干热灭菌相关温度与时间一览表
干热灭菌相关温度与时间一览表

对开门百级净化热风循环风箱:1、通常采用250℃,时间不低于45分钟;2、在250度时,1min ,FH=;3、那么45分钟,其FH值就是*45=;4、那么不是大于基本要求FH等于1000吗?5、2000年版药典就是不低于30分钟(FH=*30=),那么为什么会在2005年版做如此修改?6、是因为这里的250度是腔体内温度,不是物品内部或表面温度,也不是最低点温度,所以将其时间提高15分钟!所谓:“温度不够时间补,补的是时间”隧道烘箱:1、通常采用320度,时间不低于4分钟;2、其在300度(公认温度,低于通常,真是!),1分钟,FH=,3、那么4分钟,FH值就是*4=1022;4、那么为什么会通常用320度呢?原因同上,瓶底温度在320度时,瓶内温度才会达到300度."要提高产量,得提高温度,温度不均匀更要提高温度,补的是温度“一补时间,一补温度,中国人真是大补呀!原因不说,乱补一气,反正设备不会说话,而我们有的不是效率,而是时间。
干热灭菌FH值计算SDA不知为何?FH大于1400.准确地说是1364.干热标准温度为170C,Z为54.F(T)=10^((T-170)/Z)F(320)=10^((320-170)/54)=600,也就是说:320C 1分钟相当于标准温度(170C)600分钟320C,10MIN,F(H)=6000(5995).隧道烘箱设定320度,但不能全程达到320C,我们公司的是5分钟.以我做过的经验.,F(H)在2000到3000之间.为什么呢?正常的计算是要把长升温100度开始与降温至100度结束,均应计入FH值的计算,所以,FH值的计算是一个倒扣的船形集合,即使低于药典正常标准,仍能达到FH值大于1000,但这计算起来有点麻烦.从资料报道,国外生产能满足灭菌去热原要求,其FH≥1000。
根据中国2000版药典的要求和公式计算,认为空瓶FH≥ 1400为宜。
干热灭菌--去热源 USP、CP、EP关于干热去热原的温度时间要求
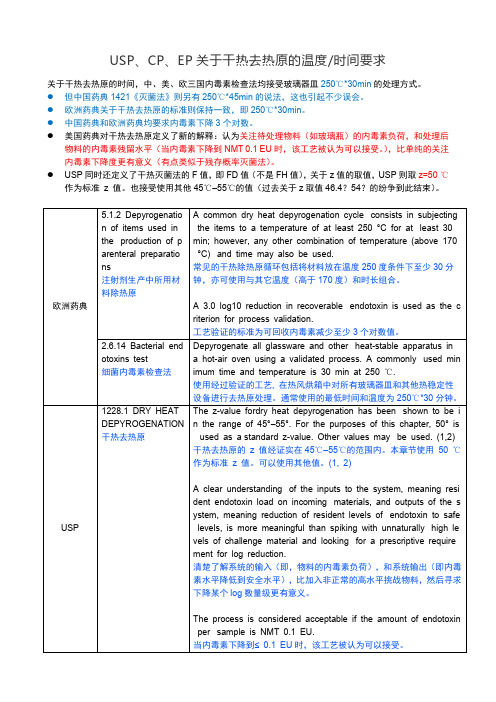
USP、CP、EP关于干热去热原的温度/时间要求
关于干热去热原的时间,中、美、欧三国内毒素检查法均接受玻璃器皿250℃*30min的处理方式。
●但中国药典1421《灭菌法》则另有250℃*45min的说法,这也引起不少误会。
●欧洲药典关于干热去热原的标准则保持一致,即250℃*30min。
●中国药典和欧洲药典均要求内毒素下降3个对数。
●美国药典对干热去热原定义了新的解释:认为关注待处理物料(如玻璃瓶)的内毒素负荷,和处理后
物料的内毒素残留水平(当内毒素下降到NMT 0.1 EU时,该工艺被认为可以接受。
),比单纯的关注内毒素下降度更有意义(有点类似于残存概率灭菌法)。
●USP同时还定义了干热灭菌法的F值,即FD值(不是FH值),关于z值的取值,USP则取z=50 ℃
作为标准z 值。
也接受使用其他45℃–55℃的值(过去关于z取值46.4?54?的纷争到此结束)。
USP,EP、中国药典干热灭菌条件对照表
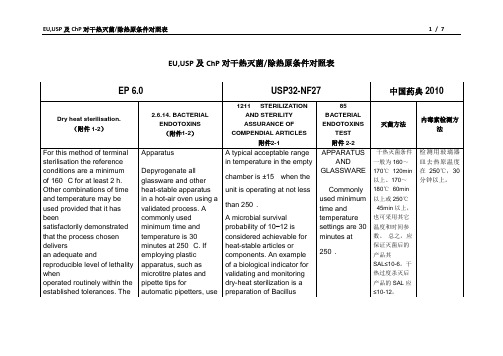
EU,USP及ChP对干热灭菌/除热原条件对照表附件1 -1EP6.0 5.1.1. METHODS OF PREPARATIONOF STERILE PRODUCTS)Dry heat sterilisation. For this method of terminal sterilisation the reference conditions are a minimumof 160 °C for at least 2 h. Other combinations of timeand temperature may be used provided that it has beensatisfactorily demonstrated that the process chosen deliversan adequate and reproducible level of lethality whenoperated routinely within the established tolerances. Theprocedures and precautions employed are such as to give anSAL of 10− 6 or better.Dry heat sterilisation is carried out in an oven equipped withforced air circulation or other equipment specially designedfor the purpose. The steriliser is loaded in such a way thata uniform temperature is achieved throughout the load.Knowledge of the temperature within the steriliser duringthe sterilisation procedure is usually obtained by means oftemperature-sensing elements inserted into representativecontainers together with additional elements at thepreviously established coolest part of the loaded steriliser.The temperature throughout each cycle is suitably recorded.Where a biological assessment is carried out, this is obtainedusing a suitable biological indicator (5.1.2).Dry heat at temperatures greater than 220 °C is frequentlyused for sterilisation and depyrogenation of glassware. Inthis case demonstration of a 3-log reduction in heat resistantendotoxin can be used as a replacement for biologicalindicators (5.1.2)附件1-2 2.6.14. BACTERIAL ENDOTOXINSThe test for bacterial endotoxins is used to detect orquantify endotoxins of gram-negative bacterial originusing amoebocyte lysate from horseshoe crab (Limuluspolyphemus or Tachypleus tridentatus). There are3 techniques for this test : the gel-clot technique, whichis based on gel formation ; the turbidimetric technique,based on the development of turbidity after cleavage of anendogenous substrate ; and the chromogenic technique,based on the development of colour after cleavage of asynthetic peptide-chromogen complex.The following 6 methods are described in the presentchapter :Method A. Gel-clot method: limit testMethod B. Gel-clot method: semi-quantitative testMethod C. Turbidimetric kinetic method182 See the information section on general monographs (cover pages) EUROPEAN PHARMACOPOEIA 6.0 2.6.14. Bacterial endotoxinsMethod D. Chromogenic kinetic methodMethod E. Chromogenic end-point methodMethod F. Turbidimetric end-point methodProceed by any of the 6 methods for the test. In the eventof doubt or dispute, the final decision is made based uponmethod A unless otherwise indicated in the monograph.The test is carried out in a manner that avoids endotoxincontamination.ApparatusDepyrogenate all glassware and other heat-stable apparatusin a hot-air oven using a validated process. A commonly usedminimum time and temperature is 30 minutes at 250 °C. Ifemploying plastic apparatus, such as microtitre plates andpipette tips for automatic pipetters, use apparatus shownto be free of detectable endotoxin and of interfering effectsfor the test.NOTE: In this chapter, the term ‘tube’ includes all types ofreceptacles, for example microtitre plate wells.indicators (5.1.2).附件2-11211STERILIZATION AND STERILITY ASSURANCE OF COMPENDIAL ARTICLESThis informational chapter provides a general description of the concepts and principles involved in the quality control of articles that must be sterile. Any modifications of or variations in sterility test procedures from those described under Sterility Tests 71should be validated in the context of the entire sterility assurance program and are not intended to be methods alternative to those described in that chapter.Within the strictest definition of sterility, a specimen would be deemed sterile only when there is complete absence of viable microorganisms from it. However, this absolute definition cannot currently be applied to an entire lot of finished compendial articles because of limitations in testing. Absolute sterility cannot be practically demonstrated without complete destruction of every finished article. The sterility of a lot purported to be sterile is therefore definedin probabilistic terms, where the likelihood of a contaminated unit or article is acceptably remote. Such a state of sterility assurance can be established only through the use of adequate sterilization cycles and subsequent asepticprocessing, if any, under appropriate current good manufacturing practice, and not by reliance solely on sterility testing. The basic principles for validation and certification of a sterilizing process are enumerated as follows:1. Establish that the process equipment has capability of operating within therequired parameters.2. Demonstrate that the critical control equipment and instrumentation arecapable of operating within the prescribed parameters for the processequipment.3. Perform replicate cycles representing the required operational range of theequipment and employing actual or simulated product. Demonstrate that the processes have been carried out within the prescribed protocol limits andfinally that the probability of microbial survival in the replicate processescompleted is not greater than the prescribed limits.4. Monitor the validated process during routine operation. Periodically as needed,requalify and recertify the equipment.5. Complete the protocols, and document steps (1) through (4) above.METHODS OF STERILIZATIONIn this informational chapter, five methods of terminal sterilization, including removal of microorganisms by filtration and guidelines for aseptic processing, are described. Modern technological developments, however, have led to the use of additional procedures. These include blow-molding (at hightemperatures), forms of moist heat other than saturated steam and UVirradiation, as well as on-line continuous filling in aseptic processing. Thechoice of the appropriate process for a given dosage form or componentrequires a high level of knowledge of sterilization techniques and information concerning any effects of the process on the material being sterilized.1Dry-Heat SterilizationThe process of thermal sterilization of Pharmacopeial articles by dry heat is usually carried out by a batch process in an oven designed expressly for that purpose. A modern oven is supplied with heated, filtered air, distributeduniformly throughout the chamber by convection or radiation and employing a blower system with devices for sensing, monitoring, and controlling the critical parameters. The validation of a dry-heat sterilization facility is carried out in a manner similar to that for a steam sterilizer described earlier. Where the unit is employed for sterilizing components such as containers intended forintravenous solutions, care should be taken to avoid accumulation ofparticulate matter in the chamber. A typical acceptable range in temperature in the empty chamber is ±15when the unit is operating at not less than 250.In addition to the batch process described above, a continuous process is frequently employed to sterilize and depyrogenate glassware as part of an integrated continuous aseptic filling and sealing system. Heat distribution may be by convection or by direct transfer of heat from an open flame. The continuous system usually requires a much higher temperature than cited above for the batch process because of a much shorter dwell time. However, the total temperature input during the passage of the product should be equivalent to that achieved during the chamber process. The continuous process also usually necessitates a rapid cooling stage prior to the aseptic filling operation. In the qualification and validation program, in view of the short dwell time, parameters for uniformity of the temperature, and particularly the dwell time, should be established.A microbial survival probability of 10–12 is considered achievable forheat-stable articles or components. An example of a biological indicator for validating and monitoring dry-heat sterilization is a preparation of Bacillus subtilis spores. Since dry heat is frequently employed to render glassware or containers free from pyrogens as well as viable microbes, a pyrogen challenge, where necessary, should be an integral part of the validation program, e.g., by inoculating one or more of the articles to be treated with 1000 or more USP Units of bacterial endotoxin. The test with Limulus lysate could be used to demonstrate that the endotoxic substance has been inactivated to not more than 1/1000 of the original amount (3 log cycle reduction). For the test to be valid, both the original amount and, after acceptable inactivation, the remaining amount of endotoxin should be measured. For additional information on the endotoxin assay, see Bacterial Endotoxins Test 85.附件2-2 85BACTERIAL ENDOTOXINS TESTAPPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validated process.2Commonly used minimum time and temperature settings are 30 minutes at 250. If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, use only that which has been shown to be free of detectable endotoxin and not to interfere with the test. [note—In this chapter, the term “tube” includes any other receptacle such as a micro-titer well.]。
灭菌的标准温度和时间

灭菌的标准温度和时间
灭菌的标准温度和时间是由微生物学和生物医学研究确定的,以确保医疗器械、药品和其他医疗用品的安全性和有效性。
不同的灭菌方法有不同的标准温度和时间。
例如,压力蒸汽灭菌的标准温度通常为121°C至134°C,灭菌时间通常为15分钟至30分钟,具体时间取决于物品的种类和大小。
过氧化氢等离子灭菌的标准温度通常为60°C至80°C,灭菌时间通常为10分钟至30分钟,具体时间取决于物品的种类和大小。
环氧乙烷灭菌的标准温度通常为54°C至70°C,灭菌时间通常为20分钟至60分钟,具体时间取决于物品的种类和大小。
除了压力蒸汽、过氧化氢等离子和环氧乙烷灭菌外,还有其他的灭菌方法,如紫外线消毒、干热灭菌、化学消毒等。
每种灭菌方法的标准温度和时间都有所不同,因此在进行医疗器械、药品和其他医疗用品的灭菌时,需要遵循相关的行业标准和规定。
干热灭菌法的基本原理

干热灭菌法的基本原理干热灭菌法是常用的一种灭菌方法,其基本原理是利用高温高压的干热条件来杀死细菌和病毒等微生物。
在医疗、食品、制药等领域中,干热灭菌法被广泛应用。
本文将介绍干热灭菌法的基本原理及其应用。
一、干热灭菌法的基本原理干热灭菌法的基本原理是利用高温高压来杀死微生物。
干热灭菌法的温度范围通常在160℃-180℃之间,持续时间为1-2小时。
在这个温度范围内,微生物细胞内的蛋白质、核酸和膜结构等都会发生变性和破坏,从而导致微生物的死亡。
干热灭菌法的灭菌效果与温度、时间和湿度等因素有关。
温度越高、时间越长、湿度越低,灭菌效果越好。
因此,在干热灭菌过程中,需要严格控制温度、时间和湿度等参数,以确保灭菌效果。
二、干热灭菌法的应用干热灭菌法在医疗、食品、制药等领域中被广泛应用。
下面将分别介绍其应用。
1. 医疗领域医疗领域中,干热灭菌法常用于灭菌器械、注射器、输液器等医疗器械。
干热灭菌法可以有效地杀死各种微生物,包括细菌、病毒和真菌等,从而避免交叉感染的发生。
此外,干热灭菌法还可以杀死一些难以灭活的微生物,如芽孢等。
2. 食品领域食品领域中,干热灭菌法常用于灭菌罐头、干货、蜜饯等食品。
干热灭菌法可以有效地杀死食品中的微生物,从而延长食品的保质期。
此外,干热灭菌法还可以杀死食品中的寄生虫和病毒等,保障食品安全。
3. 制药领域制药领域中,干热灭菌法常用于灭菌药品、试剂和原料等。
干热灭菌法可以有效地杀死微生物,从而保证药品的纯度和质量。
此外,干热灭菌法还可以避免药品中添加防腐剂,减少药品的副作用。
三、干热灭菌法的优缺点干热灭菌法有以下优点:1. 干热灭菌法可以杀死各种微生物,包括细菌、病毒和真菌等。
2. 干热灭菌法可以杀死一些难以灭活的微生物,如芽孢等。
3. 干热灭菌法可以避免使用化学消毒剂,从而避免对环境的污染。
4. 干热灭菌法可以避免药品中添加防腐剂,减少药品的副作用。
干热灭菌法也有以下缺点:1. 干热灭菌法需要较高的温度和时间,从而增加了成本和时间成本。
医疗器械湿热灭菌参数

医疗器械湿热灭菌参数医疗器械在使用过程中,容易受到微生物的污染和传播,使用不当甚至会对患者造成危害。
医疗器械的湿热灭菌变得愈发重要。
本文将介绍湿热灭菌的一些参数,以及参数的重要性。
一、灭菌温度湿热灭菌的最佳温度为121°C。
在这个温度下,只需要15-20分钟便能够灭除菌群。
当温度比121°C高时,灭菌时间可以缩短。
若温度过高则有可能会损坏器械。
灭菌温度不能随意调节。
二、灭菌时间灭菌时间是实现灭菌的重要参数之一。
通常情况下,湿热灭菌时间为15-20分钟。
但不同机型和灭菌物品所需的时间有所不同。
灭菌时间太短则无法杀灭菌群,如果时间过长则对器械的材质有可能会产生损伤或影响器械原有的性能。
三、湿度湿度对湿热灭菌也是非常重要的参数。
通常情况下,湿度要求在60%-80%之间。
湿度过低会使器械表面变干,细菌的繁殖速度会加快。
合适的湿度会使细菌繁殖受到限制。
四、压力一般来说,湿热灭菌时不需要加压。
对于带有空气的容器或器械,需要在灭菌时进行压力控制,以保证器械内部的温度和湿度均匀分布。
五、真空度对于不耐高温和潮湿的衣物和器械,使用真空干燥灭菌能够避免器械暴露在高温潮湿中所带来的损伤。
真空度要求在40-80kPa之间,过高或过低皆不利于灭菌。
六、灭菌剂量灭菌剂量是指灭菌时所加入的消毒剂的质量。
通常情况下,灭菌剂量与器械的尺寸和难度成正比。
较小的器械需要的消毒剂较少,较大的器械需要的消毒剂较多。
七、氧气浓度湿热灭菌的过程需要一定量的氧气。
如果氧气的浓度太低,会降低湿热灭菌的效果。
在灭菌的过程中必须保证氧气的充分供应。
以上的几个参数对于湿热灭菌是非常重要的。
正确合理地调节这些参数,可以确保器械在湿热灭菌过程中受到最小的损害,并达到杀菌的效果。
这对于保障医院工作效率、减少病人感染风险具有非常重要的作用。
进一步需要注意的是,灭菌过程中使用的消毒剂种类也是非常关键的。
目前,临床上常用的消毒剂有乙烯氧化物(EO)、过氧乙酸(PAA)、臭氧(O3)、氯气(Cl2)和氧化氢(H2O2)等。
干热灭菌原理

干热灭菌原理干热灭菌是一种常用的微生物灭活方法,其原理是利用高温对微生物进行灭活。
在干热灭菌过程中,微生物的细胞膜和细胞壁会受到高温的破坏,从而导致微生物的死亡。
干热灭菌主要适用于一些耐热性较强的物品,如玻璃器皿、金属器具等。
下面将详细介绍干热灭菌的原理及其应用。
首先,干热灭菌的原理是利用高温对微生物进行灭活。
在高温下,微生物的蛋白质会发生变性,细胞膜和细胞壁也会受到破坏,从而导致微生物的死亡。
干热灭菌的温度通常在160-180摄氏度之间,持续一定时间,可以有效地灭活大多数微生物,包括细菌、真菌和病毒。
其次,干热灭菌的原理还包括氧化作用。
在高温下,微生物的细胞内的生物分子会受到氧化作用的影响,从而导致微生物的死亡。
此外,高温还会影响微生物的代谢过程,使其无法正常生长和繁殖,最终导致微生物的死亡。
干热灭菌的应用范围非常广泛。
首先,医疗器械和药品是干热灭菌的重要应用领域之一。
一些耐热性较强的医疗器械和药品,如手术器械、注射器等,常常采用干热灭菌的方法进行消毒。
其次,食品工业也广泛应用干热灭菌技术。
一些食品原料和包装材料,如香料、干果、玻璃瓶等,都可以通过干热灭菌来保持其品质和安全性。
此外,实验室和科研机构中的玻璃器皿、金属器具等也常常采用干热灭菌的方法进行消毒。
总之,干热灭菌是一种常用的微生物灭活方法,其原理是利用高温对微生物进行灭活。
干热灭菌的应用范围非常广泛,包括医疗器械和药品、食品工业、实验室和科研机构等领域。
通过对干热灭菌原理的深入了解,可以更好地应用这一技术,保障物品的安全性和品质。
干热灭菌的工艺条件

干热灭菌是一种利用高温和干燥的条件来杀灭微生物的方法。
其工艺条件包括以下几个方面:
温度:通常使用高温进行灭菌,常见的温度范围是160°C至180°C。
具体的温度选择取决于被灭菌物品的特性和要求。
时间:灭菌的时间取决于温度和被灭菌物品的特性。
通常,较高的温度可以在较短的时间内完成灭菌,而较低的温度则需要更长的时间。
空气流动:确保在灭菌过程中有适当的空气流动,以促进热传递和保持温度均匀。
相对湿度:干热灭菌需要在低湿度条件下进行,通常要求相对湿度低于30%。
包装:被灭菌物品需要以适当的方式包装,以确保其在灭菌过程中不受外界污染。
值得注意的是,具体的干热灭菌工艺条件可能因不同的应用和要求而有所变化。
在实际操作中,建议参考相关的行业标准、制药工艺要求或专业指导,以确保干热灭菌的有效性和安全性。
干热灭菌相关温度与时间一览表

对开门百级净化热风循环风箱:1、通常采用250℃,时间不低于45分钟;2、在250度时,1min ,FH=30.3;3、那么45分钟,其FH值就是30.3*45=1363.5;4、那么不是大于基本要求FH等于1000吗?5、2000年版药典就是不低于30分钟(FH=30.3*30=909.1),那么为什么会在2005年版做如此修改?6、是因为这里的250度是腔体内温度,不是物品内部或表面温度,也不是最低点温度,所以将其时间提高15分钟!所谓:“温度不够时间补,补的是时间”隧道烘箱:1、通常采用320度,时间不低于4分钟;2、其在300度(公认温度,低于通常,真是!),1分钟,FH=255.5,3、那么4分钟,FH值就是255.5*4=1022;4、那么为什么会通常用320度呢?原因同上,瓶底温度在320度时,瓶内温度才会达到300度."要提高产量,得提高温度,温度不均匀更要提高温度,补的是温度“一补时间,一补温度,中国人真是大补呀!原因不说,乱补一气,反正设备不会说话,而我们有的不是效率,而是时间。
干热灭菌FH值计算SDA不知为何?FH大于1400.准确地说是1364.干热标准温度为170C,Z为54.F(T)=10^((T-170)/Z)F(320)=10^((320-170)/54)=600,也就是说:320C 1分钟相当于标准温度(170C)600分钟320C,10MIN,F(H)=6000(5995).隧道烘箱设定320度,但不能全程达到320C,我们公司的是5分钟.以我做过的经验.,F(H)在2000到3000之间.为什么呢?正常的计算是要把长升温100度开始与降温至100度结束,均应计入FH值的计算,所以,FH值的计算是一个倒扣的船形集合,即使低于药典正常标准,仍能达到FH值大于1000,但这计算起来有点麻烦.从资料报道,国外生产能满足灭菌去热原要求,其FH≥1000。
根据中国2000版药典的要求和公式计算,认为空瓶FH≥ 1400为宜。
干热灭菌法

干热灭菌法集团文件版本号:(M928-T898-M248-WU2669-I2896-DQ586-M1988)干热灭菌法一、通过使用干热空气杀灭微生物的方法叫干热灭菌。
一般是把待灭菌的物品包装就绪后,放入电烘箱中烘烤,即加热至160~170℃维持1~2h。
二、干热灭菌法常用于空玻璃器皿、金属器具的灭菌。
凡带有胶皮的物品,液体及固体培养基等都不能用此法灭菌。
三、灭菌方法:1、玻璃器皿等在灭菌前必须经正确包裹和加塞,以保证玻璃器皿于灭菌后不被外界杂菌所污染。
常用玻璃器皿的包扎和加塞方法如下:平皿用纸包扎或装在金属平皿筒内;三角瓶在棉塞与瓶口外再包以厚纸,用棉绳以活结扎紧,以防灭菌后瓶口被外部杂菌所污染;吸管以拉直的曲别针一端放在棉花的中心,轻轻捅入管口,松紧必须适中,管口外露的棉花纤维统一通过火焰烧去,灭菌时将吸管装入金属管筒内进行灭菌,也可用纸条斜着从吸管尖端包起,逐步向上卷,头端的纸卷捏扁并拧几下,再将包好的吸管集中灭菌。
2、干燥箱灭菌将包扎好的物品放入干燥烘箱内,注意不要摆放太密,以免妨碍空气流通;不得使器皿与烘箱的内层底板直接接触。
将烘箱的温度升至160~170℃并恒温1~2h,注意勿使温度过高,超过170℃,器皿外包裹的纸张、棉花会被烤焦燃烧。
如果是为了烤干玻璃器皿,温度为120℃持续30分钟即可。
温度降至60~70℃时方可打开箱门,取出物品,否则玻璃器皿会因骤冷而爆裂。
用此法灭菌时,绝不能用油、蜡纸包扎物品。
3、火焰灭菌直接用火焰灼烧灭菌,迅速彻底。
对于接种环,接种针或其它金属用具,可直接在酒精灯火焰上烧至红热进行灭菌。
此外,在接种过程中,试管或三角瓶口,也采用通过火焰而达到灭菌的目的。
湿热灭菌一、?煮沸消毒法注射器和解剖器械等均可采用此法。
先将注射器等用纱布包好,然后放进煮沸消毒器内加水煮沸。
对于细菌的营养体煮沸约15~30min,对于芽孢则需煮沸约1~2h。
二、高压蒸汽灭菌法高压蒸汽灭菌用途广,效率高,是微生物学实验中最常用的灭菌方法。
USP、CP、EP关于干热去热原的温度时间要求

USP 、CP 、EP 关于干热去热原的温度时间要求关于干热去热原的时间,中、美、欧三国内毒素检查法均接受玻璃器皿250℃*30min 的处理方式。
• 但中国药典1421《灭菌法》则另有250℃*45min 的说法,这也引起不少误会。
• 欧洲药典关于干热去热原的标准则保持一致,即250℃*30min。
• 中国药典和欧洲药典均要求内毒素下降3个对数。
• 美国药典对干热去热原定义了新的解释:认为关注待处理物料(如玻璃瓶)的内毒素负荷,和处理后物料的内毒素残留水平(当内毒素下降到NMT 0.1 EU 时,该工艺被认为可以接受。
),比单纯的关注内毒素下降度更有意义(有点类似于残存概率灭菌法)。
• USP 同时还定义了干热灭菌法的F 值,即FD 值(不是FH 值),关于z 值的取值,USP 则取z=50 ℃作为标准 z 值。
也接受使用其他45℃–55℃的值(过去关于z 取值46.4?54?的纷争到此结束)。
欧洲药典 5.1.2 Depyrogenation of items used in the production of parenteral preparations注射剂生产中所用材料除热原 A common dry heat depyrogenation cycle consists insubjecting the items to a temperature of at least 250 °C for at least 30 min; however, any other combination of temperature (above 170 °C) and time may also be used.常见的干热除热原循环包括将材料放在温度250度条件下至少30分钟,亦可使用与其它温度(高于170度)和时长组合。
A 3.0 log10 reduction in recoverable endotoxin isused as the criterion for process validation.工艺验证的标准为可回收内毒素减少至少3个对数值。
干热空气灭菌法

干热空气灭菌法
干热空气灭菌法是一种常见的消毒方法,它指的是将物品放置在
高温干热环境中,利用高温将微生物完全杀灭的方法。
其优点在于操
作简单,不需要特定的化学药品,同时可以完全消灭所有细菌、病毒
和真菌。
干热空气灭菌法的基本原理是利用高温杀灭微生物,通过制造高
温环境,可获得灭菌的效果。
对于不同的物品,其灭菌的温度和时间
是不一样的。
一般情况下,温度需要达到160度以上,时间需要持续
30分钟以上才能有效地杀灭大部分病菌。
在使用干热空气灭菌法时,需要注意以下事项:首先,应确保物
品表面没有残留物,否则会影响灭菌效果。
其次,在灭菌的过程中,
应注意不同物品的材质,不同材质的物品需要不同温度和时间。
最后,使用干热空气灭菌法时,需要遵循一定的操作规范,如缓慢升温、均
匀加热、保持温度等。
干热空气灭菌法广泛应用于医疗和食品行业。
医疗领域中,这种
方法可以用于灭菌手术器械、血液袋等,以确保手术过程中的安全性。
在食品行业中,这种方法可用于灭菌需要长时间保存的食品,包括干果、坚果、茶叶等。
总之,干热空气灭菌法是一种非常有效的灭菌方法,应用广泛,
在医疗和食品行业中都具有重要作用。
使用这种方法时,我们需要注
意物品的材质和温度,以获得最好的消毒效果,并准确掌握操作规范,确保消毒过程的安全和有效性。
干热灭菌相关温度与时间一览表

干热灭菌相关温度与时间一览表对开门百级净化热风循环风箱:1、通常采用250℃,时间不低于45分钟;2、在250度时,1min ,FH=;3、那么45分钟,其FH值就是*45=;4、那么不是大于基本要求FH等于1000吗?5、2000年版药典就是不低于30分钟(FH=*30=),那么为什么会在2005年版做如此修改?6、是因为这里的250度是腔体内温度,不是物品内部或表面温度,也不是最低点温度,所以将其时间提高15分钟!所谓:“温度不够时间补,补的是时间”隧道烘箱:1、通常采用320度,时间不低于4分钟;2、其在300度(公认温度,低于通常,真是!),1分钟,FH=,3、那么4分钟,FH值就是*4=1022;4、那么为什么会通常用320度呢?原因同上,瓶底温度在320度时,瓶内温度才会达到300度."要提高产量,得提高温度,温度不均匀更要提高温度,补的是温度“一补时间,一补温度,中国人真是大补呀!原因不说,乱补一气,反正设备不会说话,而我们有的不是效率,而是时间。
干热灭菌FH值计算SDA不知为何?FH大于1400.准确地说是1364.干热标准温度为170C,Z为54.F(T)=10^((T-170)/Z)F(320)=10^((320-170)/54)=600,也就是说:320C 1分钟相当于标准温度(170C)600分钟320C,10MIN,F(H)=6000(5995).隧道烘箱设定320度,但不能全程达到320C,我们公司的是5分钟.以我做过的经验.,F(H)在2000到3000之间.为什么呢?正常的计算是要把长升温100度开始与降温至100度结束,均应计入FH值的计算,所以,FH值的计算是一个倒扣的船形集合,即使低于药典正常标准,仍能达到FH值大于1000,但这计算起来有点麻烦.从资料报道,国外生产能满足灭菌去热原要求,其FH≥1000。
根据中国2000版药典的要求和公式计算,认为空瓶FH≥ 1400为宜。
USP,EP、中国药典干热灭菌条件对照表1
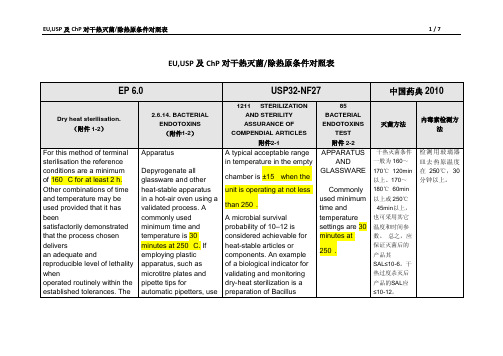
EU,USP及ChP对干热灭菌/除热原条件对照表附件1 -1EP6.0 5.1.1. METHODS OF PREPARATIONOF STERILE PRODUCTS)Dry heat sterilisation. For this method of terminal sterilisation the reference conditions are a minimumof 160 °C for at least 2 h. Other combinations of timeand temperature may be used provided that it has beensatisfactorily demonstrated that the process chosen deliversan adequate and reproducible level of lethality whenoperated routinely within the established tolerances. Theprocedures and precautions employed are such as to give anSAL of 10− 6 or better.Dry heat sterilisation is carried out in an oven equipped withforced air circulation or other equipment specially designedfor the purpose. The steriliser is loaded in such a way thata uniform temperature is achieved throughout the load.Knowledge of the temperature within the steriliser duringthe sterilisation procedure is usually obtained by means oftemperature-sensing elements inserted into representativecontainers together with additional elements at thepreviously established coolest part of the loaded steriliser.The temperature throughout each cycle is suitably recorded.Where a biological assessment is carried out, this is obtainedusing a suitable biological indicator (5.1.2).Dry heat at temperatures greater than 220 °C is frequentlyused for sterilisation and depyrogenation of glassware. Inthis case demonstration of a 3-log reduction in heat resistantendotoxin can be used as a replacement for biologicalindicators (5.1.2)附件1-2 2.6.14. BACTERIAL ENDOTOXINSThe test for bacterial endotoxins is used to detect orquantify endotoxins of gram-negative bacterial originusing amoebocyte lysate from horseshoe crab (Limuluspolyphemus or Tachypleus tridentatus). There are3 techniques for this test : the gel-clot technique, whichis based on gel formation ; the turbidimetric technique,based on the development of turbidity after cleavage of anendogenous substrate ; and the chromogenic technique,based on the development of colour after cleavage of asynthetic peptide-chromogen complex.The following 6 methods are described in the presentchapter :Method A. Gel-clot method: limit testMethod B. Gel-clot method: semi-quantitative testMethod C. Turbidimetric kinetic method182 See the information section on general monographs (cover pages) EUROPEAN PHARMACOPOEIA 6.0 2.6.14. Bacterial endotoxinsMethod D. Chromogenic kinetic methodMethod E. Chromogenic end-point methodMethod F. Turbidimetric end-point methodProceed by any of the 6 methods for the test. In the eventof doubt or dispute, the final decision is made based uponmethod A unless otherwise indicated in the monograph.The test is carried out in a manner that avoids endotoxincontamination.ApparatusDepyrogenate all glassware and other heat-stable apparatusin a hot-air oven using a validated process. A commonly usedminimum time and temperature is 30 minutes at 250 °C. Ifemploying plastic apparatus, such as microtitre plates andpipette tips for automatic pipetters, use apparatus shownto be free of detectable endotoxin and of interfering effectsfor the test.NOTE: In this chapter, the term ‘tube’ includes all types ofreceptacles, for example microtitre plate wells.indicators (5.1.2).附件2-11211STERILIZATION AND STERILITY ASSURANCE OF COMPENDIAL ARTICLESThis informational chapter provides a general description of the concepts and principles involved in the quality control of articles that must be sterile. Any modifications of or variations in sterility test procedures from those described under Sterility Tests 71should be validated in the context of the entire sterility assurance program and are not intended to be methods alternative to those described in that chapter.Within the strictest definition of sterility, a specimen would be deemed sterile only when there is complete absence of viable microorganisms from it. However, this absolute definition cannot currently be applied to an entire lot of finished compendial articles because of limitations in testing. Absolute sterility cannot be practically demonstrated without complete destruction of every finished article. The sterility of a lot purported to be sterile is therefore definedin probabilistic terms, where the likelihood of a contaminated unit or article is acceptably remote. Such a state of sterility assurance can be established only through the use of adequate sterilization cycles and subsequent asepticprocessing, if any, under appropriate current good manufacturing practice, and not by reliance solely on sterility testing. The basic principles for validation and certification of a sterilizing process are enumerated as follows:1. Establish that the process equipment has capability of operating within therequired parameters.2. Demonstrate that the critical control equipment and instrumentation arecapable of operating within the prescribed parameters for the processequipment.3. Perform replicate cycles representing the required operational range of theequipment and employing actual or simulated product. Demonstrate that the processes have been carried out within the prescribed protocol limits andfinally that the probability of microbial survival in the replicate processescompleted is not greater than the prescribed limits.4. Monitor the validated process during routine operation. Periodically as needed,requalify and recertify the equipment.5. Complete the protocols, and document steps (1) through (4) above.METHODS OF STERILIZATIONIn this informational chapter, five methods of terminal sterilization, including removal of microorganisms by filtration and guidelines for aseptic processing, are described. Modern technological developments, however, have led to the use of additional procedures. These include blow-molding (at hightemperatures), forms of moist heat other than saturated steam and UVirradiation, as well as on-line continuous filling in aseptic processing. Thechoice of the appropriate process for a given dosage form or componentrequires a high level of knowledge of sterilization techniques and information concerning any effects of the process on the material being sterilized.1Dry-Heat SterilizationThe process of thermal sterilization of Pharmacopeial articles by dry heat is usually carried out by a batch process in an oven designed expressly for that purpose. A modern oven is supplied with heated, filtered air, distributeduniformly throughout the chamber by convection or radiation and employing a blower system with devices for sensing, monitoring, and controlling the critical parameters. The validation of a dry-heat sterilization facility is carried out in a manner similar to that for a steam sterilizer described earlier. Where the unit is employed for sterilizing components such as containers intended forintravenous solutions, care should be taken to avoid accumulation ofparticulate matter in the chamber. A typical acceptable range in temperature in the empty chamber is ±15when the unit is operating at not less than 250.In addition to the batch process described above, a continuous process is frequently employed to sterilize and depyrogenate glassware as part of an integrated continuous aseptic filling and sealing system. Heat distribution may be by convection or by direct transfer of heat from an open flame. The continuous system usually requires a much higher temperature than cited above for the batch process because of a much shorter dwell time. However, the total temperature input during the passage of the product should be equivalent to that achieved during the chamber process. The continuous process also usually necessitates a rapid cooling stage prior to the aseptic filling operation. In the qualification and validation program, in view of the short dwell time, parameters for uniformity of the temperature, and particularly the dwell time, should be established.A microbial survival probability of 10–12 is considered achievable forheat-stable articles or components. An example of a biological indicator for validating and monitoring dry-heat sterilization is a preparation of Bacillus subtilis spores. Since dry heat is frequently employed to render glassware or containers free from pyrogens as well as viable microbes, a pyrogen challenge, where necessary, should be an integral part of the validation program, e.g., by inoculating one or more of the articles to be treated with 1000 or more USP Units of bacterial endotoxin. The test with Limulus lysate could be used to demonstrate that the endotoxic substance has been inactivated to not more than 1/1000 of the original amount (3 log cycle reduction). For the test to be valid, both the original amount and, after acceptable inactivation, the remaining amount of endotoxin should be measured. For additional information on the endotoxin assay, see Bacterial Endotoxins Test 85.附件2-2 85BACTERIAL ENDOTOXINS TESTAPPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validated process.2Commonly used minimum time and temperature settings are 30 minutes at 250. If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, use only that which has been shown to be free of detectable endotoxin and not to interfere with the test. [note—In this chapter, the term “tube” includes any other receptacle such as a micro-titer well.]。
湿热灭菌和干热灭菌的原理

湿热灭菌和干热灭菌的原理
湿热灭菌和干热灭菌分别是通过湿热和干热的条件来灭菌的方法。
它们的原理如下:
湿热灭菌的原理:湿热灭菌是利用高温和一定的湿度,通过破坏菌体的蛋白质和核酸结构,达到杀灭细菌的作用。
具体过程包括:
1. 温度:通常在121°C下进行湿热灭菌,这是因为该温度下
的蒸汽压能够达到大气压,保证必要的温度和压力条件。
2. 压力:高压蒸汽可以提高水的沸点,以达到高温的目的。
高压蒸汽在灭菌器中产生,维持一定的压力和温度。
3. 时间:菌体灭活需要一定的时间,通常持续灭菌时间为15-30分钟。
干热灭菌的原理:干热灭菌是通过高温和干燥的条件,使细菌失去生活能力。
具体过程包括:
1. 温度:通常在160-180°C下进行干热灭菌,这是由于高温下
的细胞内的蛋白质凝固,细胞壁和细胞膜破裂,从而灭活细菌。
2. 时间:由于干热灭菌需要更高的温度,灭菌时间较长,通常为1-2小时。
3. 干燥:在干热灭菌过程中,干燥的环境有助于杀灭细菌,因为水分对于细胞的正常功能至关重要。
通过低水分环境,细菌细胞失去水分,无法正常生长和复制。
总的来说,湿热灭菌和干热灭菌通过高温破坏细菌的蛋白质、
核酸结构和细胞膜等,达到灭菌的目的。
湿热灭菌利用高温和一定湿度,干热灭菌则是通过高温和干燥条件。
湿热灭菌和干热灭菌相比干热灭菌穿透力强传导快对吗

湿热灭菌和干热灭菌相比干热灭菌穿透力强传导快对吗
热灭菌与湿热灭菌的比较干热湿热对物品影响适用对象作用温度作用时间杀菌能力空气烤焦金属玻璃与其他不畏焦化物品高(160-400℃)长(1-5小时)较差水和蒸汽濡湿(皮革损坏)棉织品水液等不畏湿热物品低(60-134℃)短(4-60分钟)较强湿热与干热各有特点,互相很难完全取代,但总的说来,湿热的消毒效果较干热好,所以使用也普遍。
湿热较干热消毒效果好的原因有三;①蛋白质在含水多时易变性,含水量多,愈易凝固;②湿热穿透力强,传导快。
③蒸汽具有潜热,当蒸汽与被灭菌的物品接触时,可凝结成水而放出潜热,使湿度迅速升高,加强灭菌效果。
第1页共1页。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
干热灭菌相关温度与时
间一览表
标准化管理处编码[BBX968T-XBB8968-NNJ668-MM9N]
对开门百级净化热风循环风箱:
1、通常采用250℃,时间不低于45分钟;
2、在250度时,1min ,FH=;
3、那么45分钟,其FH值就是*45=;
4、那么不是大于基本要求FH等于1000吗?
5、2000年版药典就是不低于30分钟(FH=*30=),那么为什么会在2005年版做如此修改?
6、是因为这里的250度是腔体内温度,不是物品内部或表面温度,也不是最低点温度,所以将其时间提高15分钟!所谓:“温度不够时间补,补的是时间”
隧道烘箱:
1、通常采用320度,时间不低于4分钟;
2、其在300度(公认温度,低于通常,真是!),1分钟,FH=,
3、那么4分钟,FH值就是*4=1022;
4、那么为什么会通常用320度呢原因同上,瓶底温度在320度时,瓶内温度才会达到300度."要提高产量,得提高温度,温度不均匀更要提高温度,补的是温度“
一补时间,一补温度,中国人真是大补呀!原因不说,乱补一气,反正设备不会说话,而我们有的不是效率,而是时间。
干热灭菌FH值计算
SDA不知为何FH大于1400.准确地说是1364.
干热标准温度为170C,Z为54.
F(T)=10^((T-170)/Z)
F(320)=10^((320-170)/54)=600,也就是说:320C 1分钟相当于标准温度(170C)600分钟320C,10MIN,F(H)=6000(5995).
隧道烘箱设定320度,但不能全程达到320C,我们公司的是5分钟.
以我做过的经验.,F(H)在2000到3000之间.为什么呢正常的计算是要把长升温100度开始与降温至100度结束,均应计入FH值的计算,所以,FH值的计算是一个倒扣的船形集合,即使低于药典正常标准,仍能达到FH值大于1000,但这计算起来有点麻烦.
从资料报道,国外生产能满足灭菌去热原要求,其FH≥1000。
根据中国2000版药典的要求和公式计算,认为空瓶FH≥ 1400为宜。
通过计算:当FH≥1400, 安瓿在300℃时的暴露(灭菌)时间必须大于。
因此,对连续式隧道灭菌烘箱的灭菌程序为:300℃时, 大于4min即能满足工艺要求。
温度设备验证合格判断标准
摘自《GMP验证指南2003》
干热灭菌
评价干热灭菌过程的相对灭菌能力的F0值是将时间与温度条件的改变折算成170℃时的相当时间,同时设定Z值为20℃即FH值。
BP1993年版规定仅以灭菌为最终目的的干热灭菌系统,必须保证其最小的FH值大于 170℃60min。
干热除热原一般在连续法中(如隧道灭菌除热原系统)采用的温度常常高于或等于300℃,在间歇法中(如干热灭菌柜)采用的温度常常高于或等于220℃,但必须保证其暴露实际温度和时间相当于250℃超过
30min。
(GMP验证指南2003 P248)
一般情况下,腔室内各点的温度值均不应低于设备自身控制系统的温度值。
美国药典USPXXIII指出,对于运行温度高于250℃的干热灭菌、除热原系统而言其灭菌腔室内空载热分布的可接受的合格范围为±15℃。
(GMP验证指南2003 P253)
灭菌程序赋予被灭菌品标准干热时间FH≥30,灭菌温度系数Z值在干热灭菌灭菌时取值为20℃,在去除热原时,Z值取值为54℃(GMP验证指南2003 P319)
对于验证方案,由于各个公司的要求不一样,因此方案也有很大不同,有的只是为了看一看灭菌柜的温度大致情况,做到心中有数就行了,有的要和国际接轨,验证按国家GMP或美国FDA标准严格验证,一些外资企业光做空载热分布就做二十多个循环,因为不同的探头码放方式可能有不同的冷点,而满载实验更复杂,不同的装载量对冷点的影响是很大的。
验证方案应包括被验证设备生产厂家,设备号、型号;验证设备的名称、型号、证书(NIST精度可追溯)、校正设备等,探头布置图,验证总结,操作主管等相关人员。
湿热灭菌
无菌粉针湿热灭菌系统的验证:无菌分装用的胶塞、设备零部件、工具、容器、及无菌服等一般都是经过高压灭菌器灭菌后转移到无菌生产区使用的。
标准:在最大负载状态下,热穿透试验的结果达到最冷点灭菌物品的暴露时间为121℃
≥15min,即F0≥15。
(P255-266)
建议灭菌程序:
无菌工作服 121℃ 35min
T形胶塞、铝盖 121℃ 40min
可拆洗的大容量注射剂及小针配制罐等 121℃ 40min
过滤器 121℃ 40min
(P354)
热分布试验应用至少10支或10支以上经过校正的标准热电偶在空载状态下连续进行3次或3次以上试验,以证明空载灭菌器腔室内各点(包括最冷点)的温度在每次灭菌程序运行过程中的差值≤±1℃.(P256)
当产品达到灭菌温度直至冷却开始的过程中,温度变化应当保持在±0.5℃以内。
对实验数据作统计分析,最冷点和腔室平均温度间差值应不超过±2.5℃,如果超过说明设备性能太差或存在某种故障。
有好的热分布状态,冷点的F0值和产品F0平均值之间的差值不超过(P352-354)
温度干井的加热温度分别设定为:灭菌温度-10℃、灭菌温度、灭菌温度+10℃,测量记录的时间间隔为10S。
热分布均匀性,灭菌工艺规定的阶段时间是对瓶内药业进行升温、保温、降温的整个灭菌过程中,在灭菌柜内部任何一点的温度都应达到工艺规定的温度。
特别是恒温阶段,温差应≤0.5℃.(药品GMP验证教程P273)
FH值是指干热灭菌中的杀菌热力强度(分),是参照基准温度T0=170℃下的标准干热灭菌时间。
FH的计算公式如下:
FH= 计算灭菌时,Z取20℃;除热源,Z取54℃。