系审核前,企业应有至少三个月的质量体系运行记录,并完成 4〕认证机构发生认证产品测试通知单给认证机构认可的试验室,〔LVD 〕测试和电磁兼容性〔EMC 测试.测试中假设由现不合格,由企业改下后重新测试,直到测试合格为 止.测试结束,试验室由具试验报告. WOR /式可以任意编辑13、流程填写CE 认证申请书A 签订咨询委托协议书产品通过CB LVD/生物相容性 /测量性能等检测 通过ISO13485体系认证 审评 整改/补充根据欧盟对产品的分类伽玛刀属于第nb类,其CE 认证程序和内容如下:1-2次内部质量体系审核.试验室将对申请认证的产品进行低电压 申请CE认证机构受理缴费通知5〕企业编写申请认证产品的技术文件档案〔简称TCF文件〕.上述试验报告也作为TCF文件内容之一.TCF文件是申请CE认证的制造商向CE认证机构提交的一份重要文件,它是认证机构审核发证的重要依据.编制TCF文件必须全部使用英文.TCF文件包括七个方面的内容:①简介;②产品的规格表达;③设计之主要档案内容;④风险分析及评估;⑤测试报告及临床诊断资料;⑥文件设计的管制;⑦产品申请的声明宣言c 6〕认证机构对企业的ISO9000+ISO13485质量体系和TCF文件进行初审.初审后认证机构将指由质量体系和TCF文件中存在的问题,企业应据此完善质量体系和TCF文件.7〕认证机构对企业的ISO9000+ISO13485质量体系和TCF文件进行正式审核.8〕正式审核通过后,认证机构将与企业签订框架协议,明确取得CE证书后各方应遵循原那么和产品使用专业资料整理分享WOR/式可以任意编辑CE标志的范围,以及用投诉的处理方法.然后颁发ISO9000+ISO13485质量体系认证证书和CE标志证书.一般来说,从企业申请认证到认证机构颁发证书大约需要半年到一年的时间.〔五〕自行申请流程1、分析器械及特点,确定它是否在欧盟的3个医疗器械指令的范围内;2、确认适用的根本要求;指令规定,任何医疗器械必须满足相关指令中所规定的预期用途,所以对制造商来说,首先要做的而且是最重要的事情就是确认所有的适用于其产品的根本条件.3、确认任何有关的欧洲协调标准;协调标准是由欧洲标淮委员会〔CEN和欧洲电气技术委员会〔CENELEC利定的公布在欧盟官方杂志上的标准,对于某种医疗器械来说,可能有多种协调标准适用于它,因此在确认哪些协调标准适用于某种产品对应十分仔细.4、产品分类;根据指令附录IX的分类规那么,医疗器械分成4类,即I、IIa、IIb和III类,不同类型的产品,具获得CE标志的途径〔符合性评价程序〕不同,因此对制造商来说,如何准确地确定其产品的类型,是十分关键的.5、保证产品满足根本要求或协调标准的要求并且使证据文件化〔技术文档的整理〕;制造商应能提由充分的证据〔如,由认证机构或其他检测机构依据协调标准进行的检测等〕来证实产品符合根本要求.6、确定相应的符合性评价程序;如附图I所示,对于IIa、IIb和III类医疗器械的制造商来说,存在着如何选择符合性评价程序途径的问题.主要的区别是选择型式试验的方式,还是选择质量体系的方式,这两种途径各有其特点,制造商应根据自己的实际情况选择最为适合的途径.7、选择认证机构;对于IIa、IIb和III类医疗器械,以及无菌的或具有测量功能的I类医疗器械,应选择一个认证机构并进行符合性评价程序.在欧盟官方杂志上公布的认证机构名单上,对每个认证机构可以从事的医疗器械认证范围以及可进行的符合性评价程序途径都有严格的规定,制造商在选择认证机构时,必须非常谨慎,预防造成不必要的损失.8、起草符合性声明并加贴“ CE〞认证标志;可以说符合性声明是最重要的文件.每一种器械必须包括医疗器械指令的附录中所描述的符合性声明.〔六〕怎样选择认证机构1、认证执构是一个由欧盟认可的审核和认证机构,它可从事医疗器械指令的附录中所描述的一种或多种符合性评价程序.认证机构必须位于欧盟的某个成员国内.2、选择认证机构是制造商面临的极其关键的问题之一.为了能够有效地工作应和认证机构建立长期和密切的联系.应对非常慎重地选择“伙伴〞所花费的时间和财力应被认为是一项公司未来的投资.3、一般未说,制造商在选择认证机构的时候,应考虑以下因素:⑴医疗器械认证方面的经验;⑵所熟悉的医疗器械的范围;⑶拥有的专业特长,如电磁兼容、软件等;⑷与一些委托方的关系及委托方的资格;⑸被授权的医疗器械认证范围;专业资料整理分享WOR/式可以任意编辑⑹被授权的可进行的符合性评价程序;⑺对已有证书的态度;⑻费用;⑼地点和工作语言;4、对于国内的医疗器械制造商来说,选择认证机构时应考虑那些在国际上有相当的知名度、并在同行业中为许多已认证的厂家所迭择的认证机构,如“CE0483'公告公司等,这样可以少走弯路.5、检测单位应为认证机构成认的单位,根据各认证机构不同而不同,有些单位成认17025体系认证在中国注册,有些不成认.6、例子(1)TUV做CE在行业内比拟有名气,但收费超级高,一般预付70%.(2)ITC也在做CE,预付30%成认检测机构的范围很广,收费相对低.四、CE标志的使用1、力口贴"CE"标志的相关要求(1) “CE'标志无处不在,由新方法指令所涉及的所有医疗器械产品在投放市场前都必须加贴“CE'标志.所有在欧洲市场销售的产品上市前都要求加贴“CE'标志;" CE'标志必须加贴在显要位置上;CE合格标记由词首大写字母"CE'构成,组成CE标记的两个字符应根本等高,且字符的纵向尺寸最小不得小于5mm“CE’标志最低高度不得少于5mm如有必要,其缩小或扩大应按比例进行.它说明产品符合欧洲医疗器械指令的唯一标志.(2)厂商根据指令关于使用CE标志应通过何种合格评定模式的要求、合格评定的原那么和93/465/EEC号理事会指令,在八种认证模式中选取适宜的模式.(3)根据指令要求采取自我评定或申请第三方评定或强制申请欧共体通知程序认可认证机构评定后,编制制造商自我评定的一致性声明和(或)认可认证机构的CE证书,作为可以或准许使用CE标志的前提条件.(4)由制造商按有关指令规定在通过规定模式的合格评定后,自行制作或加附CE标志及有关指令规定的附加信息.(5)有关指令规定应在CE标志部位,接着加附认可认证机构的识别编号时,应由执行合格评定的认可认证机构自行加附,或授权制造商或其在欧共体的代理商负责.对特别危险的产品,指令中规定由强制性认可认证机构进行产品样品试验和(或)质量体系认可的,均应先取得评定认可,才能获准使用CE标志.2、医疗器械产品的"CE"标志的获取(1)任何医疗器械生产企业,假设其想获得其产品的欧洲上市,只要其根据其生产的产品对应的医疗器械CE指令进行认证准备,获CE认证证书时就是其CE'标志的获取和正式使用之时.实际上也就是通过合得“格评定形式证实产品符合指令的根本要求,但必须由第三方进行测试或认证方可.(2)合格评定可8种根本方法相应组合进行,即生产内部限制、EEM式检验、符g型式要求、生由产质量保证、产品质量保证、产品验证、单件验证及正式质量保证.每个新方法指令中都规定了适用的合格评定程序的范围和内容.通常情况下,合格评定程序在设计和生产阶段发挥作用,有的模式只涉及生产阶段, 有的模式涉及到设计和生产两个阶段,其目的就是要求制造商采取一切必要的举措保证其产品合格.产品符合协调标准或经过适当的合格评定程序,即可加贴"CE"标志.(3)合格评定活动是由指令机构完成,其首要任务就是依据指令中规定的根本要求进行合格评定,以保证加贴"CE〞标志的产品符合相关指令中的相关程序.五、产品分类专业资料整理分享WOR/式可以任意编辑1、强制性认证产品分类,共22种;(1)燃气炉具AppliancesBurningGaseousFuels(AppliGas)(2)载人的索道装置CablewayInstallationstoCarryPersons(3)低电压电气设备LowVoltageElectricalEquipment(4)建筑产品ConstructionProducts(5)使用于具有爆炸性环境中的设备和防护系统(6)民用爆破器材ExplosivesforCivilUses(8)家用电冰箱或电冷柜HouseholdRefrigerators&Freezers(9)升降机Lift(10)机械Machinery(11)航海设备MarineEquipment(12)(普通)医疗器械MedicalDevices(13)主动可植入医疗器械ActiveImplantableMedicalDevices(14)体外诊断医疗器械InVitroDiagnosticMedicalDevices(15)非自动称量仪器Non-automaticWeighingInstruments(16)无线电及电信终端设备RadioEquipment&TelecommunicationsTerminalEquipment(R&TTE)(17)个人防护设备PersonalProtectiveEquipment(PPE)(18)简单压力容器SimplePressureVessels(19)压力设备PressureEquipment(20)休闲用船只RecreationalCraft(21)玩具Toys(22)跨欧洲高速列车系统Trans-EuropeanConventionalRailSystem、专业资料整理分享WOR/式可以任意编辑升降设备95/16/EC「1999年7月1日|家用制冷器具96/57/EC1999年9月3日承压设备97/23/EC"一2022年5月29日通信设备98/13/EEC92年11月6日/95年5月1日体外诊断医疗器械98/79/EC2022年12月7日无线电、电信终端设备99/5/EC2000年4月8日空中索道2000/9/EC2022年5月3日环境噪音设备2000/14/EC2022年1月30日荧光灯镇流器2000/55/EC2000年10月8日3、医疗器械产品分类(D医疗器械指令附录九中详定18条规那么,按医疗产品的危险程度,将产品分为I类、na类、n b类、田类.第I类产品要加贴CE标志,可采取自行宣告的方式.即厂商编制产品的技术文件档案,同时自行按有关EN标准对产品进行测试或委托有水平的试验室进行测试合格.第na类、第n b类、第in类产品要加贴CE标志,那么必须由欧盟指定的验证机构验证.欧盟还规定,这几类产品获得CE认证的先决条件是制造厂需能过ISO9000+ISO13485质量体系认证,取ISO9000+ISO13485质量体系认证证书,且证书的颁发单位应为欧盟得认可的认证机构.ISO9000+ISO13485质量体系认证和CE认证可同时进行,但CE证书必须待ISO9000+ISO13485 质量体系认证通过后,方可予以颁发4、医疗器械产品分类规那么(1)规那么应用由器械的预期使用目的决定;(2)如果器械是和其它器械配合使用,分类规那么分别适用于每种器械;(3)附件可以和其它一起使用的器械分开单独分类;(4)启动或影响某种器械的软件与器械属于同一类型.5、医疗器械的预期用途在产品分类中起着至关重要的作用,即使同一产品的分类亦会由于其预期用途的不同而有重大不同例如:(1)不灭菌纱布片:I类申报机构六、CE 的相关信息专业资料整理分享灭菌伤口清洁纱布片:I*类带X 线灭菌伤口清洁纱布片:IIa 类 灭菌烧伤用纱布片:IIb 类 灭菌心脏手术用纱布片:III 类⑵ (3) (4) (5) 级级(测量功能) 级(灭菌)a 级b 级设计阶段自我符合声明自我符合声明自我符合声明生产阶段 自我符合声明 申报机构 申报机构申报机 构 申报机申报机构。
EN标准

欧盟有三个指令和医疗设备相关,分别是MDD(医用电气设备)指令IVD(体外诊断设备)指令和AIMM(主动式植入式设备)指令根据指令的不同,产品的协调标准也是不同的.因为我接触的医疗器械没有及到植入式设备,所以主要对其它两个指令进行大致的说明.一:MDD指令,协调标准就是EN60601-1-2,目前的最新版本好象是2005而国内对应的行业标准是YY0505:2005,本来是要07年4月强制执行的,但是估计要延期;这一类产品主要是那些监护仪心电图机超声血压仪等等二:IVD指令,协调标准是EN 61326-1:2006,专标EN 61326-2-6:2006,其中EN 61326-1是实验室用设备的一个标准.目前国内相对应的标准是推荐的GB/T 18268, 当然国标对应的是欧盟的很早的版本, 所以测试项目上有一点小小的差异.这一类产品主要是血液细胞分析仪尿液分析仪生化分析仪酶标仪洗板机ISE等.因为IVD设备属于实验室用,所以它所处的电磁环境相对较好,测试要求也较MDD设备为低.00000000000000000000000000000000000000000000000000000000000000000 EN292.1-1991机械安全---基本观念,设计总原则---第一部分:基本术语学和方法论.pdfEN294-1992机器的安全性.防止上肢触及危险位置的安全距离.pdfENV12610-1997医疗信息学.医用产品标识.pdfENV12612-1997医疗信息学.健康护理管理信息交换用信息.pdfENV13607-2000医疗卫生信息.医疗处方信息交换用文电.pdfENISO11073-10101-2005健康信息学.床旁检测医疗设备通信.第10101部分_术语.pdfEN928-1995玻璃容器内诊断系统.玻璃容器内诊断用医疗产品EN29001,EN46001,EN29002以及....pdfENISO14971-2000医疗装置.医疗装置风险管理应用.pdfENISO13485-2003质量体系.医疗装置.ENISO9001应用的特殊要求.pdfENISO13488-2000质量体系.医疗装置.ENISO9002应用的特殊要求.pdfEN980-2003医疗设备标签用图形符号(包括修改件A1-1999).pdfENISO10993-1-2003医疗器械的生物评定.第1部分_评定和试验.pdfENISO10993-10-2002医疗器械的生物评定.第10部分_刺激与延迟式超灵敏性试验.pdf ENISO10993-11-1995医疗装置生物学评定.第11部分_系统毒性试验.pdfENISO10993-18-2005医疗器械的生物学评价第18部分:化学品材料的标识.pdfENISO10993-4-2002医疗器械的生物评定.第4部分_与血液相互作用的选择试验.pdf ENISO10993-8-2000医疗装置的生物评价.第8部分_生物试验用标准物质的选择和合格鉴定.pdfEN60601-1-1-2001医疗电气设备.第1部分_安全性的一般规定.1.补充标准_医疗电气系统安全规则.pdfEN60601-1-1990医疗电气设备.第1部分_安全性的一般规定.pdfEN60601-1-4-1996医疗电气设备.第1-4部分_安全性的一般要求.补充标准_程序化电气医疗系统.pdfEN60601-1-8-2004医用电气设备第1-8部分_安全通用要求并列标准_医用电气设备和医用电气系统中的报警系统的.pdfEN60601-2-25-1995医疗电气设备.第2-25部分_心电图安全的特殊要求.pdfEN60601-2-34-2000医疗电器设备.第2部分_直接血压监视设备安全的特殊要求..pdfEN60601-1-2-2001医用电气设备.第1-2部分_通用安全要求.补充标准_电磁兼容性.要求和试验.pdfEN60601-1-6-2004医用电气设备.第1-6部分_安全的通用要求.附属标准_使性..pdfEN60601-2-49-2001医用电气设备.第2-49部分_多功能病人监测设备安全的特殊要求.pdf EN60601-2-4-2003医用.pdfEN46001-1996质量保证体系.医疗器械.采用ENISO9001时的特殊要求.pdfEN46003-1999质量体系.医疗设备.ENISO9003实施的特殊要求.pdfEN1041-1998医疗产品生产者提供的信息.pdf 000000000000000000000000000000000000000000000000000000000000000000000000EMC&PCB Layout│PCB layout中的走線策略.pdf│开关电源抗干扰问题研究.pdf│解析几种有效开关电源电磁干扰抑制.pdf│高速PCB板的电源布线设计.pdf│├─EMC的培训资料│第一章基本概念.ppt│第三章电磁屏蔽技术.ppt│第二章地线干扰与接地技术.ppt│第五章PCB的电磁兼容设计.ppt│第四章干扰滤波技术.pptEN980-2003医疗设备标签用图形符号(包括修改件A1-1999).pdfEN60601-1-1-2001医疗电气设备.第1部分_安全性的一般规定.1.补充标准_医疗电气系统安全规则.pdfEN60601-1-1990医疗电气设备.第1部分_安全性的一般规定.pdfEN60601-1-4-1996医疗电气设备.第1-4部分_安全性的一般要求.补充标准_程序化电气医疗系统.pdfEN60601-1-8-2004医用电气设备第1-8部分_安全通用要求并列标准_医用电气设备和医用电气系统中的报警系统的.pdfEN60601-2-25-1995医疗电气设备.第2-25部分_心电图安全的特殊要求.pdfEN60601-2-34-2000医疗电器设备.第2部分_直接血压监视设备安全的特殊要求..pdfEN60601-1-2-2001医用电气设备.第1-2部分_通用安全要求.补充标准_电磁兼容性.要求和试验.pdfEN60601-1-6-2004医用电气设备.第1-6部分_安全的通用要求.附属标准_使性..pdfEN60601-2-49-2001医用电气设备.第2-49部分_多功能病人监测设备安全的特殊要求.pdf EN60601-2-4-2003医用.pdfEN1041-1998医疗产品生产者提供的信息.pdf。
欧洲的医疗器械标准

欧洲医疗器械标准一、通用安全和性能要求欧洲医疗器械标准(EU Directive)对医疗器械的通用安全和性能要求做出了规定,要求医疗器械必须满足相关要求,以确保其安全性和有效性。
这些要求包括:1. 医疗器械必须能够达到预定的性能指标,并且在使用过程中不会产生危害人体健康或增加感染的风险。
2. 医疗器械必须具备安全的使用性能,包括无菌、无热原、无毒性等。
3. 医疗器械的设计和制造必须考虑到易于清洁、消毒和保养,以及在使用过程中易于操作和控制。
4. 医疗器械必须能够经受常规使用过程中的各种应力作用,并能够在出现故障时发出警报。
二、医疗器械的设计和制造要求欧洲医疗器械标准对医疗器械的设计和制造要求做出了规定,以确保其符合通用安全和性能要求。
这些要求包括:1. 医疗器械的设计必须考虑到其预定用途和使用环境,并能够满足相关标准和法规的要求。
2. 医疗器械的制造必须采用高质量的材料和工艺,以确保其性能可靠、稳定和持久。
3. 医疗器械的制造必须遵循严格的质量控制程序,以确保每个生产环节的准确性和可靠性。
4. 医疗器械的制造必须进行严格的测试和检验,以确保其符合相关标准和法规的要求。
三、医疗器械的试验方法欧洲医疗器械标准对医疗器械的试验方法做出了规定,以确保其符合通用安全和性能要求。
这些试验方法包括:1. 物理性能试验:包括尺寸、重量、材料和构造等方面的试验。
2. 化学性能试验:包括成分分析、稳定性、毒性等方面的试验。
3. 生物学性能试验:包括细胞毒性、遗传毒性等方面的试验。
4. 电器安全性能试验:包括耐压、泄漏电流、接地等方面的试验。
5. 灭菌和消毒性能试验:包括对医疗器械进行灭菌和消毒的性能试验。
6. 临床试验:在欧盟范围内进行临床试验,以证明医疗器械的安全性和有效性。
7. 环境条件试验:包括对医疗器械进行温度、湿度、大气压力等环境条件的试验。
8. 电磁兼容性试验:包括对医疗器械进行电磁兼容性的试验。
9. 包装和标识的检验:包括对医疗器械的包装和标识进行检查和检验。
欧洲医疗器械CE认证制度介绍

针对体外诊断医疗器械的 特殊要求和CE认证程序。
由欧洲标准化组织(CEN )和欧洲电工标准化组织 (CENELEC)制定的与医 疗器械相关的技术标准, 为制造商提供技术指导和 支持。
04
CE认证的监管与执行
监管机构的职责与权力
01
制定和更新医疗器械CE 认证的技术标准和指南 。
02
评估和监督认证机构的 工作,确保其按照法规 和标准进行认证。
03
对违反CE认证规定的企 业采取执法措施,包括 罚款、撤销认证等。
04
协调欧洲各国监管机构 之间的合作,确保CE认 证制度的一致性和有效 性。
市场监督与执法措施
01
02
03
04
对市场上销售的医疗器械进行 抽查和评估,确保其符合CE
认证要求。
对不符合CE认证要求的医疗 器械采取下架、召回等措施,
防止其继续流通。
证书颁发
认证机构向申请人颁发CE认证证书,证书上注明认证范围、有效期等信息。申请人获得CE认证证书后,即可在 欧盟市场上销售其医疗器械产品。
03
CE认证的技术要求与标准
医疗器械的分类与定义
医疗器械分类
根据风险等级和使用目的,医疗器械被分为I类、IIa类、IIb类和III类。
定义
医疗器械是指用于预防、诊断、治疗、缓解人类疾病、损伤或残疾的设备、器具、器材、材料或其他 物品,无论单独使用还是组合使用。
法规与标准
欧盟成员国根据指令制定相应的法规和标准,以确保产品符合指令的基本要求。 医疗器械的相关法规和标准包括医疗器械法规(Medical Device Regulation, MDR)等。
CE认证的主管机构与职责
主管机构
在欧盟层面,欧洲委员会(European Commission)负责 监督和管理CE认证制度。在各成员国,则有指定的认证机构 (Notified Bodies)负责进行具体的认证工作。
欧盟医疗器械指令的基本要求及协调标准

欧盟医疗器械指令的基本要求及协调标准欧盟医疗器械指令的基本要求及协调标准
医疗器械的基本要求是指令中的核心部分,在三个医疗器械指令的附录Ⅰ中均列出了该指令所适用的医疗器械的基本要求内容,这些基本要求项目涵盖了产品的各个方面,包括通用要求和针对不同种类医疗器械的特殊要求。
例如,在医疗器械指令(EC-Directive 93/42/EEC)附录Ⅰ中共有十四条基本要求,包括六条通用要求和八条特殊要求。
六条通用要求的主要内容是:(奥咨达医疗器械咨询)
1.器械必须是安全的。
器械带来的风险与受益比必须在可以接受的范围内。
2.器械在设计时必须考虑安全因素,应采用公认的技术。
将器械的风险消除或降低到最小,如无法排除风险则需设置保护措施,并将保护措施失效后的残余风险通知使用者。
3.器械必须达到制造商规定的性能。
4. 在器械的使用寿命内,器械的安全性和有效性必须得到保证。
(只专注于医疗器械领域)
5.器械的安全性和有效性必须在合理的运输、储存条件下不受影响。
6. 器械在使用中带来副作用必须在可接受的范围内。
欧盟医疗器械标准(二)2024

欧盟医疗器械标准(二)引言概述:欧盟医疗器械标准(二)主要涵盖了医疗器械的监管制度、分类和评估、技术文件要求、标识和包装以及市场监管等关键内容。
本文将分为五个大点来详细阐述这些内容,帮助读者更好地了解欧盟对医疗器械的标准和要求。
正文:一、医疗器械的监管制度1. 欧盟医疗器械监管机构的职责和作用2. 欧盟医疗器械监管的法律法规与指令3. 医疗器械的注册与许可制度4. 欧盟医疗器械的市场准入要求5. 医疗器械的监测和追溯体系二、医疗器械的分类和评估1. 医疗器械分类的原则和标准2. 欧盟医疗器械评估的程序和要求3. 医疗器械临床评价的指导原则4. 高风险医疗器械的特殊评估方法5. 新型医疗器械的技术评估和小型企业的特殊情况三、技术文件要求1. 医疗器械技术文件的结构和内容2. 医疗器械技术文件准备和更新的要求3. 技术文件中的临床评价和性能评估要求4. 技术文件的审评和审查程序5. 技术文件修改和变更管理的规定四、标识和包装1. 医疗器械的标识要求和标识内容2. 标志性标识和附加标识的要求3. 医疗器械包装的设计原则和要求4. 标识和包装的相关法规和指南5. 医疗器械标识和包装的监管和合规要求五、市场监管1. 欧盟医疗器械市场监管的目标和职责2. 医疗器械市场监管体系的建立和运行3. 医疗器械的风险管理和退市措施4. 医疗器械召回和报告的规定5. 市场监管措施的执行和处罚制度总结:欧盟医疗器械标准(二)从监管制度、分类和评估、技术文件要求、标识和包装、市场监管等五个大点详细说明了欧盟对医疗器械的标准和要求。
了解欧盟标准的适用范围和具体要求对从事医疗器械行业的企业和制造商来说至关重要,以确保他们的产品能够符合相关的法规和标准,进入欧盟市场并得到认可。
欧洲的医疗器械质量体系标准

欧洲的医疗器械质量体系标准欧洲的医疗器械质量体系标准涉及多个方面,包括法规、认证标准、质量管理体系等。
本文将介绍欧洲医疗器械质量体系标准的一般框架,以及其中一些重要的标准和认证程序。
在欧洲,医疗器械的质量体系标准主要由欧洲联盟制定和监管。
欧盟在医疗器械质量管理方面有一套严格的法规框架,其中最重要的是欧洲医疗器械指令(Medical Devices Directive, MDD)和欧洲医疗器械监管法规(Medical Devices Regulation, MDR)。
MDD于1993年发布,对医疗器械的生产、销售和使用提供了法律指导。
这一指令要求所有在欧洲市场销售的医疗器械必须符合特定的质量标准,并经过预先确定的程序认证。
MDD后来经过多次修订,其中包括将一些高风险类别的医疗器械纳入MDD的审查范围。
而MDR则于2017年发布,计划从2021年开始实施。
MDR在MDD的基础上进行了全面更新,强调更严格的质量控制和监管要求。
MDR要求所有的医疗器械都需要进行重新认证,审查程序变得更加复杂和严格。
这一法规还要求医疗器械制造商要对其产品质量管理体系进行全面的检查和审查。
除了指令和法规,欧洲还制定了一系列的标准和认证程序,以确保医疗器械的质量和安全。
ISO 13485是医疗器械质量管理体系的国际标准,被广泛应用于欧洲。
这一标准要求医疗器械制造商建立和维护有效的质量体系,以确保产品符合客户要求和适用的法规要求。
ISO 13485还对医疗器械的生命周期进行了管理,包括设计开发、生产、销售、安装和售后服务等各个阶段。
另外,欧洲还有针对特定类型医疗器械的标准,例如心脏起搏器、人工关节等。
这些标准通常由相关行业协会或国家标准化机构制定,并经过欧盟的认可。
这些标准在欧洲市场销售的相关医疗器械中具有法律效力,对产品性能、使用说明、安全指导等进行了详细规定。
此外,医疗器械的认证程序也是欧洲医疗器械质量体系中的重要组成部分。
欧盟医疗器械指令9342EEC指令

10
第1章 定义和范围 附件: ‘accessory’ means an article which whilst not being a device is intended specifically by its manufacturer to be used together with a device to enable it to be used in accordance with the use of the device intended by the manufacturer of the device;
生效日期:2010-03-21
2
第1章 定义和范围
93/42/EEC指令不适用于: - A. 98/79/EC 指令涉及的体外诊断器械 - B. 90/385/EEC 指令涉及的有源植入式器械 - C. 65/65/EEC指令涉及的药品,包括涉及的89/381/EEC指 令的来源于血 液的药品 - D. 76/768/EEC 指令涉及的化妆品 - E. 人血、人血制品、人血浆或人血细胞,或在投放市场时含有这种血制 品、血浆或细胞的器械 - F. 人体移植物、人体组织或细胞,或含有人体组织或细胞或由其衍生的 制品 - G. 动物移植物或动物组织或细胞,除非器械是利用不能存活的动物组织 或从动物组织中衍生的不能存活的产品制造的
15
第1章 定义和范围 预期用途: 指产品说明或标签或宣传资料载明的, 使用器械应取得的 功效.
‘intended purpose’ means the use for which the device is intended according to the data supplied by the manufacturer on the labelling, in the instructions and/or in promotional materials;
欧洲医疗器械法规简介欧洲医疗器械法规综述

评价方法与标准
安全性评价
通过对医疗器械的设计、制造、使用等全过程进行风险评估,确定其可能产生的危害和风险,并采取相应的风险控制 措施,以确保医疗器械在使用过程中的安全性。
有效性评价
通过对医疗器械的临床试验、性能评估等数据进行综合分析,评价其对于治疗或诊断疾病的实际效果,以确定医疗器 械的有效性和可靠性。
欧洲医疗器械监管机构协作网络
各成员国监管机构之间建立协作网络,共同推进 医疗器械监管工作。
信息共享平台
建立欧洲范围内的医疗器械信息共享平台,实现 监管信息的实时交流和共享。
3
联合行动与决策
针对重大或跨国的医疗器械问题,各监管机构可 以采取联合行动和决策,确保问题得到及时有效 解决。
05
医疗器械的安全性与有效性评价
体外诊断医疗器械指令(IVDD)
规定了体外诊断医疗器械的性能和安全要求,以及相应的符合性评估 程序。
新法规(MDR)
对医疗器械的监管更加严格,强调全生命周期管理和临床数据的收集 与分析。
监管机构及职责
欧洲药品管理局(EMA)
负责协调和监督欧洲药品和医疗器械的监管工作,提供科学建议 和决策支持。
国家药品监管机构
各成员国设立的国家药品监管机构,负责在本国范围内实施医疗器 械法规,并开展相关的监督和检查工作。
欧盟委员会
负责制定和修订医疗器械法规,监督法规的实施情况,并协调各成 员国之间的监管合作。
法规的历史与发展
初始阶段
20世纪70年代,欧洲开始制定医疗器 械法规,以保障公众的健康和安全。
发展阶段
新法规的实施
法规概述
处罚和制裁
对于违反法规要求的行为,将采取相应的 处罚和制裁措施,包括罚款、撤销注册证 、禁止销售等。
欧盟医疗器械的指令包括哪些

欧盟医疗器械的指令包括哪些欧盟医疗器械的指令包括哪些医疗器械指令(EC-Directive 93/42/EEC)由23项条款和12个附录组成,其主要内容为:1、定义和范围(Definitions,scope)2、上市与投入使用(Placing on the market and putting into service)该条款中规定制造商需采取所有必要的措施,确保医疗器械在依照设计的目的安装、维护和使用时不会危及患者、使用者或相关人员的安全及健康。
3、基本要求(Essential requirements)该条款中规定医疗器械必须符合指令附录Ⅰ中的基本要求。
(奥咨达医疗器械咨询)4、医疗器械的自由流通和特殊用途的医疗器械(Free movement,devices intended for special purposes)该条款中规定各成员国不能对符合指令规定的临床研究用器械、定制器械和带有CE标记的医疗器械产品设置流通障碍。
同时规定定制器械、参展器械和临床研究用器械在使用时可无需带有CE标记。
5、可参考的标准(Reference to standards)6、标准与技术法规委员会(Committee on Standards and Technical Regulations)该条款规定依据83/189/EEC号指令第五条所设立的委员会应协助欧盟委员会工作。
7、医疗器械委员会(Committee on Medical Devices)该条款规定依据90/385/EEC号指令第六条第二项所设立的委员会应协助欧盟委员会工作。
(只专注于医疗器械领域)8、保护条款(Safeguard clause)该条款中规定了成员国对被发现不符合指令要求的医疗器械产品的处理措施。
旨在最大限度保护患者、使用者及相关人员的安全与健康。
9、分类(Classification)该条款中规定医疗器械划分为四类,具体分类标准参考附录Ⅸ中内容。
欧盟医疗器械标准

欧盟为消除各成员国间的贸易壁垒,逐步建立成为一个统一的大市场,以确保人员、服务、资金和产品(如医疗器械)的自由流通。
在医疗器械领域,欧盟委员会制定了三个欧盟指令,以替代原来各成员的认可体系,使有关这类产品投放市场的规定协调一致。
这三个指令分别是:1.有源植入性医疗器械指令(AIMD,90/335/EEC),适用于心脏起搏器,可植入的胰岛素泵等有源植入性医疗器械。
AIMD于1993年1月1日生效。
过渡截止期为1994年12月31日,从1995年1月1日强制实施。
2.活体外诊断器械指令(IVD),适用于血细胞计数器,妊娠检测装置等活体外诊断用医疗器械。
该指令目前仍在起草阶段,可能于1998年末或1999年初正式实施。
3.医疗器械指令(Medical Devices Direc-tive,93/42/EEC),适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械(敷料、一次性使用产品、接触镜、血袋、导管等);以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。
该指令已于1995年1月1日生效,过渡截止日期为1998年6月13日从1998年6月14日起强制执行。
上述指令规定,在指令正式实施后,只有带有CE标志的医疗器械产品才能在欧盟市场上销售。
医疗器械CE认证(Medical Devices Direc-tive,93/42/EEC)介绍MDD是目前欧洲可见到的最为全面的医疗器械方面的规定,在该指令中,共有23个条款和12个附录。
其重要部分包括在以下条款中:第1条款:本指令适用于医疗器械及其附件第第2条款:成员国必须确保投放其市场和使用的医疗器械是安全的。
第3条款:所谓“安全”的器械应满足附录1中的基本要求。
第4条款:带有CE标志的医疗器械可在欧盟自由流通。
特殊条款(附录和X)允许使用无CE标志客户定制产品及临床研究的产品。
第5条款:符合协调标准的医疗器械被认为满足基本要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
5.器械的安全性和有效性必须在合理的运输、储存条件下不受影响。
6. 器械在使用中带来副作用必须在可接受的范围内。
1.器械必须是安全的。器械带来的风险与受益比必须在可以接受的范围内。
2.器械在设计时必须考虑安全因素,应采用公认的技术。将器械的风险消除或降者。
3.器械必须达到制造商规定的性能。
4. 在器械的使用寿命内,器械的安全性和有效性必须得到保证。(只专注于医疗器械领域)
欧盟医疗器械指令的基本要求及协调标准
医疗器械的基本要求是指令中的核心部分,在三个医疗器械指令的附录Ⅰ中均列出了该指令所适用的医疗器械的基本要求内容,这些基本要求项目涵盖了产品的各个方面,包括通用要求和针对不同种类医疗器械的特殊要求。
例如,在医疗器械指令 (EC-Directive 93/42/EEC)附录Ⅰ中共有十四条基本要求,包括六条通用要求和八条特殊要求。六条通用要求的主要内容是:(奥咨达医疗器械咨询)