上市后临床跟踪管理程序
上市后监督程序

上市后监督程序版本号:A0生效日期:2020年11月09日页码:第1页,共4页修订记录日期2020.11.09版本A0申请单号修改记录新版发行修改人/审批会签记录部门总经理会签人部门管理者代表技术部生产部品质部批准:会签人人力资源部会签采购部销售部文件形成编制:分发范围与受控记录总经理管理者代表人力资源分发受控部技术部生产部品质范围:记录:部采购部销售部1.目的:上市后监视程序文件编号:QP-35版本号:A0生效日期:2020年11月09日页码:第2页,共4页为获得、评审、分析及跟踪产品上市后阶段的经验特制定本程序。
2.范围:适用于本公司所有产品上市后的监管。
3.定义:上市后监视(PMS):医疗东西上市后监视是对投放市场的产品获取质量、安全或机能等信息的活动。
CAPA:纠正和预防措施。
4.职责:品质部负责履行此程序。
5.程序:5.1上市后监督系统是本公司质量体系的一部分,用于获取并评审上市后信息经验。
初本文件外,上市后监督系统还包括《顾客信息反馈控制程序》、《警戒系统控制程序》及《纠正和预防措施控制程序》等文件。
5.2上市后监督系统基于从厂外收集的信息(如顾客抱怨、销售人员的反馈、主管当局的报告、相关文献等)并对其进行分析。
5.3上市后监视可能获取的信息:有一些材料和反馈信息可以从上市后监视体系中获得。
下列信息可能无法所有提供,按照实践情况决定其优先性。
1)生产中发觉的问题;2)产品质量改进;3)风险分析(或其他)切实其实认;4)对长期性能/可靠性和/或慢性适应性的认识;5)对性能改变趋势的认识;6)对不同使用人群中的性能差异的认识;7)用途的反馈;8)使用说明书的反馈;9)使用者培训需求的反馈;10)同其他器械配合使用的反馈;11)顾客满意度的反馈;12)医疗器械警戒报告;上市后监视程序13)对器械无用方式的认识;14)市场持续发展能力的反馈。
文件编号:QP-35版本号:A0生效日期:2020年11月09日1)专家组;2)顾客调查;3)顾客抱怨和保证声明;4)预览文献;5)间接或通过销售人员反馈到公司的抱怨之外的用户反馈;6)东西追踪的登记;7)培训进程当中使用者的反馈;8)其它团体(例如主管当局);9)媒体;10)本公司或竞争对手的类似器械的经验;11)内部测试;12)失效模式分析。
药品上市后研究流程和要点

药品上市后研究流程和要点药品上市后的研究流程和要点通常包括以下几个阶段:1. 药品安全性监测:药品上市后需要持续进行安全性监测,收集和评估药品使用过程中可能出现的不良反应和不良事件。
2. 药品疗效评价:药品上市后需要进一步评价其疗效和临床应用效果。
这包括进行临床研究,收集科学、系统的临床数据来评价药物的疗效和安全性,以确认其在实际临床应用中的价值。
3. 药品副作用评估:研究人员需要评估药品可能的副作用和不良反应,并进行安全性评估。
这可以通过临床试验和自发报告事件来获得。
4. 药品的效应监控:药品上市后需要建立系统的监控措施,监测药品的效果和效应。
这包括对药品的疗效、安全性和用药策略等方面的监测和评估。
5. 药品信息更新:药品上市后需要不断更新药品的信息,包括药物说明书、警示信息等,以提供给医生、患者和其他相关人员参考。
在进行药品上市后研究时,需要注意以下几个要点:1. 临床试验的设计:药品上市后需要进行科学合理的临床试验,以评估其疗效和安全性。
试验的设计应当严谨,并符合相关的法规和指导意见。
2. 临床试验的监管:药品上市后的临床试验需要获得监管机构的批准和监督。
研究人员需要遵守相关的法规和规定,保证试验的科学性和道德性。
3. 安全性监测措施:药品上市后需要建立有效的安全性监测措施,收集和评估药品使用过程中的不良反应和不良事件,并及时采取措施解决可能的安全问题。
4. 药品信息披露:药品上市后需要及时披露药品的相关信息,如副作用、禁忌症等,以提高医生和患者的知情权和选择权。
5. 综合评价和决策:基于药品上市后的研究结果和监测数据,监管机构需要对药品的疗效和安全性进行综合评价,并作出相应的决策,如修订药物说明书、增加警示等。
同时,医生和患者也需要根据这些数据和评价结果进行合理用药决策。
CE上市后临床跟踪控制程序

CE上市后临床跟踪控制程序编制人/日期:审核人/日期:批准人/日期:修订页目录1.目的 (4)2.范围 (4)3.职责 (4)4.程序 (4)5.术语及定义 (4)6.相关文件 (6)7.相关记录 (6)1.目的根据MDR中附录XIV、MEDDEV 2.12-2第2版《医疗器械上市后临床跟踪研究指南》的要求,规范公司的带CE标识的医疗器械是否进行上市后临床跟踪研究以及如何进行研究,特拟定本文件。
2.范围适用于本公司医疗器械产品的CE上市后临床跟踪的制定与管理。
3.职责3.1总经理负责CE上市后临床跟踪(PMCF)计划及评估报告的批准。
3.2管理者代表负责CE上市后临床跟踪(PMCF)计划及评估报告的审核。
3.3研发部及临床专家负责CE上市后临床跟踪(PMCF)计划及上市后临床跟踪评估报告的编制。
3.4质管部负责CE上市后临床跟踪(PMCF)计划及报告的归档管理,及有关法规的更新。
3.5各相关部门按分配执行。
4.术语及定义(来源于MEDDEV 2.12-2第2版)上市后临床跟踪研究(Post Market Clinical Follow-Up Study):在医疗器械获得CE标记后进行的研究,旨在回答与根据其批准的标签适用时,器械的临床安全性或性能(即残余风险)相关的具体问题5.程序5.1CE上市后临床跟踪(PMCF)的基本要求:上市后临床跟踪(PMCF)是更新临床评价报告的持续过程,并应加入制造商CE上市后监督计划(Post-market Surveillance plan)中,详见《CE上市后监督控制程序》。
上市后临床跟踪(PMCF)的执行应当遵循PMCF计划中的规定并记录的方法。
5.2可以进行上市后临床跟踪研究的情况包括:-创新,例如器械的设计、材料、物质、操作原理、技术或医学适应症是新颖的;-已经完成了上市前临床评估和重新认证的产品发生产品或其预期用途重大变化时;-与产品相关的高风险,如产品的设计、材料、零部件、侵入性及临床程序相关的风险;-高风险的解剖位置;-高风险目标人群,如儿科、老人;-疾病的严重程度/治疗的难易程度;-概括临床调查结果的能力问题;-长期未解决安全性和性能方面的问题;-以往临床调查的结果,包括不良事件或上市后监督活动;-对以前未研究的可能影响利益/风险比例子群的识别,如在不同组群的髋关节植入物;-在上市前合理的随访时间尺度与产品预期寿命之间存在差异情况下的继续验证;-来自文献或其他类似市场器械的数据来源的风险;-与其他医疗产品或治疗的相互作用;-暴露于更大量和更多样化人群的临床使用者时,对器械的安全性和性能进行验证;-出现与设备的安全性或其他性能相关的新信息;-获得CE标志是基于等同性证明的基础上的。
医疗技术临床应用追踪管理制度

医疗新技术临床应用追踪管理制度为进一步规范医院医疗技术临床应用和完善新技术的准入、评估,保障医疗安全,提高医疗质量和医疗技术水平,根据卫生部《医疗技术临床应用管理办法》(卫医政发[2009第18号])结合我院的实际,特制定本制度。
第一章医疗新技术临床应用追踪管理第一条医教科作为主管部门,对于全院的医疗新技术临床应用进行全程管理和评价,制定医院新技术新项目管理档案。
对新技术实施过程中存在的问题进行分析,并提出指导性建议或意见,及时发现医疗技术风险,并敦促相关科室及时采取相应措施,以避免医疗技术风险或将其降到最低限度。
第二条医疗新技术实施过程中,各级人员必须严格执行技术规范、操作规程及各项规章制度,服从科室管理。
科主任、项目负责人应认真组织、严格把关、定期进行质量监控,检查实施情况,及时发现各种问题并予以有效的解决。
第三条在新技术新项目临床应用过程中,应充分尊重患者的知情权和选择权,并注意保护患者安全,及时履行告知义务。
主管医师应向患者或其委托人详细交待病情,重点交待新技术对于患者的适应性、效益性和可能存在的风险及费用情况,尊重患者及委托人意见,在征得其同意并在《知情同意书》上签字后方可实施。
第四条各科室在开展新技术临床应用过程中做好应用记录和总结分析工作,完善疗效的评价分析,应当(1)认真记录病历资料,随访观察疗效;(2)定期总结病历,每年对新技术实施情况进行评估,填写《新技术、新项目开展情况追踪登记表》,《追踪登记表》中详述开展例数、疗效、经济及社会效益、质量评价等;(3)年终将本年度开展的新技术病例进行分析总结上报,医教科针对汇总情况进行有重点的抽查核实。
第五条经医院评估,符合先进性、安全性等要求的技术项目鼓励继续开展,不符合先进性、安全性等要求的技术项目,医教科根据评估结论决定该技术院内停止使用。
第二章医疗新技术临床应用的暂停、评估与停用、复用第六条医疗新技术应用过程中,出现不良后果或技术问题时,有关人员必须采用措施保证医疗安全并及时向科主任、项目负责人报告。
医疗器械GMP程序文件-上市后临床跟踪控制程序
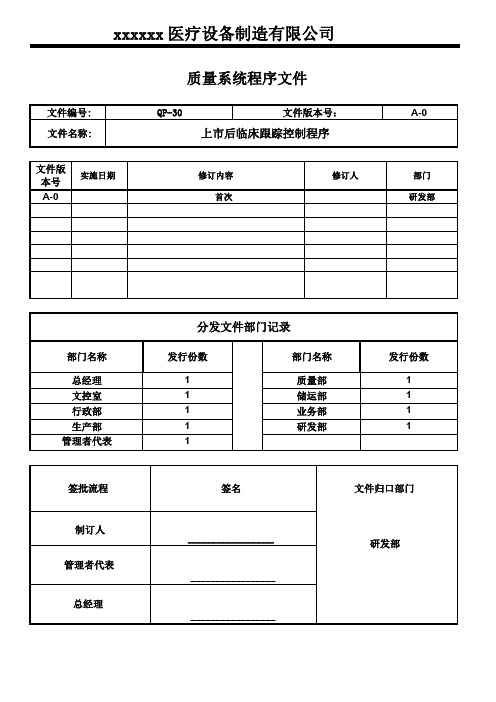
xxxxxx医疗设备制造有限公司质量系统程序文件文件编号:QP-30文件版本号:A-0文件名称:上市后临床跟踪控制程序文件版实施日期修订内容修订人部门本号A-0首次研发部分发文件部门记录部门名称发行份数部门名称发行份数总经理1质量部1文控室1储运部1行政部1业务部1生产部1研发部1管理者代表1签批流程签名文件归口部门制订人_________________研发部管理者代表_________________总经理_________________上市后临床跟踪控制程序发布日期页码第1页共4页1.目的识别和调查与使用投放市场的医疗器械有关的剩余风险,通过系统化的上市后临床随访研究(PMCF)进行调查和评估,确保其投放市场后的器械的长期安全性和性能2.范围本程序规定对上市后临床跟踪研究进行汇编的职责、工作程序、内容和要求。
本程序适用于采用CE标志有关产品上市后临床跟踪研究3.职责3.1研发部负责上市后临床跟踪计划的汇编工作;3.2研发部收集汇总上市后临床数据、进行上市后临床数据评估、维护上市后临床数据库及编制评审上市后临床报告;3.3质量部参与上市后临床跟踪报告评审,组织事故分析评审。
4.工作程序4.1定义:4.1.1临床评价:与医疗器械有关并用来验证器械根据制造商预期使用的临床安全和性能的临床资料的评估和分析。
4.1.2临床数据:临床数据是由医疗器械的使用生成的安全和/或性能信息。
4.1.3临床证据:与医疗器械有关的临床资料和临床评价报告。
4.1.4临床调查:在一个或多个人体受试者上进行的任何系统调查或研究,用于评估医疗器械的安全和/或性能。
4.1.5上市后临床随访(PMCF)的研究:在器械获得CE标识之后的一项研究,用以回答按照已批准的标签使用的器械的临床安全性或性能(即剩余风险)的具体问题。
4.1.6PMCF计划:对于符合93/42/EEC指令,加贴CE标识的医疗器械投放市场后,由制造商设立的文件化的,前瞻性的,组织化的方法和步骤收集其临床资料,目标是在整个医疗器上市后临床跟踪控制程序发布日期页码第2页共4页械的预期寿命期间,被识别风险的可接受性下,确认临床效果和安全性,在事实证据的基础上,探测新出现的风险。
产品上市后跟踪管理程序(ISO13485)

产品上市后跟踪管理程序(ISO13485)1.0⽬的为了识别和调查本公司的医疗器械产品上市销售后的医疗器械在使⽤过程中所具有的风险,特制定本程序。
2.0范围适⽤于本公司所有已经上市销售的医疗器械产品的上市后的临床监督。
3.0依据根据MEDDEV 2.12-2 进⾏。
4.0权责管理者代表:负责组织制定上市临床跟踪的计划及组织实施。
总经理:负责批准上市后临床跟踪的计划及为上市后临床跟踪提供保障和资源。
研发部、品质部、业务部:配合进⾏产品上市后临床跟踪计划及措施的实施。
5.0程序5.1 上市后临床跟踪策划:5.1.1针对公司的每⼀种上市后的医疗器械产品都应该策划,由公司管理者代表组织制订。
5.1.2在制订上市后临床跟踪计划时可以通过以下⽅式或渠道进⾏:对所有收到的投诉及不良事件的数据,系统的进⾏评审。
对制造商和发布的⽂献提供的所有信息来源进⾏评估。
对上市后的性能进⾏监测。
对上市前试验的病⼈的预期跟踪观察在器械已投放市场后,对⼀群有代表性病⼈进⾏前瞻性研究的⽅式进⾏对患者或使⽤者进⾏开放式的调查。
5.1.3这个计划需要考虑以下内容:临床调查的结果,包括识别的不良事件。
器械的平均预期寿命。
器械制造商的声明。
声明的等同性能。
适⽤的新信息。
当实施上市后临床跟踪时,须依据产品的⽤户⼿册,执⾏预产品预期暗⽰的⽤途。
对上市后的临床研究必须考虑到相应国家的法规。
5.1.4 上市后临床跟踪计划制定后由总经理负责批准。
5.2 上市后临床跟踪的实施:在上市后临床跟踪计划被批准后,由管理者代表根据划的内容组织相关⼈员进⾏按计划实施。
当实施PMCF时,须依据产品的⽤户⼿册,执⾏预产品预期暗⽰的⽤途。
对上市后的临床研究必须考虑到相应国家的法规。
在跟踪期间,应考虑到产品暗⽰的平均预期寿命,本公司各医疗器械产品的使⽤寿命具体见产品的设计⽂件或说明书。
5.3 信息评价:管理者代表应组织相关⼈员对收集到的信息进⾏及时评价及定期总结评价,并应保持评价的结果。
MEDDEV2.122上市后临床跟踪指南中英文对照版

EUROPEAN COMMISSIONDG ENTERPRISEDirectorate GGUIDELINES ON POST MARKET CLINICAL FOLLOW-UP上市后临床跟踪指南The present Guidelines are part of a set of Guidelines relating to questions of application of EC-Directives on medical devices. They are legally not binding. The Guidelines have been carefully drafted through a process of intensive consultation of the various interested parties (competent authorities, Commission services, industries, other interested parties) during which intermediate drafts were circulated and comments were taken up in the document. Therefore, this document reflects positions taken by representatives of interested parties in the medical devices sector.本准则是一个有关的欧共体指令对医疗设备的应用问题指引的一部分。
他们在法律上没有约束力。
该指引已审慎草拟通过各有关方面(主管机关,委员会的服务,工业,其他有关各方)在此期间,中间草案分发和评论的文件采取了密集的磋商进程。
因此,这份文件反映了有关各方的代表在该领域采取的医疗设备的位置。
医药产品上市工作流程梳理

医药产品上市工作流程梳理
标题:医药产品上市工作流程梳理
一、研发阶段
1. 研发构思:首先,医药公司根据市场需求和科研趋势,提出新药或改良药物的研发理念。
2. 前期研究:进行实验室研究,包括化合物筛选、药理毒理研究等,以确定候选药物。
3. 临床前试验:在动物身上进行试验,评估药物的安全性和有效性。
二、申请临床试验阶段
1. 注册申请:准备详细的临床试验方案和相关资料,向药品监督管理部门提交临床试验申请。
2. 审评审批:药品监管部门对申请进行审查,批准后方可进行临床试验。
三、临床试验阶段
1. 临床试验I期:主要评估药物的安全性,通常在少量健康志愿者中进行。
2. 临床试验II期:扩大样本量,评估药物的疗效和初步确定剂量。
3. 临床试验III期:在更大规模人群中进行,验证药物的有效性和安全性。
四、上市申请阶段
1. 数据整理:汇总所有临床试验数据,编制药品注册申报资料。
2. 上市申请:向药品监管部门提交新药上市申请。
3. 审评审批:药品监管部门对申报资料进行全面审查,包括质量、疗效、安全性等方面。
五、上市后监测
1. 上市许可:获得批准后,产品可以上市销售。
2. 市场推广:进行药品的市场推广和销售。
3. 后期监测:持续跟踪药品在市场上的使用情况,收集不良反应信息,并定期向药品监管部门报告。
4. 药品再评价:根据市场反馈和新的科研成果,对药品进行再评价,可能需要调整使用说明或进行再研发。
以上就是医药产品从研发到上市的基本流程,每个步骤都需要严格遵守相关法规和标准,确保产品的质量和安全性。
ce.上市后监管控制程序

1.目的依据欧盟MDR EU2017-745法规第Ⅶ章上市后监管、警戒和市场监管第83条,84条,85条的要求,通过定期收集售后有关产品使用后的质量信息,分析可能存在的质量问题,并采取适当的纠正和预防措施。
2.适用范围适用于本公司的产品和同行业同类产品,在国内外售后的信息收集与处理。
3.职责3.1销售部:负责收集国外售后产品的质量信息,并提出报告。
3.2管理者代表:负责收集国内外官方对医疗器械的质量信息和法规要求,并提出报告,负责客户投诉信息的汇总,组织评审各部门提供的质量信息报告,提出处理意见,并组织实施。
3.3质量部:协助本公司范围内的调查。
3.5总经理:负责批准对信息的处理意见。
4.内容4.1建立上市后监督系统4.1.1上市后监督系统的形式应包括:处理投诉和警械系统、顾客反馈管理、用户和患者调查、文献评论、上市后的临床随访等。
4.1.2上市后监督系统可视为对产品可能出现的风险和长期的安全和性能评价。
在识别这种新出现的风险,应考虑到下面的标准:a)该产品采用新的设计,材料,作准则,技术,或者是新的医学特征,严重的疾病;b)敏感目标人群c)已上市类似产品存在的风险d)确认在一个CE前临床评价可接受的风险,应长期和/或监测更多的临床人员。
e)临床使用时间和产品的预期寿命的差别。
4.2上市后监督系统计划4.2.1产品上市后应由市场部负责编制《产品上市后监督计划》。
4.2.2《产品上市后监督计划》可以采取病人后续的观察形式,包括市场前的评审,和/或产品已投放市场后有代表性病人的前瞻性研究。
4.2.3《产品上市后监督计划》将要考虑到以下内容:a)临床调查,包括确定的不良事件结果b)产品的平均预期寿命c)产品制造商声明d)声明的性能e)适当可用的新信息4.3上市后监督系统的实施4.3.1实施上市后监督系统时,必须始终依据产品的预定适用范围内按使用说明书使用。
4.3.2对上市后的临床研究必须考虑到相应国家的法规。
新药上市的临床试验和监管要求

新药上市的临床试验和监管要求随着科学技术的不断发展和医疗需求的增加,新药研发成为一项重要的任务。
然而,新药的研发上市不仅需要经过严谨的临床试验,还需要遵守监管机构的要求。
本文将探讨新药上市的临床试验和监管要求。
一、临床试验临床试验是新药研发过程中的重要环节。
它涉及到人类的安全与健康,因此需要遵循一系列的规定和程序。
临床试验主要包括以下几个阶段:1. 前期研发阶段:在这个阶段,研究人员首先进行动物实验,评估药物的毒性和安全性。
只有在动物实验结果表明药物在一定剂量下没有严重的不良反应后,才能进入下一阶段。
2. 临床试验设计:在设计临床试验时,需要明确试验的目标、方法以及研究对象的选择。
根据药物类型和疾病特点,临床试验可以分为Ⅰ期、Ⅱ期和Ⅲ期。
Ⅰ期试验主要评估药物的耐受性和安全性,Ⅱ期试验主要评估药物的疗效和剂量,Ⅲ期试验则是在大规模病人中进行的,旨在验证药物的治疗效果和安全性。
3. 试验过程和数据分析:在进行临床试验时,需要遵守严格的伦理准则和法律法规。
试验过程中应进行严格的数据记录和分析,确保试验结果的可信性和有效性。
研究人员应及时报告试验中发现的任何不良反应,并根据需要采取相应的措施保护试验对象的权益和安全。
二、监管要求为了保证新药的质量和安全性,各国都设立了相应的监管机构,对新药的上市进行严格监管。
监管主要包括以下几个方面:1. 新药申请:在新药研发完成后,生产企业需要向监管机构提交申请,申请将新药列入上市审批。
申请应包括药物的成分、性质以及临床试验结果等详细信息。
监管机构将根据申请的完整性和准确性决定是否受理。
2. 临床试验数据评估:监管机构在审批过程中将对药物的临床试验数据进行评估。
评估主要关注药物的疗效和安全性,确保药物的治疗效果显著且不会对人体造成严重的不良反应。
评估结果将对是否批准上市起到重要的决定作用。
3. 上市准许和监管后审查:审批通过后,监管机构将发放新药的上市准许。
然而,监管并不止于此。
上市后临床跟踪控制程序

上市后临床跟踪控制程序(ISO13485-2016)1.0 PURPOSEThe purpose of this work instruction is to define the process to determine and document whether a post-market clinical follow-up study is required forTDI Foot/Ankle Array 8ch medical devices bearing the CE mark. The process will lead to a determination of whether a post-market clinical follow-up study is required and provide guidance for post-market clinical monitoring requirements if a study is not required.2.0 SCOPEThe work instruction applies to all medical device businesses and sites operating under the TDI Foot/Ankle Array 8ch Healthcare Quality Management System.Only medical devices bearing the CE Mark will be required to follow this work instruction.3.0 REFERENCES3.1. External References3.1.1. LawsCouncil Directive 93/42/EEC of 14 June 1993 concerning medical devices including amendments through 05 September 20073.1.2.Guidance DocumentsEuropean Commission Enterprise-Directorate-General MEDDEV 2.12-2 Guidelines on Post Market Clinical Follow-Up dated May 2004 MEDDEV 2.7.1 Rev.3 guidelines on medical device-clinical evaluation-a guide for manufacturers and notified bodies dated April 2009GHTF Post-Market Clinical Follow-Up Studies; SG5(PD)N4R7 (Proposed document 23 July 2008)GHTF Clinical Investigations; SG5(PD)N3R7 (20 January 2008)4.0 ROLES AND RESPONSIBILITIESImportant: When a title of a position is listed in this work instruction, it relates to that position or its equivalent.Below are the roles and responsibilities discussed within this document. Table 错误!文档中没有指定样式的文字。
药品上市后研究操作规程

药品上市后研究操作规程药品上市后研究操作规程一、引言药品上市后的研究是为了进一步评估药物的疗效和安全性,提供更为全面的临床实践依据。
本操作规程旨在明确药品上市后研究的组织、实施、监测与管理等相关事项,确保研究的科学性和可信度。
二、研究范围药品上市后研究涵盖以下内容:1. 药物的长期疗效观察;2. 药物在特定人群中的疗效和安全性评价;3. 药物的新适应症、新用法和新用量的疗效和安全性评价;4. 药物的中长期使用风险评估;5. 药物的药效学和药代动力学的进一步研究;6. 药物在特定亚群人群中的疗效和安全性评价等。
三、研究计划和申报1. 研究计划应根据药物的特点和上市后信息反馈形成,明确研究目的、研究设计和方法、研究人群、研究期限等内容。
2. 研究计划应由申请人提出,并提交给国家药监部门进行审批。
3. 研究计划的申报材料应包括研究方案、研究者资格和经验、数据收集和分析的计划等。
四、研究实施和监测1. 研究实施应遵循临床实践指南和相关伦理原则,确保研究的科学性和可靠性。
2. 研究应由具有相关临床经验和研究背景的研究机构或研究者进行,并按照研究计划的要求进行。
3. 研究者应及时收集、记录和整理研究数据,并按照规定的流程进行数据分析和统计。
4. 研究过程中应进行临床监测,包括随访、检查、实验室检验等,以及不良反应和事件的监测和报告。
五、研究结果和报告1. 研究完成后,应由研究者进行数据分析和结果解读,并形成研究报告。
2. 研究报告应包括研究背景和目的、研究设计和方法、参与者和样本规模、数据收集和分析、主要的研究结果和结论等内容。
3. 研究报告应准确、完整地反映研究的实际情况,并应提交给国家药监部门进行评审和审核。
六、研究管理和结果应用1. 国家药监部门应加强对药品上市后研究的管理和监督,确保研究的严谨性和可靠性。
2. 研究结果应及时公布,供临床医生和患者参考和使用。
3. 研究结果应作为指导临床实践和药物使用的重要依据,并纳入相关的临床指南和规范中。
临床研究中的病例追踪流程

临床研究中的病例追踪流程病例追踪是指在临床研究过程中对患者进行长期观察和记录,以获取疾病的发展、治疗效果等相关信息。
病例追踪的流程包括研究计划、患者招募、数据收集和分析等多个环节。
本文将介绍临床研究中的病例追踪流程,并探讨其重要性。
一、研究计划在进行病例追踪研究前,研究者首先需要制定详细的研究计划。
这包括确定研究的目的、研究设计、样本大小计算、数据采集工具等。
研究计划应该详细描述病例追踪的整个过程,确保每个环节都能得到充分考虑和准备。
二、患者招募患者招募是病例追踪研究中的重要步骤。
研究者可以通过多种途径招募患者,如医院门诊、社区健康中心等。
在招募患者时,研究人员应该向患者详细说明研究的目的、方法、风险和利益,并取得他们的知情同意。
三、数据收集数据收集是病例追踪的核心环节。
研究人员需要定期对患者进行随访,并记录相关的临床信息、生化指标、影像学结果等。
为了确保数据的准确性和一致性,研究人员还需要进行培训,统一数据采集的标准和方法。
在进行数据收集时,研究人员还需要保护患者隐私和个人信息的安全。
他们应该遵守伦理规范,确保患者的隐私不受侵犯,并采取必要的措施保护数据的安全性。
四、数据分析数据分析是病例追踪研究的关键步骤。
研究人员需要对收集到的数据进行整理、归类和统计分析。
他们可以使用适当的统计方法,比如生存分析、回归分析等,来研究疾病的发展、治疗效果等因素。
数据分析的结果将为临床研究提供重要的依据和参考。
研究人员应该对分析结果进行科学解读,并撰写相应的研究报告或论文。
五、结果解读和报告根据病例追踪研究的结果,研究人员可以对疾病的发展、治疗效果等进行解读和讨论。
他们可以比较不同治疗方案的优劣,分析影响疾病预后的因素,并提出相应的建议。
最后,研究人员应该将研究结果进行报告。
他们可以通过学术期刊发表论文,或者在学术会议上进行口头报告,与其他研究人员和临床医生分享他们的研究成果。
结论病例追踪在临床研究中具有重要的作用。
欧盟MDR临床评价和上市后临床跟踪

欧盟MDR临床评价和上市后临床跟踪欧盟MDR(Medical Device Regulation)是一项新的医疗器械法规,在2024年5月26日正式实施。
MDR为欧盟成员国监管机构提供了更为严格的要求,旨在确保医疗器械的安全性和有效性,并提高对患者的保护。
其中包括MDR对临床评价和上市后临床跟踪的规定。
首先,MDR对临床评价提出了更为严格的要求。
根据新法规,制造商需要进行一系列的临床评价来证明其产品的安全性和有效性。
临床评价应基于最新的医学和科学知识,并遵循科学方法和伦理原则。
此外,评价还应评估产品与患者的兼容性,以及产品在实际使用中的性能和效果。
临床评价的核心是临床试验,这是一项研究设计,旨在评估医疗器械在患者身上的安全性和有效性。
根据MDR的要求,试验应以符合道德要求的方式进行,并采取必要的措施保护参与者的权益。
此外,试验结果应具有科学和统计学的可信性,并进行详细的数据分析和结果报告。
除了临床评价,MDR还要求制造商进行上市后的临床跟踪。
这意味着一旦产品获得市场准入,制造商需要跟踪产品在实际使用中的性能和安全性,并及时收集和分析相关数据。
制造商还应与患者、医生和其他相关利益相关方进行沟通,以了解产品在实际使用中的效果和问题,并采取相应的改进措施。
总之,欧盟MDR对临床评价和上市后临床跟踪提出了更为严格的要求。
制造商需要进行临床评价以证明产品的安全性和有效性,并使用科学方法和伦理原则进行临床试验。
同时,制造商还需要进行上市后的临床跟踪,及时收集和分析相关数据,并与相关利益相关者进行沟通和合作。
这些规定旨在确保医疗器械的安全性和有效性,提高对患者的保护。
pmcf法规

pmcf法规
PMCF法规是指欧盟的新医疗器械法规(MDR)中对医疗器械的上市后临床跟踪(PMCF)的要求。
PMCF是持续更新的临床评价过程,应在制造商的上市后监督计划中予以设计体现,旨在为器械的临床评价提供最新数据,确保器械获批上市后,其安全性和性能将持续获得监督。
根据MDR 86(1)条规定,PSUR基于PMCF,因此,除I类产品外所有类别的器械都需要PMCF。
器械制造商在设计和运行PMCF研究时,应牢记五个主要目标:在整个预期使用寿命内确认器械的安全性和性能;识别以前未知的副作用并监测已明确的副作用和禁忌症;根据事实证据确定和分析风险;确保MDR附件I(1,9)提到的收益风险比是持续可接受的;识别器械可能的系统误用或标签外使用,以验证器械的预期使用目的是否正确(如果有很多标签外使用,说明之前制定的预期使用可能有误)。
PMCF计划应包括以下信息:上市后临床随访的一般方法和程序;应用PMCF的具体方法和程序,例如评估合适的注册或临床研究;上述方法和程序的适当性的理由;参考临床调查报告和风险管理;上市后临床随访要解决的具体目标;评估与同等或类似设备相关的临床数据;参考任何相关的CS(通用规范),制造商使用的协调标准以及PMCF的相关指南;PMCF 活动的详细且有充分理由的时间表;定期安全更新报告。
需要注意的是,PMCF法规仅是欧盟医疗器械法规的一部分,完整的法规还包括上市后监督计划和定期安全更新报告等要求。
如果你想了解更多关于PMCF法规的信息,可以参考相关的法律法规文件或咨询专业人士。
医疗器械上市后监督管理程序

1、目的适当使用和执行上市后临床跟踪研究,来解决与剩余风险相关的问题,及时修改产品标签,说明书的内容及技术文件,确保产品的持续安全有效,满足欧盟MEDDEV 2.12/2 rev2(《GUIDELINES ON MEDICAL DEVICES:POST MARKET CLINICAL FOLLOW-UP STUDIES A GUIDE FOR MANUFACTURERS AND NOTIFIED BODIES》January 2012发布)的要求。
作为产品质量体系的一部分,一个适当的市场监督程序,对上市产品识别使用中风险识别和风险研究的关键,特制定本程序。
2、范围适用于本公司带有CE标识的医疗器械产品的销售后的临床数据跟踪。
3、职责3.1 管理者代表:负责组织落实产品符合MDD 93/42/EEC 的要求。
3.2 工程部:负责按本程序及相关法规制定《上市后的临床跟踪计划》及相关文件,并根据汇总上市后的临床数据,评价现有文件的适合性并修改相关的文件。
3.3 品管部:负责按本程序及相关法规对文件归档保存,负责产品相关的标签的控制。
3.3 业务部:负责审核《上市后的临床跟踪计划》,并在计划批准后按文件要求执行。
3.4 总经理:负责对《上市后的临床跟踪计划》的批准。
4、定义4.1 临床调查:在一个或更多的人类受试者上进行的任何系统性的调查或研究,从而评估医疗设备的安全性和性能。
4.2 上市后临床跟踪研究:跟随着设备的CE标记而执行的研究,其意图是回答与设备依照批准的标签使用时的临床安全性和性能(如剩余风险)相关的特定问题。
4.3 PMCF计划:制造商建立的备用证明文件的、前瞻性的、有组织的方法和程序,其根据与特定设计档案一致的CE标记产品的使用或属于相同亚类或MDD93/42/EEC指令中定义的一般设备组的一组医疗设备的使用来收集临床数据。
它的目标是在医疗设备的预期生命期中巩固临床表现和安全性和经鉴定的风险的可接受性,以及在事实证据的基础上探测产生的风险。
上市后临床跟踪协议

上市后临床跟踪协议
上市后临床跟踪协议是指在医疗器械或药物上市后,进行后续临床研究以跟踪产品的安全性和有效性的协议。
该协议用于了解产品在实际临床使用中的表现,以确保产品的长期安全性和有效性,并提供进一步的临床数据支持。
上市后临床跟踪协议通常由制造商或申请者与监管机构(如药品监管部门)共同制定,其中包括具体的研究目标、方法、样本规模、研究时间、数据收集与分析等内容。
协议还明确了责任分工和监控机制,以监督和评估后续临床研究的进行。
上市后临床跟踪协议的目的是提供可靠的临床数据,以便评估产品的长期安全性和有效性,并及时发现任何安全性问题或不良事件。
这些数据可以用于更新产品标签,指导医生和患者的使用,以及支持后续的临床决策和监管活动。
上市后临床跟踪协议在药物和医疗器械领域都是常见的做法。
对于一些高风险或新型技术的产品,其上市后临床跟踪协议可能会更加详细和严格,以确保产品的安全性和有效性。
需要注意的是,上市后临床跟踪协议是一项长期的工作,需要耗费大量时间和资源。
然而,通过对产品进行长期的跟踪研究,可以提供对产品性能的更全面和可靠的评估,增强人们对产品的信任和安全性的保障。
CE认证上市后临床跟踪指南

CE认证上市后临床跟踪指南EUROPEAN COMMISSIONDG ENTERPRISEDirectorate GUnit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES: Guidance document MEDDEV 2.12-2May 2004GUIDELINES ON POST MARKET CLINICAL FOLLOW-UP上市后临床跟踪指南The present Guidelines are part of a set of Guidelines relatin g to questions of application of EC-Directives on medical devices. They are legally not binding. The Guidelines have been carefully drafted through a process of inte nsive consultation of the various interested parties (competent authorities, Commission services, industries, other interested pa rties) during which intermediate drafts were circulated and com ments were taken up in the document. Therefore, this documen t reflects positions taken by representatives of interested parties in the medical devices sector.本准则是一个有关的欧共体指令对医疗设备的应用问题指引的一部分。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
上市后临床跟踪控制程序文件编号: QP-29版本:A/0生效日期:页码: 19编制:审核:批准:1.PURPOSEThe purpose of this work instruction is to define the process to determine and document whether a post-market clinical follow-up study is required forTDI Foot/Ankle Array 8ch medical devices bearing the CE mark. The process will lead to a determination of whether a post-market clinical follow-up study is required and provide guidance for post-market clinical monitoring requirements if a study is not required.2.SCOPEThe work instruction applies to all medical device businesses and sites operating under the TDI Foot/Ankle Array 8ch Healthcare QualityManagement System.Only medical devices bearing the CE Mark will be required to follow this work instruction.3.REFERENCES3.1.External ReferenceswsCouncil Directive 93/42/EEC of 14 June 1993 concerning medical devicesincluding amendments through 05 September 20073.1.2.Guidance DocumentsEuropean Commission Enterprise-Directorate-General MEDDEVGuidelines on Post Market Clinical Follow-Up dated May 2004MEDDEV guidelines on medical device-clinical evaluation-a guidefor manufacturers and notified bodies dated April 2009GHTF Post-Market Clinical Follow-Up Studies; SG5(PD)N4R7 (Proposeddocument 23 July 2008)GHTF Clinical Investigations; SG5(PD)N3R7 (20 January 2008)4.ROLES AND RESPONSIBILITIESImportant:When a title of a position is listed in this work instruction,it relates to that position or its equivalent.Below are the roles and responsibilities discussed within this document. Table 4-1: Roles and ResponsibilitiesProvide consultation to the Product Regulatory AffairsRepresentative in determining for a given project/productwhether a post-market clinical follow-up study is requiredProvide consultation to the Product Regulatory AffairsRepresentative to determine if an equivalent device existsProvide consultation to the Product Regulatory AffairsRepresentative in identifying emerging risks for the medicaldeviceProvide consultation to the Research Manager or designee todetermine the type of post-market clinical follow-up study tobe implemented, if applicableDetermine for a give project/product whether a post-marketclinical follow-up study is requiredDetermine if an equivalent device existsIdentify potential emerging risksReview risk assessmentComplete the Post-Market Clinical Follow-Up Justification Formregarding decision to perform a studyComplete the Post-Market Clinical Follow-Up Plan form thatdetails the post-market clinical follow-up planDetermine how often clinical data must be reviewedReview and approve the clinical evaluation performed by theResearch Manager or designeeProvide consultation to the Research Manager to determine thetype of post-market clinical follow-up study to be implemented,if applicableTable 4-1: Roles and Responsibilities Role ResponsibilityResearch Manager or designee Provide consultation to the Product Regulatory Affairs Representative in determining for a given project/product whether a post-market clinical follow-up study is required Provide consultation to the Product Regulatory Affairs Representative to determine if an equivalent device exists Provide consultation to the Product Regulatory Affairs Representative to identify potential emerging risksReview the Post-Market Clinical Follow-Up Justification form and Post-Market Clinical Follow-Up Plan form to confirm the decisions regarding the need for a post-market clinical follow-up study and clinical follow-upDetermine how often clinical data must be reviewedDetermine the type of post-market clinical follow-up study to be implemented, if applicableReview new data . literature, adverse events, complaints, etc,) and determine if a post-market clinical follow-up study is necessary based on new information (clinical evaluation)Medical Affairs Representative Review the Post-Market Clinical Follow-Up Justification form and Post-Market Clinical Follow-Up Plan form to confirm the decisions regarding the need for a post-market clinical follow-up study and clinical follow-upReview and approve the clinical evaluation performed by the Research Manager or designee5.WORK INSTRUCTIONPost-market clinical monitoring is an essential element in establishing long term safety follow-up data and possible emergent risks for medical devices. These risks and data cannot adequately be detected andcharacterized by relying solely on pre-market clinical investigations.Post market clinical monitoring may include a combination of several strategies:P roduct complaint reviewP ost-market event reporting review of users and patientsL iterature reviewP ost-market clinical follow-up studies (PMCFS)This work instruction was created to determine when a PMCFS is necessary to maintain an adequate post-market surveillance system, as required bythe Medical Device Directive 93/42/ECC (MDD) as amended by MDD 2007/47/EC.It will also provide guidance on the post-market clinical monitoringrequirements if a PMCFS is not required.Figure 5-1: High-Level Process Overview for Post-Market Clinical Follow-UpPMCFSDetermination5.1.General Requirements5.1.1.Prior to M3 sign-off, the Product Regulatory Affairs Representative inconsultation with the Research Manager or designee and the DesignEngineering and/or Engineering Representative shall determine for agiven project/program whether a PMCFS is required. They shall alsodetermine the post-market clinical follow-up plan.5.1.2.A PMCFS may not be required for products for which medium/long-termclinical performance and safety is already known from previous use of the device or where other appropriate post-market surveillanceactivities would provide sufficient data to address the risks.5.2.Determining the Type of Post-Market Clinical Follow-UpRequiredPost-market clinical monitoring shall have one of two outcomes, (1) PMCFS required or (2) no PMCFS required.The need for a PMCFS shall be based on a combination of several factors detailed in this section.5.2.1.The Product Regulatory Affairs Representative in consultation with theResearch Manager or designee and Design Engineering and/or Engineering Representative shall determine whether an equivalent device exists.Equivalence shall be demonstrated in all the essential characteristics precisely defined below. Equivalence means:ClinicalUsed for the same clinical condition or purpose;Used at the same site in the body;Used in similar population (including age, anatomy,physiology);Have similar relevant critical performance according toexpected clinical effect for specific intended useTechnicalUsed under similar conditions of use;Have similar specifications and properties;Be of similar design;Use similar deployment methodsHave similar principles of operationBiologicalSame or similar use of materials in contact with human tissuesor body fluids5.2.2.Products for which the medium/long term clinical performance and safetyis already known from previous use of the device, or from fullytransferable experience with equivalent devices shall not require aPMCFS.NOTE:If the device quoted as the “equivalent” requires a PMCFS, then the new product shall be subject to the same requirement.5.2.3.The need for a PMCFS shall be determined based on the identification ofresidual risks that may impact the risk/benefit ratio. A study shouldalways be considered for devices where the identification of possible emerging risks and the evaluation of long term safety and performance are essential. The Product Regulatory Affairs Representative inconsultation with the Research Manager or designee and Design Engineering and/or Engineering Representative shall identify such emerging risk, the following criteria should be taken into account:innovation, ., where the design of the device, the materials, theprinciples of operation, the technology or the medical indicationsare novel;high risk anatomical locations ., heart, central nervous system,etc.);severity of disease/treatment challenges;sensitivity of target population ., infants, children, pregnantwomen, etc.);identification of an acceptable risk during the pre-CE clinicalevaluation, which should be monitored in a longer term and/orthrough a larger population;well known risks identified from the literature or similarmarketed devices;discrepancy between the pre-market follow-up time scales and theexpected life of the product;5.2.4.A properly conducted risk analysis is essential in determining whatclinical evidence may be needed for a particular device. Any risks identified as an “unacceptable” risk at the conclusion of thedevelopment process shall require a PMCFS. A study should also beconsidered for risks identified as “acceptable” or “risk mitigation required” if the device meets any of the other char acteristicsidentified in 5.2.1 and The risk assessment shall be performedaccording to the Risk Management Procedure. The Product RegulatoryAffairs Representative shall review the risk assessment.5.2.5.The Product Regulatory Affairs Representative shall complete the PostMarket Clinical Follow-Up Study Determination Form (Appendix A) once the decision regarding the need for a study has been determined.NOTE:This form may also be used as a guide in making the determination about the need to perform a PMCFS.5.2.6.The Product Regulatory Affairs Representative shall complete thePost-Market Clinical Follow-Up Plan (Appendix B) that details the plan for post-market clinical follow-up.5.2.7.The Research Manager or designee and Medical Affairs Representative shallreview the Post-Market Clinical Follow-Up Justification Form and ThePost-Market Clinical Follow-Up Plan to confirm the decisions regarding post-market clinical monitoring.5.3.No Post Market Clinical Follow-Up Study Required5.3.1.If it was determined that no PMCFS is required (based on section ,post-market clinical monitoring is still required for the medical device.5.3.2.Justification regarding the decision not to perform a PMCFS must beclearly documented and maintained in the design history/technical file (see 5.2.5).5.3.3.Post-Market Clinical Monitoring Requirements (minimum)5.3.3.1.At a minimum, the following post-market clinical monitoringactivities shall be completed according to TDI Foot/Ankle Array 8chestablished procedures/work instructions. These elements will beinputs into the Post-Market Literature Evaluation and Market Analysis Report.Review of product complaints according to Complaint HandlingProcedureReview of post market adverse events according to Post Market EventReporting ProcedureLiterature review according to TDI Foot/Ankle Array 8ch Evaluationof Clinical Data to Support CE Marking Work Instruction .5.3.3.2.Review of product complaints, post market adverse events and theliterature review shall be completed at the intervals specified in Table 5-1. The timing outlined provides the minimum requirements. TheProduct Regulatory Affairs Representative and/or the Research Manager or designee can determine that clinical data shall be reviewed more often.Table 5-1: Timing for Review of Clinical Data based on Medical Device ClassDevice Classification Timing for review of clinical data (minimum)Class I AnnuallyClass IIa, IIb At a minimum annually, should consider moreoftenClass III Semi-annually . twice a year), should considermore often5.3.3.3.At the interval outlined in Table 5-1, the Research Manager ordesignee shall complete a literature review and analysis of post-market experiences . complaints and adverse events) and re-evaluate if a PMCFSneeds to be conducted based on this data. The Post Market Literature Evaluation and Market Analysis Conclusion form (Appendix D) shall becompleted and maintained as part of the device’s designhistory/technical file. The Product Regulatory Affairs Representative and Medical Affairs Representative shall review and approve thisdocument.NOTE:The literature review shall be executed according to the Evaluation of Clinical Data to Support CE Marking Work Instruction, section . However, the following forms/templates shall be used in place of those specified in this work instruction:a.Instead of using The Literature Evaluation Plan template referenced, usethe Post Market Literature Evaluation and Market Experience Plan form(Appendix C)b.Instead of using The Literature Evaluation Report and Conclusion template,use the Post-Market Literature Evaluation and Market Analysis Report andConclusion form (Appendix D)5.4.Post Market Clinical Follow-Up Study Required5.4.1.If it was determined that a PMCFS is required, in addition to therequirements listed under 5.3.3, studies such as extended follow-up of patients enrolled in the pre-market trials, prospective study of arepresentative subset of patients after the device is placed on the market, or an open registry may be performed.5.4.2.The PMCFS shall be carried out in accordance with TDI Foot/Ankle Array8ch’s Research Involving Human Subjects Procedure5.4.3.The Research Manager or designee in consultation with the RegulatoryAffairs Representative and the Design Engineering and/or EngineeringRepresentative will determine the type of PMCFS that will be implemented.5.4.4.The study should take into account the following:Results of the clinical investigation including adverse eventsidentifiedAverage life expectancy of the deviceThe claims made by the manufacturer for the devicePerformances for which equivalence is claimedNew information becoming available5.4.4.1.At the interval outlined in Table 5-1, the Research Manager ordesignee shall complete a literature review and analysis of post-market experiences . complaints and adverse events) and review the ongoingresults/data of the PMCFS. The Post Market Literature Evaluation and Market Analysis Conclusion form (Appendix D) shall be maintained as partof the device’s design history/technical file. The Product Regulatory Affairs Representative and Medical Affairs Representative shall review and approve this document.NOTE:The literature review shall be executed according to the Evaluation of Clinical Data to Support CE Marking Work Instruction, section . However, the following forms/templates shall be used in place of those specified in this work instruction:a.Instead of using The Literature Evaluation Plan template referenced, usethe Post Market Literature Evaluation and Market Experience Plan form(Appendix C)b.Instead of using The Literature Evaluation Report and Conclusion template,use the Post-Market Literature Evaluation and Market Analysis Report andConclusion form (Appendix D)5.5.Elements of a post-market clinical follow-up study5.5.1.Post-market clinical follow-up studies are performed on a device withinits intended use/purpose(s) according to the instructions for use.5.5.2.A PMCFS shall include the elements defined in the Writing ClinicalInvestigational Plans and Protocols Work Instruction.5.5.3.The objective(s) of a PMCFS should be stated clearly and should addressthe residual risk(s) identified. It should be formulated to address one or more specific questions relating to the clinical safety or performance of the device.5.5.4.Post-market clinical follow-up studies should be designed to address theobjective(s) of the study. The design may vary based on the objective(s) and should be scientifically sound to allow for valid conclusions to be drawn.5.5.5.The study design can take several forms, for example:t he extended follow-up of patients enrolled in pre-marketinvestigations;a new clinical investigation;a review of data derived from a device registry;a review of relevant retrospective data from patients previouslyexposed to the device.t he analysis plan including any interim reporting; andp rocedures for early study termination.5.5.6.The data and conclusions derived from the PMCFS are used to provideclinical evidence to support the post-market surveillance program. This process may result in the need to reassess whether the device continuesto comply with the Essential Principles. Such assessments may result in corrective or preventive actions.6.APPENDIX6.1.Appendix A: Post-Market Clinical Follow-Up StudyDetermination6.2.Appendix B: Post-Market Clinical Follow-Up Plan6.3.Appendix C: Post Market Literature Evaluation and MarketExperience Analysis Plan6.4.Appendix D: Post-Market Literature Evaluation and MarketAnalysis Report and Conclusion。