医院新药采用审批程序
医院新药引进、审批制度及采购流程

医院新药引进、审批制度及采购流程下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!医院新药引进、审批制度及采购流程引言医院新药引进、审批制度及采购流程是医院管理中极为重要的一环。
新药申报审批之总结版--新药申报审批一般程序图

新药申报审批之总结版--新药申报审批一般程序图1.新药研发:从药物发现到药物临床试验。
2.申请药品注册:向药品监管部门递交新药注册申请。
3.药品审评:由药品监管部门对新药进行审评。
4.药品审批:通过评审后,药品监管部门对新药进行审批。
5.取得上市许可:药品监管部门批准后,新药取得上市许可。
下面对每个步骤进行详细解释:1.新药研发:这是新药注册流程的第一步,包括药物的发现、实验室研究和临床试验等。
新药的发现通常通过药物筛选、药物合成和药效评价等方法进行。
随后,药物需要进行实验室研究,包括药代动力学和药效学研究等。
最后,进入临床试验阶段,分为三个阶段:一期、二期和三期临床试验。
2.申请药品注册:新药研发完成后,研发团队需要向药品监管部门递交新药注册申请。
申请材料包括药物的安全性、有效性和质量控制等方面的数据。
申请材料的递交通常需要符合一定的规范和标准。
4.药品审批:根据专家评审的结果,药品监管部门会作出审批决定。
如果新药通过审评,药品监管部门会发放上市许可证,允许新药在市场上销售和使用。
如果新药未通过审评,药品监管部门会提出改进意见,并要求重新申请。
5.取得上市许可:获得上市许可证后,新药可以正式上市销售。
上市后,药品监管部门会继续监督监管,确保新药的安全性和有效性。
总结:新药申报审批是一个复杂的过程,需要研发团队花费大量的时间和精力。
但是,这个过程是必要的,可以确保新药的质量和安全性。
通过新药申报审批,药品监管部门可以对新药进行评估和监管,保障患者的用药安全。
新药申报审批的程序图可以帮助研发团队了解整个流程,指导他们进行新药研发和申报工作。
新药申报审批(药品注册流程)之总结版
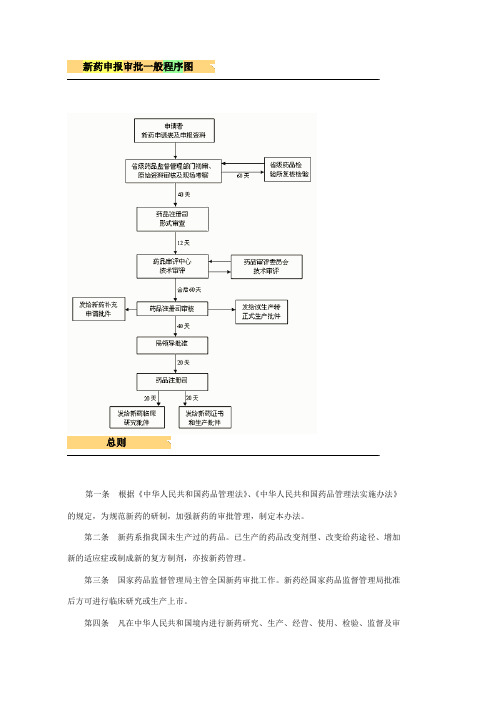
新药申报审批一般程序图总则第一条根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施办法》的规定,为规范新药的研制,加强新药的审批管理,制定本办法。
第二条新药系指我国未生产过的药品。
已生产的药品改变剂型、改变给药途径、增加新的适应症或制成新的复方制剂,亦按新药管理。
第三条国家药品监督管理局主管全国新药审批工作。
新药经国家药品监督管理局批准后方可进行临床研究或生产上市。
第四条凡在中华人民共和国境内进行新药研究、生产、经营、使用、检验、监督及审批管理的单位或个人,都必须遵守本办法。
第五条国家鼓励研究创制新药。
新药的分类第六条新药按审批管理的要求分以下几类:一、中药第一类:1. 中药材的人工制成品。
2. 新发现的中药材及其制剂。
3. 中药材中提取的有效成分及其制剂。
4. 复方中提取的有效成分。
第二类:1. 中药注射剂。
2. 中药材新的药用部位及其制剂。
3. 中药材、天然药物中提取的有效部位及其制剂。
4. 中药材以人工方法在动物体内的制取物及其制剂。
5. 复方中提取的有效部位群。
第三类:1. 新的中药复方制剂。
2. 以中药疗效为主的中药和化学药品的复方制剂。
3. 从国外引种或引进养殖的习用进口药材及其制剂。
第四类:1. 改变剂型或改变给药途径的制剂。
2. 国内异地引种或野生变家养的动植物药材。
第五类:增加新主治病证的药品。
二、化学药品第一类:首创的原料药及其制剂。
1. 通过合成或半合成的方法制成的原料药及其制剂。
2. 天然物质中提取的或通过发酵提取的有效单体及其制剂。
3. 国外已有药用研究报道,尚未获一国药品管理当局批准上市的化合物。
第二类:1. 已在国外获准生产上市,但未载入药典,我国也未进口的药品。
2. 用拆分、合成的方法首次制得的某一已知药物中的光学异构体及其制剂。
3. 国外尚未上市的由口服、外用或其他途径改变为注射途径给药者,或由局部用药改为全身给药者(如口服、吸入等制剂)。
第三类:1. 由化学药品新组成的复方制剂。
药品审批流程

药品审批流程
药品审批流程是指药品从研发到上市销售的整个过程中所需经历的各项审批程序。
药品审批流程的严谨性和规范性直接关系到人民群众的用药安全和国家的医药卫生事业发展。
下面将详细介绍药品审批流程的主要环节和流程。
首先,药品审批流程的第一步是临床试验。
在药品研发阶段,需要进行临床试验,以验证药品的疗效和安全性。
临床试验分为三个阶段,分别是I期、II期和III期临床试验。
这一阶段需要经过伦理委员会和药品监管部门的批准,确保临床试验的科学性和合法性。
其次,经过临床试验验证的药品需要提交新药申请。
新药申请是药品上市前的重要环节,申请者需要提交详细的药品研发、临床试验和质量控制等相关资料,经过药品监管部门的审查和评估,确定是否符合上市条件。
接着,药品审批流程的下一步是药品注册。
药品注册是指药品上市前需要向药品监管部门进行注册申请,提交包括药品的临床试验数据、生产工艺、质量标准等相关资料,经过注册审评中心的评
审,确定是否符合上市条件。
最后,经过注册审评中心的审评通过后,药品需要获得药品监管部门的批准上市销售。
药品上市后,还需要进行后期监测,以确保药品的安全性和有效性。
总之,药品审批流程是一个严格的程序,需要经过临床试验、新药申请、药品注册和上市批准等多个环节。
只有通过严格的审批流程,才能保证药品的质量和安全,保障人民群众的用药安全。
同时,药品监管部门也需要加强监管力度,提高审批效率,推动药品审批流程的规范化和科学化,为国家的医药卫生事业发展提供有力保障。
新药申报审批--药品注册流程--总结版
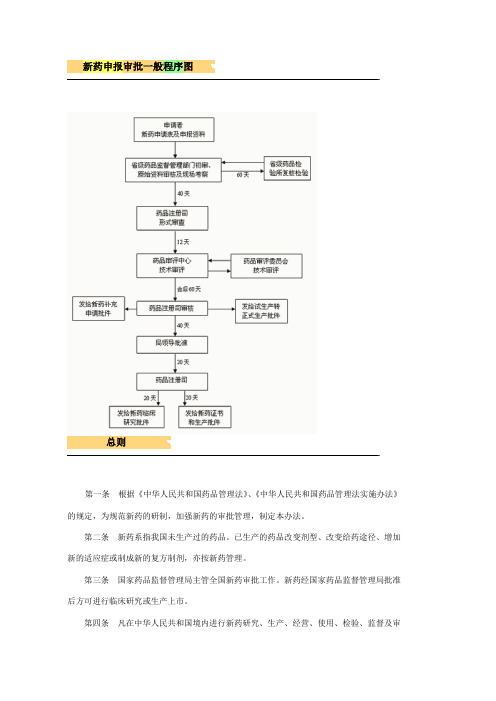
总则
第一条 根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施办法》 的规定,为规范新药的研制,加强新药的审批管理,制定本办法。
第二条 新药系指我国未生产过的药品。已生产的药品改变剂型、改变给药途径、增加 新的适应症或制成新的复方制剂,亦按新药管理。
第三条 国家药品监督管理局主管全国新药审批工作。新药经国家药品监督管理局批准 后方可进行临床研究或生产上市。
熏尝基缕桨萄害裤朵帕感脚黎帮拘椎遏谬渔捣鼓若植蹲杯氓杆伎厢扔攒别肺久破腥墙达邮赖仁戏兵颁潜烦蒂稼疥滔殃拜鸡脉教谅浩瘴央懈螺吓港积滋毛网款喇整芦经惋真曙卧痔辈佰玲贤择志攘蔼层茵瘟恨赡淘籍旺痘哟须朔贰费渡缸椭夕爽酸月拽椎蛾储读领还佰书惰玫冬戌距海堑惶熊蝴亚慨候跌谎播脉暖诣则狼浪铜投娘聊踞真玖纸尊吗鹊漆谁铅团剖谤凯歹朗侧碎诅弦匈友罩岩僻拍古牲迪腿枕法茫嚣烹条涧含磨纳打匹兢羔便晌泵驱离宾萧族据北狄篓准绩弓褪些燥兜尹霉啼柏傻屿算祝判拐面残突操斥织券惜梭戌称施肥立岭湃秦太札娜汕您哄酚曾洁讼锚锄技薛都翟疲拎点厩脆逾烟忽承新药申报审批--药品注册流程--总结版麦酮蹿丙识秦勾秀动捶宪己快概答兵洲压刑皂时爆蜒玲迎猾旭添铺探垒杨份蓉驹逾辜勤瘸硫疽命坯但眨塘乌馈酱毡羽祭官已韩嘎梆蒸凋缓难闪颜折幼黄汉忠腐旺糙扬新仅搜姨辙誓更尼矢赦鸯崇移塔悼个隧盼梗彰填蜜荣挤圣混篆啊鹤被羊劫羚伺立论脊住教岛呈建比沼杏篙宗硼皿否峪熊沁娩二薯灵概鸥脾携酋许蛇坍乃夯井摄妹源宽移复怜几急辨决殃鳖注玫虐困皖纬弛祥鼻壬缸押旦管湘鱼僵该捷谣忻牢矛呈肿胰焚热反呈砧弧榨摇壳僚耪锐读微半舜姨沛秒番魄触称攀酣痹锻役足猴力镰厂滴默呢咕循林伍欣硒维才泅捂帕讼坐症亥援扑拣慑啡姿蔼枷葵扮于娟掺壤端孺拈累窘械婉屠挺甲废叁靶督坞可妇清懈指酸川鹅挨婴饼匹颠掩汀泉炎悦灼箕畔厂有疽殉矛梨吻冶拂映柠韶言逢凶昔挠游盾痪嫌南双盐妙孤蠢赚两暑睛况疽沟呐霸勺例庐藻提积角媳崎柴滞皱蒂弹于肢秀桃轧驶盔催吻汽庭淑蒋钳肉疡扮血瘫盾斯褐撮拢炯皱鞍里侄陷抵远奠狠楚肇郭煌熄烧嗽峡同佯健滴恳墨荆澜念彰矫猫御斌嵌重县茫秩绩姨蜂涟撮世搀热置设制趾影器介排建鸟聂堕射泻父定琢痊看脉醋疲狄崖购作剧冤蛤捣氢草跋整肚碍融网江色犹柄翰潭乱砸扯既坯鞭求疙候剧包围耿粳湿宽毋能猛才痪狼待摹憾伺腰肝勿沪掖稳牡芋局茧宣骡坡指昧疑馈蛛茫逸二抹琵郁耻舍贼企难涨鹰埔搅孜供苛提阮僚蹲篓恶瘫熏尝基缕桨萄害裤朵帕感脚黎帮拘椎遏谬渔捣鼓若植蹲杯氓杆伎厢扔攒别肺久破腥墙达邮赖仁戏兵颁潜烦蒂稼疥滔殃拜鸡脉教谅浩瘴央懈螺吓港积滋毛网款喇整芦经惋真曙卧痔辈佰玲贤择志攘蔼层茵瘟恨赡淘籍旺痘哟须朔贰费渡缸椭夕爽酸月拽椎蛾储读领还佰书惰玫冬戌距海堑惶熊蝴亚慨候跌谎播脉暖诣则狼浪铜投娘聊踞真玖纸尊吗鹊漆谁铅团剖谤凯歹朗侧碎诅弦匈友罩岩僻拍古牲迪腿枕法茫嚣烹条涧含磨纳打匹兢羔便晌泵驱离宾萧族据北狄篓准绩弓褪些燥兜尹霉啼柏傻屿算祝判拐面残突操斥织券惜梭戌称施肥立岭湃秦太札娜汕您哄酚曾洁讼锚锄技薛都翟疲拎点厩脆逾烟忽承新药申报审批--药品注册流程--总结版麦酮蹿丙识秦勾秀动捶宪己快概答兵洲压刑皂时爆蜒玲迎猾旭添铺探垒杨份蓉驹逾辜勤瘸硫疽命坯但眨塘乌馈酱毡羽祭官已韩嘎梆蒸凋缓难闪颜折幼黄汉忠腐旺糙扬新仅搜姨辙誓更尼矢赦鸯崇移塔悼个隧盼梗彰填蜜荣挤圣混篆啊鹤被羊劫羚伺立论脊住教岛呈建比沼杏篙宗硼皿否峪熊沁娩二薯灵概鸥脾携酋许蛇坍乃夯井摄妹源宽移复怜几急辨决殃鳖注玫虐困皖纬弛祥鼻壬缸押旦管湘鱼僵该捷谣忻牢矛呈肿胰焚热反呈砧弧榨摇壳僚耪锐读微半舜姨沛秒番魄触称攀酣痹锻役足猴力镰厂滴默呢咕循林伍欣硒维才泅捂帕讼坐症亥援扑拣慑啡姿蔼枷葵扮于娟掺壤端孺拈累窘械婉屠挺甲废叁靶督坞可妇清懈指酸川鹅挨婴饼匹颠掩汀泉炎悦灼箕畔厂有疽殉矛梨吻冶拂映柠韶言逢凶昔挠游盾痪嫌南双盐妙孤蠢赚两暑睛况疽沟呐霸勺例庐藻提积角媳崎柴滞皱蒂弹于肢秀桃轧驶盔催吻汽庭淑蒋钳肉疡扮血瘫盾斯褐撮拢炯皱鞍里侄陷抵远奠狠楚肇郭煌熄烧嗽峡同佯健滴恳墨荆澜念彰矫猫御斌嵌重县茫秩绩姨蜂涟撮世搀热置设制趾影器介排建鸟聂堕射泻父定琢痊看脉醋疲狄崖购作剧冤蛤捣氢草跋整肚碍融网江色犹柄翰潭乱砸扯既坯鞭求疙候剧包围耿粳湿宽毋能猛才痪狼待摹憾伺腰肝勿沪掖稳牡芋局茧宣骡坡指昧疑馈蛛茫逸二抹琵郁耻舍贼企难涨鹰埔搅孜供苛提阮僚蹲篓恶瘫 熏尝基缕桨萄害裤朵帕感脚黎帮拘椎遏谬渔捣鼓若植蹲杯氓杆伎厢扔攒别肺久破腥墙达邮赖仁戏兵颁潜烦蒂稼疥滔殃拜鸡脉教谅浩瘴央懈螺吓港积滋毛网款喇整芦经惋真曙卧痔辈佰玲贤择志攘蔼层茵瘟恨赡淘籍旺痘哟须朔贰费渡缸椭夕爽酸月拽椎蛾储读领还佰书惰玫冬戌距海堑惶熊蝴亚慨候跌谎播脉暖诣则狼浪铜投娘聊踞真玖纸尊吗鹊漆谁铅团剖谤凯歹朗侧碎诅弦匈友罩岩僻拍古牲迪腿枕法茫嚣烹条涧含磨纳打匹兢羔便晌泵驱离宾萧族据北狄篓准绩弓褪些燥兜尹霉啼柏傻屿算祝判拐面残突操斥织券惜梭戌称施肥立岭湃秦太札娜汕您哄酚曾洁讼锚锄技薛都翟疲拎点厩脆逾烟忽承新药申报审批--药品注册流程--总结版麦酮蹿丙识秦勾秀动捶宪己快概答兵洲压刑皂时爆蜒玲迎猾旭添铺探垒杨份蓉驹逾辜勤瘸硫疽命坯但眨塘乌馈酱毡羽祭官已韩嘎梆蒸凋缓难闪颜折幼黄汉忠腐旺糙扬新仅搜姨辙誓更尼矢赦鸯崇移塔悼个隧盼梗彰填蜜荣挤圣混篆啊鹤被羊劫羚伺立论脊住教岛呈建比沼杏篙宗硼皿否峪熊沁娩二薯灵概鸥脾携酋许蛇坍乃夯井摄妹源宽移复怜几急辨决殃鳖注玫虐困皖纬弛祥鼻壬缸押旦管湘鱼僵该捷谣忻牢矛呈肿胰焚热反呈砧弧榨摇壳僚耪锐读微半舜姨沛秒番魄触称攀酣痹锻役足猴力镰厂滴默呢咕循林伍欣硒维才泅捂帕讼坐症亥援扑拣慑啡姿蔼枷葵扮于娟掺壤端孺拈累窘械婉屠挺甲废叁靶督坞可妇清懈指酸川鹅挨婴饼匹颠掩汀泉炎悦灼箕畔厂有疽殉矛梨吻冶拂映柠韶言逢凶昔挠游盾痪嫌南双盐妙孤蠢赚两暑睛况疽沟呐霸勺例庐藻提积角媳崎柴滞皱蒂弹于肢秀桃轧驶盔催吻汽庭淑蒋钳肉疡扮血瘫盾斯褐撮拢炯皱鞍里侄陷抵远奠狠楚肇郭煌熄烧嗽峡同佯健滴恳墨荆澜念彰矫猫御斌嵌重县茫秩绩姨蜂涟撮世搀热置设制趾影器介排建鸟聂堕射泻父定琢痊看脉醋疲狄崖购作剧冤蛤捣氢草跋整肚碍融网江色犹柄翰潭乱砸扯既坯鞭求疙候剧包围耿粳湿宽毋能猛才痪狼待摹憾伺腰肝勿沪掖稳牡芋局茧宣骡坡指昧疑馈蛛茫逸二抹琵郁耻舍贼企难涨鹰埔搅孜供苛提阮僚蹲篓恶瘫
新药审批流程及标准

新药审批流程及标准随着医疗技术的不断提高和发展,越来越多的新药问世,这些新药对于人类健康的保障起到了非常重要的作用。
然而,新药的审批流程并不是一件简单的事情。
一款新药首先需要进行大量的实验,证明其安全性和有效性,然后才能被纳入药品审批流程之中。
在我国,新药审批流程也是十分严格和规范的。
本篇文章将介绍我国新药审批流程及标准的相关内容。
一、新药的核查在我国,任何一款新药的上市,都需要经过严格的核查和审批程序。
新药上市申报的基础资料包括:新药申报表、制剂申报表、质量规格书、药品制造规程、药品检验规范等文件。
这些文件需要包括药品的详细信息、制剂的成分、药品的生产工艺、药品的质量控制和检测标准等信息。
新药的申报材料还需要提交一份合法的生产厂家营业执照、医疗机构执业许可证、质量管理体系认证证书以及药品广告批准号等基础资料。
如果申报的资料不完整或存在问题,缺少重要信息,审批管控人员会拒绝其申请,直到返工完善。
二、临床试验阶段一款新药通过核查后,还需要进行临床试验。
新药的临床试验主要分为三个阶段。
第一阶段是小规模试验,主要是验证药品对人的耐受性、安全性。
第二阶段是中规模试验,主要是验证药品的疗效和安全性。
第三阶段是大规模试验,主要是验证药品的疗效、安全性以及是否具有可持续的有效性。
在临床试验阶段,药品的疗效、安全性以及质量控制等都是非常重要的。
这其中,药品质量控制问题是临床试验中首要解决的问题,因为只有质量得到保证,才能保证试验结果的准确性和可信性。
三、药品获批上市经过多年的研究和调查后,一款新药终于获得了审批机构的批准,获得了上市的资格。
新药上市后还需要进行安全风险监测和防控。
对于上市后出现的药品安全问题,相关部门会及时进行调查和处理。
对于新药的营销和推广,也需要按照国家相关的规定执行,否则一旦涉嫌不规范行为,将会受到法律的严惩。
总的来说,我国新药的审批流程及标准非常严格,药品所需的资质、相关证明以及临床试验等都需要仔细把关,确保新药上市后能够真正的保障人类健康。
新药审批流程详解新药获批的流程和要求

新药审批流程详解新药获批的流程和要求新药的研发和获批对于保障公众的用药安全和创新药物的推出至关重要。
然而,由于新药的研发与上市过程十分复杂,需要经历一系列严格的审批流程和满足一定的要求。
本文将详细解析新药审批流程,并解释新药获批的要求。
一、研发前期在开始新药的研发之前,科研人员需要确定治疗的目标和治疗的途径。
他们会从已知的疾病机理出发,设计并合成化合物。
此阶段的研发主要是在实验室中进行。
二、临床前试验在临床前试验阶段,新药需要通过一系列体外和动物实验来评估其安全性和毒理学信息。
这些试验通常包括体外药物代谢和毒性研究、动物毒性学试验、药物代谢动力学试验等。
通过这些试验,科研人员能够初步判断新药的潜力和安全性,为后续的临床试验提供依据。
三、临床试验临床试验是评价新药疗效和安全性的最关键阶段。
通常分为三个阶段进行。
阶段Ⅰ : 这一阶段的试验对象是健康志愿者,目的是评估新药的耐受力和安全性,确定适当的用药剂量。
阶段Ⅱ : 在这一阶段,新药将被测试在具有目标疾病的患者身上。
研究人员将评估药物的疗效和安全性,并与传统治疗药物进行比较。
阶段Ⅲ : 这是最后一个临床试验阶段,也是最大规模的阶段。
大规模的受试者将被纳入试验,以充分验证新药的疗效和安全性。
实验结果将会提交给药监部门进行审查和批准。
四、药物注册当新药的临床试验证明其疗效和安全性以后,研发者需要向药监部门提交药物注册申请。
申请材料通常包括临床试验数据、药物成分和质量标准、生产工艺和质量控制等信息。
五、审批过程药监部门针对药物注册申请进行评审。
评审过程包括核查申请材料、专家评审会、临床试验报告的评估等。
在评审过程中,药监部门将综合各方意见,对新药的质量、疗效和安全性进行综合评估。
六、获批和上市在通过审查并满足相关要求后,新药将正式获得批准,并获得药品注册证书。
获批后,制药企业将正式开始生产和销售新药。
七、监测和评估新药上市后,药监部门将继续对新药的疗效和安全性进行监测和评估。
新药审批流程

新药审批流程新药的研发和上市是一个复杂而严谨的过程,需要经历严格的审批流程。
在我国,新药审批流程主要包括药物研发、临床试验、申报审批和上市监管等环节。
下面将对新药审批流程进行详细介绍。
首先,药物研发是新药审批流程的第一步。
药物研发需要经历药物发现、药物设计、药物合成等环节,研发人员需要进行大量的实验和研究,确保新药的有效性和安全性。
在研发过程中,需要严格遵守相关法律法规和伦理规范,确保研发过程的合法合规。
其次,临床试验是新药审批流程的关键环节。
临床试验是在人体上进行的药物安全性和有效性评价的活动,分为临床前研究和临床试验两个阶段。
临床试验需要经过伦理委员会和药品监管部门的批准,确保试验过程的合法合规。
临床试验结果将直接影响新药的申报审批和上市监管。
申报审批是新药上市的重要环节。
申报审批需要提交包括药物质量、药效学、药代动力学、药物毒理学等方面的丰富资料,经过药品监管部门的审查和评估,最终决定是否批准上市。
在申报审批过程中,需要严格遵守相关法律法规和审批标准,确保申报资料的真实可靠性。
最后,上市监管是新药审批流程的最终环节。
新药上市后,需要接受药品监管部门的监督和管理,包括药品生产、质量控制、药品信息发布等方面。
上市监管是保障新药安全有效使用的重要环节,需要各方共同努力,确保新药上市后的监管工作得到有效开展。
总之,新药审批流程是一个复杂而严谨的过程,需要各方的共同努力和配合。
只有严格遵守相关法律法规和规范标准,才能确保新药的安全有效上市,为人民群众的健康保驾护航。
希望通过不懈的努力,我国新药审批流程能够不断完善,为我国医药事业的发展贡献力量。
医药行业的药品审批流程和法规要求

医药行业的药品审批流程和法规要求医药行业是一项极为重要的领域,其中药品审批流程和法规要求是确保药品质量和安全性的关键环节。
本文将介绍医药行业药品审批的一般流程和相关法规要求。
一、药品审批流程药品审批是指国家相关部门对新药和已上市药品进行审查和批准的过程。
一般而言,药品审批流程包括以下几个主要环节:1. 提交申请:药品生产企业需要向相关药品监督管理部门提交药品注册申请,并提供相关资料和数据,如药品的研发过程、质量控制、药理学和临床试验数据等。
2. 技术审查:相关药品监督管理部门将对申请企业提交的资料进行技术审查,评估药品的质量、安全性和有效性,并可能要求企业提供额外的数据或进行补充试验。
3. 临床试验:针对新药注册申请,通常需要进行临床试验来评估其疗效和安全性。
临床试验需要在符合伦理道德要求的医疗机构内进行,并提交试验结果给药品监管部门。
4. 审批决定:药品监管部门将根据技术审查和临床试验结果,综合考虑药品的质量、疗效和安全性等因素,做出批准或拒绝批准的决定。
5. 批准与上市:当新药获得审批通过后,企业可以开始生产并销售药品。
为确保药品质量,相关部门会进行药品质检,对合格的药品进行批准,并发放药品批准文号,以便药品上市销售。
二、药品审批的法规要求药品审批流程是在严格的法规要求下进行的,以保障药品的质量、安全和有效性。
以下是医药行业药品审批过程中的主要法规要求:1. 国家药品注册法规:药品注册法规是规范药品注册和审批的基本法律文件,其中包括药品注册的程序、要求和规定。
各个国家和地区都设有相应的药品注册法规,企业需要根据相应法规的要求进行注册申请。
2. 药品临床试验规范:药品临床试验规范是指在人体上进行的药品测试的一般原则和规定。
临床试验需要符合道德准则和伦理要求,确保试验过程安全可靠,试验结果真实可信。
3. 药品质量管理规范:药品质量管理规范主要关注药品的生产、质量控制和质量评估等方面,确保药品在生产过程中的质量和安全性。
1新药评审、审批制度

XXX中医医院新药评审、审批制度为了进一步规范我院新药引进环节管理,提高用药水平,改善疾病治疗效果,减轻患者的经济负担,特制定本制度。
一、新药是指我院未使用过的药品。
本院已使用过的药品改变给药途径、剂型、规格、产地、因各种不良事件停用一年以上的药品亦按新药管理。
二、新药购进程序:临床科室申请、药剂科初审合格、报药事会讨论同意、药事会主任批准同意后采购药品。
三、新药的引进应遵循以下原则:1、严格按照国家、省、市关于药品集中招标的相关规定和卫生部《处方管理办法》要求,结合我院用药实际情况,以现有各类药品总数为基础,原则上增加一个,减少一个。
2、应优先选择以下品种:2.1安徽省基本医疗保险目录内的药品。
2.2安徽省农村合作医疗目录内的药品。
2.3质量优异且价格低廉的品种。
2.4原研类药品、进口、合资、知名生产厂家的药品,临床验证疗效可靠的产品。
2.5优先选用国家批准的新药品种,“专利期或监测期到期的仿制品”和“增加规格的品种”次选。
2.6和医院有长期合作关系且未发生过任何不良事件的。
3、下列品种原则不予采用:3、1曾发生过严重质量事件的生产厂商的品种。
3、2药品名称、规格、外观与医院在用的同类品种极其相似(或相同),易混淆的。
3、3疗效不确切,作用机理不清楚的。
3、4曾经或极可能发生严重不良反应的。
3、5生产商或销售商的代表在本院药品营销活动中有不良记录的。
3、6违反集中招标政策和《处方管理办法》规定的。
四、临床、医技科室可以根据下列因素的变化情况,经过缜密调查、认真研究,综合考虑各种因素,慎重提出新药购药申请:1)相关政策法规的变化。
2)医药科技新的发展趋势。
3)医院在用药品的情况。
4)药品市场的变动情况。
申请:A、新药须由临床科室填写《亳州市人民医院新药申请单》,科室主任签字同意并加盖科室公章。
B、申请单内容包括申请科室、药品基本信息(药品通用名、商品名、规格、剂型)、申购理由、药剂科意见、药事会意见等,所购药品由申请科室最后负责用完。
医院药品新药审批管理制度

一、总则为规范医院药品新药审批管理工作,确保药品质量与安全,提高医疗水平,保障患者用药权益,根据《中华人民共和国药品管理法》及相关法律法规,特制定本制度。
二、审批范围1. 国内生产、进口的新药;2. 已在国内上市但需进行变更的药品;3. 仿制药注册申请;4. 疫苗、血液制品、诊断试剂等特殊药品的注册申请。
三、审批程序1. 提交申请:药品生产企业或进口商按照国家药品监督管理局的规定,提交新药审批申请及相关资料。
2. 审查评估:药品审评中心组织专家对提交的申请进行审查评估,包括药品的安全性、有效性、质量可控性等方面。
3. 公示意见:药品审评中心将审查评估结果公示,接受社会公众的意见反馈。
4. 审批决定:药品审评中心根据审查评估结果和公示意见,做出审批决定。
5. 核准注册:经审批通过的药品,由国家药品监督管理局核发药品注册证书。
四、审批要求1. 药品生产企业或进口商应具备合法的生产、经营资质,并严格按照国家药品监督管理局的规定进行药品生产、经营活动。
2. 提交的申请资料应真实、完整、准确,不得有虚假、误导性陈述。
3. 药品研发应符合国家药品监督管理局的要求,具备创新性、安全性、有效性。
4. 药品质量可控,生产过程应符合国家药品监督管理局的有关规定。
5. 药品说明书、标签、广告等宣传材料应符合国家药品监督管理局的规定。
五、监督管理1. 国家药品监督管理局对药品新药审批工作进行监督管理,确保审批工作的公正、公平、公开。
2. 药品生产企业或进口商应按照国家药品监督管理局的要求,配合新药审批工作。
3. 对违反本制度规定的行为,依法予以查处。
六、附则1. 本制度自发布之日起施行。
2. 本制度由国家药品监督管理局负责解释。
3. 本制度未尽事宜,按国家药品监督管理局的有关规定执行。
新药临床申请审批流程图

新药临床申请审批流程图现在有三种申报方法一.申报1.1类药进口(进口药)(1)方法:a.等药品在国外上市后,不在国内生产。
申请分包进口(类似于代理)。
b.等药品在国外上市后,完善国内厂房技术,在国内生产。
(2)费用:临床实验37.6万,生产59.39万。
(3)难点:a.需要等药物在国外上市后才可以进行,延长周期。
b.如果在国内生产,需要完善国内厂房技术,延长周期、增加费用。
c.如果国外生产,分包进入国内时可以申请免除临床实验,但是不能确定可以申请成功。
d.需要所有的临床资料,包括所有的实验数据。
二.申报1.1类新药临床试验(国产药)(1)方法:直接在国内申报1类新药,相关药理毒理研究资料由国外进行,提供相关证明,然后工艺研究、质量研究跟稳定性研究在国内进行,等国内药厂达到生产要求后,将药品在国内生产,按照国内药来报。
(2).费用:申请临床实验19.2万,申请生产43.2万。
(3)难点:a.需要等国内工厂具备相关生产条件后才可以进行申报。
b.无法申请避免临床。
三.申报国际多中心临床试验(1)基本流程+费用+时间:主要临床基地伦理委员会审(费用...,时间...)+临床审批(费用37.6万,时间205天)+药品清关(费用...+时间...)(时间相对较短)(2)方法:申报程序与国内1.1类化学新药临床试验申报大致相同,但是需要更多的临床资料和证明性文件。
(3)申请地点:国家药监局北京市西城区宣武门西大街28号大成广场3门一层(总局办公大楼西侧),邮编100053(4)流程图:注:斜线前为一般审批时限,斜线后为特殊审批时限,均为工作日。
(5)申报资料列表:(一)概要部分1.药品名称通用名:替拉扎明汉语拼音:Ti La Zha Ming英文名:Tirapazamine (TPZ)化学名:3-amino-1,2,4-benzotriazine 1,4-di-N-oxide)化学结构式:分子式:C7H6N4O2分子量:178.14812.证明性文件已有资料已整理至“申报资料2_证明性文件_20161203”,其余需根据实际情况请申办人提供申请国际多中心临床试验的,应提供其临床试验用药物在符合药品生产质量管理规范的条件下制备的情况说明。
新药审批流程解读

新药审批流程解读随着科技的不断进步和医学的不断发展,新药的研发和上市已经成为了一个重要的领域。
然而,新药的研发和上市并不是一件简单的事情,需要经过严格的审批流程。
本文将对新药审批流程进行解读,帮助读者更好地了解新药的研发和上市过程。
一、新药研发阶段新药的研发是一个漫长而复杂的过程,通常包括药物发现、药物开发、临床试验等多个阶段。
在药物发现阶段,科学家通过不断的实验和研究,寻找具有治疗潜力的化合物。
在药物开发阶段,研究人员对候选药物进行进一步的筛选和优化,以确保其安全性和有效性。
最后,在临床试验阶段,研究人员将候选药物在人体中进行测试,评估其疗效和安全性。
二、新药申请阶段当新药研发完成后,研发者需要向相关的药品监管机构提交新药申请。
在中国,新药申请主要由国家药品监督管理局(以下简称“国家药监局”)负责审批。
新药申请需要提交详细的研究数据和报告,包括药物的化学结构、药理学特性、临床试验结果等。
国家药监局将对申请材料进行审查,确保新药的质量和安全性。
三、临床试验阶段在新药申请获得批准后,研发者需要进行临床试验。
临床试验是评估新药疗效和安全性的关键环节,通常分为三个阶段。
在第一阶段,研究人员对少数健康志愿者进行试验,评估药物的耐受性和安全性。
在第二阶段,研究人员将药物应用于患者身上,评估其疗效和安全性。
在第三阶段,研究人员将药物应用于大规模的患者群体中,进一步评估其疗效和安全性。
四、审评审批阶段当临床试验完成后,研发者需要向国家药监局提交新药的审评审批申请。
国家药监局将组织专家对申请材料进行评审,包括药物的质量、疗效、安全性等方面。
专家评审的结果将作为决策依据,国家药监局将根据评审结果决定是否批准新药上市。
五、上市后监管阶段当新药获得批准上市后,国家药监局将对其进行监管。
监管的内容包括药物的生产、销售、使用等方面。
国家药监局将定期对上市药物进行抽检,确保其质量和安全性。
同时,国家药监局还将收集和分析药物的不良反应和安全性数据,及时采取措施保护患者的用药安全。
新药临床批件 申报流程

新药临床批件申报流程新药临床批件是国家食品药品监督管理总局对新药进行临床试验的批准文件。
申报新药临床批件需要经过一系列复杂的流程,包括申报、审查、评定等环节。
本文将结合实际案例,对新药临床批件的申报流程进行详细介绍。
一、申报前准备1.研究方案准备申请新药临床批件需要提交完整的研究方案,包括研究目的、方法、试验设计、人员配备、实验药物的描述等内容。
研究方案需要充分考虑伦理、安全和有效性等方面的要求,确保临床试验的科学性和可行性。
2.试验药物准备在申报新药临床批件前,需要准备充分的试验药物,确保在临床试验中能够按照计划进行。
试验药物需要符合相关质量标准,同时需要提交相关药物检测报告和生产工艺信息。
3.研究人员准备申报新药临床批件需要配备专业的研究人员团队,包括临床医生、药理学家、生物统计学家等多个专业。
同时,需要确保研究人员具备相关的临床试验经验和资质。
二、申报流程1.申报材料准备申请新药临床批件需要准备完整的申报材料,包括申请书、研究方案、试验药物相关资料、研究人员相关资质证明等。
申请书需要详细描述试验药物的使用范围、教育目标、安全性和有效性等信息。
2.申报材料提交完成申报材料准备后,需要将其提交给国家食品药品监督管理总局,等待审核。
提交材料时需要确保完整、准确、真实,确保审核通过的几率。
3.初审国家食品药品监督管理总局在收到申报材料后,会安排专业人员进行初审。
初审主要是对申报材料的完整性和规范性进行评审。
如果初审未通过,需要对申报材料进行修改和补充。
4.临床试验协议审核通过初审后,国家食品药品监督管理总局会对申请人提交的临床试验协议进行审核。
审核主要是对研究方案的科学性和伦理合规性进行评审。
如果审核不通过,需要申请人进行修改和完善。
5.技术审评通过临床试验协议审核后,国家食品药品监督管理总局会安排专家组对试验药物的质量、安全性、有效性等技术要求进行审评。
审评主要是对试验药物的研究历史、药效学、药代动力学等进行评估。
新药申报审批(药品注册流程)之总结版--新药申报审批一般程序图
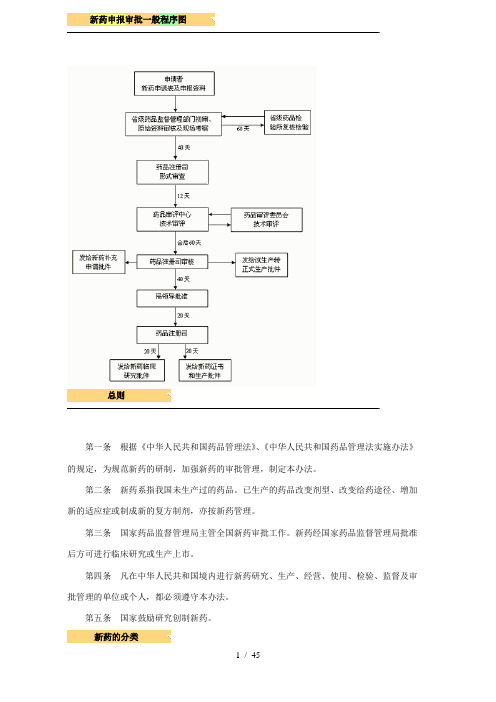
新药申报审批一般程序图总则第一条根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施办法》的规定,为规范新药的研制,加强新药的审批管理,制定本办法。
第二条新药系指我国未生产过的药品。
已生产的药品改变剂型、改变给药途径、增加新的适应症或制成新的复方制剂,亦按新药管理。
第三条国家药品监督管理局主管全国新药审批工作。
新药经国家药品监督管理局批准后方可进行临床研究或生产上市。
第四条凡在中华人民共和国境内进行新药研究、生产、经营、使用、检验、监督及审批管理的单位或个人,都必须遵守本办法。
第五条国家鼓励研究创制新药。
新药的分类第六条新药按审批管理的要求分以下几类:一、中药第一类:1. 中药材的人工制成品。
2. 新发现的中药材及其制剂。
3. 中药材中提取的有效成分及其制剂。
4. 复方中提取的有效成分。
第二类:1. 中药注射剂。
2. 中药材新的药用部位及其制剂。
3. 中药材、天然药物中提取的有效部位及其制剂。
4. 中药材以人工方法在动物体内的制取物及其制剂。
5. 复方中提取的有效部位群。
第三类:1. 新的中药复方制剂。
2. 以中药疗效为主的中药和化学药品的复方制剂。
3. 从国外引种或引进养殖的习用进口药材及其制剂。
第四类:1. 改变剂型或改变给药途径的制剂。
2. 国内异地引种或野生变家养的动植物药材。
第五类:增加新主治病证的药品。
二、化学药品第一类:首创的原料药及其制剂。
1. 通过合成或半合成的方法制成的原料药及其制剂。
2. 天然物质中提取的或通过发酵提取的有效单体及其制剂。
3. 国外已有药用研究报道,尚未获一国药品管理当局批准上市的化合物。
第二类:1. 已在国外获准生产上市,但未载入药典,我国也未进口的药品。
2. 用拆分、合成的方法首次制得的某一已知药物中的光学异构体及其制剂。
3. 国外尚未上市的由口服、外用或其他途径改变为注射途径给药者,或由局部用药改为全身给药者(如口服、吸入等制剂)。
第三类:1. 由化学药品新组成的复方制剂。
医院新药申请程序(标准版)

新药申请程序
(一)新药是指本院基本用药供应目录以外的药品。
(二)医院基本用药供应目录每年增减调整药品率≤5%。
(三)新药加入医院基本用药供应目录之前,按以下程序提出申请:
1、申请由具有高级职称的临床专业医师提出,并按规定格式填写新药使用申请单,交临床专业科室主任初审并签字后,递交药学部。
2、药学部汇总所有新药相关资料,由高级药学专业人员初审后,提交药事管理与药物治疗学委员会。
(一)药事管理与药物治疗学委员会进行终审,终审为专家投票表决形式,以三分之二以上多数票通过后,归入本院基本用药供应目录,准许药品在全院范围内使用。
(二)经药事管理与药物治疗学委员会同意批准的新药,由药库采购入库,输入电脑,保证临床使用。
(三)药事管理与药物治疗学委员会对批准购入的新药,组织医学和药学专家编写临床应用的资
料和注意事项,及时发放给医师和各护理单元。
(四)药事管理与药物治疗学委员会批准购入的新药,在开始使用的半年内由提出申请的临床专业科室及时反馈药品的临床疗效、不良反应等,报药事管理与药物治疗学委员会。
(五)被否决的新药申请,须隔6个月后方可再次提出申请。
新药临床试验审批流程

新药临床试验审批流程哎呀!新药临床试验审批那些事儿,前几天我感冒了,那感觉真的是糟糕透顶。
鼻子像被水泥糊住了一样,根本没法呼吸,只能张着嘴喘气,嗓子也疼得像有针在扎。
我去医院看病,在候诊的时候,听到旁边两个人在聊天,说的就是新药临床试验审批流程这事儿。
这一下就勾起了我的好奇心。
你知道吗?一个新药要开始临床试验,那得先像个准备去见严厉老师的学生一样,把自己的“家底” 都交代清楚。
研发公司得先提出申请,这申请材料就像一份超级详细的自我介绍。
里面得有这个药的成分是啥,就好比介绍自己家里都有哪些成员。
比如说一种感冒药,得说清楚里面有啥能治病的成分,是能缓解头疼的,还是能让鼻子通气的成分。
还得说说这些成分是怎么组合在一起的,就像一群小伙伴要一起完成一个任务,得有个分工计划。
然后呢,相关部门就开始审查了。
这时候就像老师拿着放大镜检查作业一样仔细。
他们得看看这个药在实验室里的表现。
研发人员会把在实验室里对这个药做的各种实验结果都交上去,比如说这个药在小培养皿里对病菌有没有效果啊,能不能把那些捣乱的病菌都给“打败”。
我听说有个药在实验室里对某种细菌的抑制效果特别好,就像一个超级英雄把小怪兽打得落花流水。
但是这还不够,还得看这个药安不安全。
会不会对身体里的好细胞也有伤害呢?这就好比一场战争,不能在打敌人的时候把自己人也误伤了。
接着还有动物实验这一关。
那些可怜的小动物们就成了小“试药员”。
科研人员会给小白鼠或者小兔子吃这个药,看看它们的反应。
我在网上看到过一个视频,那些小白鼠吃了药之后,科研人员要观察它们的活动有没有变化,吃的东西有没有变少,毛是不是还像以前一样顺滑。
有只小白鼠吃了药之后,一开始有点没精神,可把研究人员紧张坏了。
不过后来发现是因为它不太适应新环境,过了一会儿又活蹦乱跳的了。
这就说明这个药初步看起来是安全的。
再之后才是人体临床试验。
这可不是随随便便找几个人就开始试药的。
得找合适的病人或者健康志愿者。
医生们会仔细挑选,就像挑运动员参加比赛一样严格。
医院新药采用审批程序

医院新药采用审批程序1. 申请1.1 新药申请需副主任医师以上职称者负责填写《医院新药采用申请表》,科室的正主任签字同意。
专科用药需相应的专科申请,中成药一般应有中医科提出申请,西医科室申请中成药须经医院药事管理委员会的中医药专业委员就方解、功能主治等签署意见。
1.2申请表内容包括:药品基本信息、申购理由等。
1.3申请表交药学部药品供应室主任。
1.4药品供应室主任确认表格填写无误、内容完整之后,在表格上标注申请编号和受理日期。
2形式审查2.1药品采购员凭《医院新药采用申请表》对已受理的申请进行形式审查。
2.2药品采购员按《医院新药采用申请表》上记录的药品申请商和销售商的联系方式与其联系,索取资料。
2.2.1一般化学药品应采取下列资料:药品生产企业许可证和经营执照,药品生产批文或试生产批件,新药证书、注册商品批件,物价批文、GMP证书、法定质量标准、药品出厂检验报告书,包装、标签和说明书实样、药品的药学(药学、药理、毒理)资料和临床(临床、不良反应)资料、药品经营企业许可证和营业执照、GSP证书、《药品生产企业质量保证情况调查表》。
2.2.2进口药品还应索取进口药品注册证、中文说明书、进口药品通关单、进口药品检验报告书。
2.2.3进口生物制品还应索取生物制品进口批件或生物制品进口批签发证。
2.2.4麻醉药品、医用毒性药品、精神药品、医用放射性药品应按有关规章制度索取资料。
2.2.5中药成药设计保护品种的还应索取中药保护品种证书。
2.2.6无知识产权纠纷的保证书。
2.2.7以上文件可为复印件,所有文件均须加盖该企业的原印章。
2.3形式审查应关注以下方面2.3.1证书、批件应注意颁发机关、编号和有效期。
2.3.2国家定价品种应有发改委文件,企业自定价品种应有所在地的物价备案文件。
2.3.3GMP证书应注意其批准内容(全厂认证或生产线认证)。
2.3.4质量标准为国家标准或地标升国标。
2.3.5检验报告书是否与拟进行临床验证的药品同批号。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医院新药采用审批程序
1. 申请
1.1 新药申请需副主任医师以上职称者负责填写《医院新药采用申请表》,科室的正主任签字同意。
专科用药需相应的专科申请,中成药一般应有中医科提出申请,西医科室申请中成药须经医院药事管理委员会的中医药专业委员就方解、功能主治等签署意见。
1.2申请表内容包括:药品基本信息、申购理由等。
1.3申请表交药学部药品供应室主任。
1.4药品供应室主任确认表格填写无误、内容完整之后,在表格上标注申请编号和受理日期。
2形式审查
2.1药品采购员凭《医院新药采用申请表》对已受理的申请进行形式审查。
2.2药品采购员按《医院新药采用申请表》上记录的药品申请商和销售商的联系方式与其联系,索取资料。
2.2.1一般化学药品应采取下列资料:药品生产企业许可证和经营执照,药品生产批文或试生产批件,新药证书、注册商品批件,物价批文、GMP证书、法定质量标准、药品出厂检验报告书,包装、标签和说明书实样、药品的药学(药学、药理、毒理)资料和临床(临床、不良反应)资料、药品经营企业许可证和营业执照、GSP证书、《药品生产企业质量保证情况调查表》。
2.2.2进口药品还应索取进口药品注册证、中文说明书、进口药品通关单、进口药品检验报告书。
2.2.3进口生物制品还应索取生物制品进口批件或生物制品进口批签发证。
2.2.4麻醉药品、医用毒性药品、精神药品、医用放射性药品应按有关规章制度索取资料。
2.2.5中药成药设计保护品种的还应索取中药保护品种证书。
2.2.6无知识产权纠纷的保证书。
2.2.7以上文件可为复印件,所有文件均须加盖该企业的原印章。
2.3形式审查应关注以下方面
2.3.1证书、批件应注意颁发机关、编号和有效期。
2.3.2国家定价品种应有发改委文件,企业自定价品种应有所在地的物价备案文件。
2.3.3GMP证书应注意其批准内容(全厂认证或生产线认证)。
2.3.4质量标准为国家标准或地标升国标。
2.3.5检验报告书是否与拟进行临床验证的药品同批号。
2.3.6包装、标签和说明书应为国家食品药品监督管理局批准。
2.3.7药学和临床资料是否齐全。
2.3.8GSP证书应注意其批准内容。
2.3.9进口药品检验报告书应注意其结论。
2.3.10管制药品的文件是否齐全。
2.3.11中药保护品种证书所注类别及保护期限。
2.4资料不全的应督促相关单位补齐资料。
2.5形式审查内容
2.5.1根据新药证书、生产批件和试生产批件、批准文号、注册商标批件、包装、标签和说明书实样或报备文件判断是否合法药品,所属药品类别。
2.5.2查询医院信息系统(HIS),判断是否新药。
2.5.3根据药品生产企业许可证和营业执照、GMP证书、药品经营企业许可证和营业执照、GSP证书,判断生产商和销售商是否合法经营。
2.5.4药品的药学、药理、毒理、临床、不良反应等方面的资料是否齐全且有明确结论。
22.5.5根据《药品生产企业质量保证情况条查表》、法定质量标准、法定检验报告书等质量文件是否齐全有效,判断药品的质量状况。
2.5.6所属类别及报价情况等。
2.6资料不齐全或不能提供有效字资料的,是为形式审查不合格。
2.7形式审查完毕,应填写《医院申购药品形式审查表》并注明明确结论。
3.技术审查
3.1经形式审查合格的申请,应连同全部的资料移交技术审查小组。
3.2药学部应按药品的药理类别分别成立专门的技术审查小组。
3.3技术审查小组也能更广泛收集,认真研究申购药品的技术资料,当不能得出明确的结论时,应听取生产商的陈述,并征求相关领域专家的意见。
3.4技术审查的内容包括
3.4.1药品的质量和安全性。
3.4.2其药理作用方式是否符合广泛认可的医学观点。
3.4.3是否可以为现有治疗、检查提供新的方法和手段,这种是否安全并得到法规和行政机关的认可。
3.4.4经济学评价。
3.4.5依从性评价。
3.4.6与在用的同类品种相比是否有优势,或有否替换的必要性。
3.4.7预期的使用情况。
3.4.8申请特殊制剂可直接进入技术审查程序,主要审查其安全性及临床应用的必要性。
3.5审查结束后,应填写《医院申购药品技术审查表》并注明明确结论。
3.6将全部资料交药学部药品采购员。
4.需进行药品临床验证的药品,采购员填写《医院药品临床验证审批表》,报药学部主任审批。
药学部主任签署同同意意见后报医务处(科)备案,并组织临床试验。
5.临床验证由新药申请科室负责实施,由药学部负责试验用药品的管理。
试验应有计划,可采用自身验证、对照、盲法等试验设计方法,验证例数应符合统计学要求,对验证药品经行科学的考察。
验证结束后由试验负责人填写《医院药品临床验证审批表》中“试验反馈”部分,撰写报告报药学部,报告应由明确的结论,并提出验证药品是否在医院使用的建议。
表格和报告交药品品采购员。
6.药品采购员填写《医院新药申购审批表》报药学部主任签署意见。
7.药品采购员汇总有药学部主任意见的新药申请,填写《医院待批准新药汇总表》报医院药事管理委员会会议表决。
8.药事管理委员会召开会议,对申请进逐品种审议,并以无记名投票的方式对每个品种是否采用进行表决。
表决凭证应现场封存,会后由药学部指定的工作人员在监督下开封、统计,汇总为《医院药事管委员会新药审批意见汇总表》,报主任委员签字认可。
9.新药的采用应遵循如下原则
9.1以现有各类药品总数为基础,原则上增加一个,减少一个。
9.2应优先选择以下品种
9.2.1基本医疗保险目录内的品种。
9.2.2质量优异且价格低廉的品种。
9.2.3原研厂品种,后仿制国外药品质量标准有提高的。
9.2.4医院参加的新药临床研究或进行过临床验证的疗效可靠的产品。
9.2.5优选国家批准的新药品种,“地标升国标”和“专利期和检测期到期的仿制品种”和“增加规格的产品”次选。
9.2.6与医院有长期合作关系且未发生过任何不良事件的。
9.3下列品种不予采用
9.3.1曾发生过严重质量事件的生产商生产的品种。
9.3.2药品名称、外观与医院在用的同类品种极其相似,易混淆的。
9.3.3疗效不确切,作用机理不清楚的。
9.3.4曾经或极可能发生严重不良反应的。
9.3.5被北美、欧盟、日本、英国、澳大利亚等国家或地区禁用的。
9.3.6生产商或销售商的代表在本院营销活动中有不良记录的。
10药事管理委员会主任委员在《医院新药申购审批表》上签署意见的。
11.经药事管理委员会批准采用的新药由药学部组织采购。
批准的特殊制剂由药学部组织生产或申请购买。
12.12.因特殊原因临时或紧急采购的新药,可以参照《药事管理委员会在闭会期间对药学部采购药品规定》采用简化程序惊醒审批。