化学药BE试验备案平台介绍 共21页
化学仿制药BE备案范围和程序

附件化学药生物等效性试验备案范围和程序一、备案范围(一)属于下列情形的化学药,应当进行BE试验备案:1.仿制已上市的参比制剂,其活性成分、给药途径、剂型、规格应与参比制剂相一致。
参比制剂应为原研药品。
2.已批准在境内上市,需通过BE试验开展相应变更研究的药品。
3.已在境内上市,需通过BE试验与参比制剂进行质量和疗效一致性评价的药品。
参比制剂应为原研药或国际公认的仿制药。
(二)属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批。
1.放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品;2.细胞毒类药品;3.不适用BE试验方法验证与参比制剂质量和疗效一致的药品;4.不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品;5.注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。
二、备案程序(一)注册申请人向具有资质的药物临床试验机构提出申请,获得该机构伦理委员会的批准,并签署BE试验合同。
(二)注册申请人开展生物等效性试验前30天,应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,按要求提交备案资料。
(三)备案资料主要包括注册申请人信息、产品基本信息、处方工艺、质量研究和质量标准、参比制剂基本信息、稳定性研究、原料药、试验方案设计、伦理委员会批准证明文件等。
(四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。
(五)注册申请人在获得备案号后,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,注册申请人须说明情况;2年内未提交受试者入组试验信息的,所获得备案号自行失效。
(六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),按照试验方案开展BE试验。
药物临床试验登记与信息公示平台介绍及常见问题分析_黄钦

《赫尔 辛 基 宣 言 》是 人 体 医 学 研 究 的 国 际 伦 理 准则,其要求临床试验信息必须在首例受试者入组 前对公众公示,还要求不论阳性或阴性的试验结果 均须公示[1]。通过对试验研究信息的公示,有助于 药物研发人员了解整个行业的研发动态,了解和借 鉴他人的研发理念和技术方法。此外,对临床试验 进行登记和信息公示也是研究人员和监管部门对社
[关键词] 药物; 临床试验; 登记; 公示 [中图分类号] R95 [文献标志码] C [文章编号] 1003 - 3734( 2014) 23 - 2721 - 04
Introducing the platform for registry and publicity of drug clinical trials and analyzing the common questions in trial registry
2721 中国新药杂志 2014 年第 23 卷第 23 期
Chinese Journal of New Drugs 2014,23( 23)
这项工作开展以来,我们发现登记人在平台登 记和信息填写的具体操作中,存在一些常见的共性 问题,为此有必要对平台的设计思路和搭建过程予 以介绍,同时针对广大登记人遇到的常见问题进行 答复说明,希望帮助相关人员更好地理解和做好这 项工作。 1 平台搭建的设计思路和简要过程
子账户对其下创建试验的管理功能与主账户的 相同。但是,子账户只能对其下创建的试验进行管 理,不能查看主账户中的其他信息,包括主账户的企 业信息、联系人信息以及其他试验信息等。另外,每 个子账户只能单独管理,不能合并。 2. 1. 4 转让方进行变更试验申办者操作时找不到 该功能键以及转让方能否看到受让方填写的试验信 息 登记表中的“申办者名称”系来源于 CDE 数据 库“申请表 ”中 的 申 请 人 名 称,由 系 统 自 动 关 联,且 登记人不可修改。若发生批件转让,会导致自动关 联的申办者名称与实际批件持有人不符。为解决该 问题,系统设置了“变更试验申办者”功能。发生批 件转让时,可 以 通 过 该 功 能 将 登 记 表 内 的“申 办 者 名称”项变更为受让方名称。
一图秒懂BE备案程序_静墨舒悦
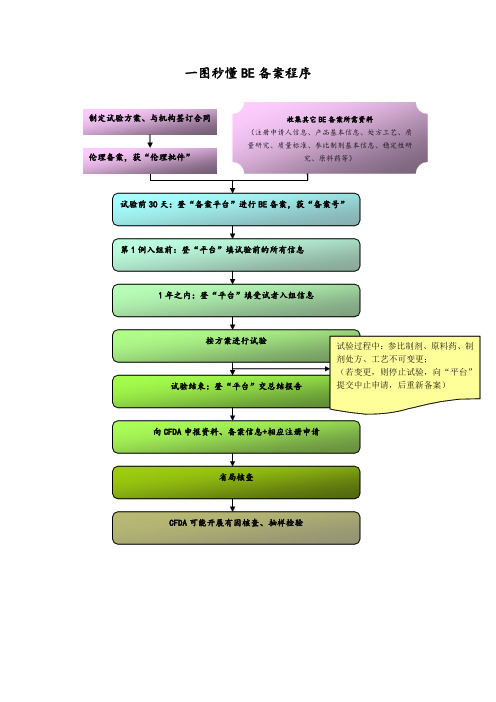
一图秒懂BE 备案程序
制定试验方案、与机构签订合同收集其它BE备案所需资料
(注册申请人信息、产品基本信息、处方工艺、质
量研究、质量标准、参比制剂基本信息、稳定性研
究、原料药等)
伦理备案,获“伦理批件”
试验前30天:登“备案平台”进行BE备案,获“备案号”
第1例入组前:登“平台”填试验前的所有信息
1年之内:登“平台”填受试者入组信息
按方案进行试验
向CFDA申报资料、备案信息+相应注册申请 试验结束:登“平台”交总结报告
省局核查
CFDA可能开展有因核查、抽样检验 试验过程中:参比制剂、原料药、制剂处方、工艺不可变更;
(若变更,则停止试验,向“平台”提交中止申请,后重新备案)。
药物临床试验登记和信息公示平台介绍 审评中心 黄钦

最新动态
On August 2, 2012, 《the Trial and Experimental Studies Transparency (TEST) Act (H.R. 62Drazen, M.D. : We can make progress in medicine only if people are willing to put themselves at risk to test new diagnostic and therapeutic approaches. To recognize and reward these participants, and in keeping with the Declaration of Helsinki, clinical trials should be conducted in the open, with full public knowledge of the question asked, the intervention tested, and the results obtained. _ ----------引自NEJM 367:9.AUG.30,2012
http://www.who.int/ictrp/trial_reg/en/index.html
2
赫尔辛基宣言的要求
A. INTRODUCTION B. PRINCIPLES FOR ALL MEDICAL RESEARCH 19. Every clinical trial must be registered in a publicly accessible database before recruitment of the first subject. 30. …… Negative and inconclusive as well as positive results should be published or otherwise made publicly available …… C. ADDITIONAL PRINCIPLES FOR MEDICAL RESEARCH COMBINED WITH MEDICAL CARE 3
化学药生物等效性试验备案范围和程序

附件化学药生物等效性试验备案范围和程序一、备案范围(一)属于下列情形的化学药,应当进行BE试验备案:1.仿制已上市的参比制剂,其活性成分、给药途径、剂型、规格应与参比制剂相一致。
参比制剂应为原研药品。
2.已批准在境内上市,需通过BE试验开展相应变更研究的药品。
3.已在境内上市,需通过BE试验与参比制剂进行质量和疗效一致性评价的药品。
参比制剂应为原研药或国际公认的仿制药。
(二)属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批。
1.放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品;2.细胞毒类药品;3.不适用BE试验方法验证与参比制剂质量和疗效一致的药品;4.不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品;5.注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。
二、备案程序(一)注册申请人向具有资质的药物临床试验机构提出申请,获得该机构伦理委员会的批准,并签署BE试验研究合同。
(二)注册申请人开展生物等效性试验前30天,应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,按要求提交备案资料。
(三)备案资料主要包括注册申请人信息、产品基本信息、处方工艺、质量研究和质量标准、参比制剂基本信息、稳定性研究、原料药、试验方案设计、伦理委员会批准证明文件等。
(四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。
(五)注册申请人在获得备案号后,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,注册申请人须说明情况;2年内未提交受试者入组试验信息的,所获得备案号自行失效。
(六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),按照试验方案开展BE试验。
食药总局:化学药生物等效性试验由审批制改为备案管理

食药总局:化学药生物等效性试验由审批制改为备案管理
中国经济网北京12 月2 日讯根据《国务院关于改革药品医疗器械审评审批制度的意见》、《国家食品药品监督管理总局关于药品注册审评审批若干政策的公告》等要求,自2015 年12 月1 日起,化学药生物等效性试验由审批制改为备案管理。
一、注册申请人应按照药品注册的相关法律法规和技术要求开展BE 试验研究,确保研究的科学性、伦理合理性及研究资料的真实性、准确性,研究过程可追溯性。
二、注册申请人如需进行化学药BE 试验,可登陆国家食品药品监督管理总局化学药BE 试验备案信息平台,按要求填写备案信息,提交备案资料,获取备案号。
三、在填写备案信息前,注册申请人需将试验方案提请承担BE 试验的药物临床试验机构伦理委员会伦理审查,并与药物临床试验机构签署BE 试验合同。
四、注册申请人需监督承担BE 试验的临床试验机构及相关责任人按试验方案组织BE 试验。
BE 试验完成后,由注册申请人向食品药品监管部门提出药品注册申请并提交相关资料。
五、各省食品药品监督管理局负责对本行政区域内的注册申请人、药物临床试验机构所开展的BE 试验进行日常监督管理,并对注册申请人完成的BE 试验数据的真实性、完整性进行核查。
核查通过后,由核查人员起草核查意见,由省级食品药品监督管理局负责同志签发报国家食品药品监督管理总局。
六、国家食品药品监督管理总局对注册申请人的备案资料进行分析和技。
仿制药质量和疗效一致性评价参比制剂与BE备案

BE试验备案和程序
2.属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批1)放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品;2)细胞毒类药品;3)不适用BE试验方法验证与参比制剂质量和疗效一致的药品;
备案范围
BE试验备案和程序
征求意见时间
2015.10.30-2015.11.20
发布时间
2016.03.18
参比制剂遴选
企业找不到且无法确定参比制剂,需开展临床有效性试验。
总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的公告(食品药品监管总局公告2016年第106号)总局药品审评中心正在拟定一致性评价临床技术指南
参比制剂遴选
公 开 公 布
公布目录
参比制剂遴选
国际公认的同种药物定义: 在欧盟、美国、日本获准上市并获得参比制剂地位的仿制药。 ---《普通口服固体制剂参比制剂选择和确定指导原则》参比制剂办理一次性进口的要求?原研地产化如何申报参比制剂?国产药品可否作为参比制剂?国内上市改剂型、改酸根、碱基的制剂,如何选择参比制剂?
企业用户备案
企业用户备案
化学药BE试验备案信息平台简介
1.承诺书 首先需要签署承诺书:查看相应的承诺书条款,清楚了解要求后,在同意签署以上承诺的地方选择“是”,点击下一步进入上传伦理委员会批件页面。
企业用户备案
企业用户备案
化学药BE试验备案信息平台简介
2.上传伦理委员会批件 填写伦理委员会批件号,并上传批件的PDF文件。上传成功后点击“下一步”进入备案申请表页面。此时备案记录已经生成,备案状态为“待提交申请表” 。
选 择 原 则
仿制药be试验备案的技术要求

仿制药BE试验备案的技术要求1. 背景介绍仿制药是指与原研药具有相同的活性成分、剂型和适应症的药物。
为了确保仿制药的质量和疗效与原研药相当,需要进行生物等效性(BE)试验。
BE试验是一种比较仿制药与原研药在体内吸收、分布、代谢和排泄等方面的相似性的试验。
2. 技术要求2.1. 设计方案•BE试验应设计为随机、双盲、交叉组设计,以确保结果的客观性和可靠性。
•BE试验应包括足够数量的受试者,以提高统计学功效。
•BE试验应根据国际通用准则进行设计,并符合相关法规和标准要求。
2.2. 受试者选择•受试者应符合以下条件:–年龄范围在18岁至45岁之间;–健康状态良好,无严重慢性疾病;–不得同时参与其他临床试验;–需签署知情同意书。
2.3. 给药和采样•给药方式应与实际临床使用方式一致。
•仿制药和原研药的给药剂量应相同。
•采样时间点应根据药物的特性和代谢动力学进行合理选择。
2.4. 分析方法•BE试验应使用合适的分析方法,如非线性混合效应模型(NLME)或线性混合效应模型(LME)等。
•分析方法应能够准确评估仿制药和原研药之间的生物等效性。
2.5. 数据分析与报告•数据分析应基于预先设定的统计分析方案进行,包括计算均值、标准差、置信区间等指标。
•结果报告应清晰、详细地描述BE试验的设计、方法、结果和结论,并包括必要的图表和统计数据。
2.6. 质量控制•BE试验的质量控制包括但不限于以下方面:–药物质量控制:确保仿制药和原研药的质量符合相关要求。
–实验室设备验证:确保实验室设备符合相关标准,并能够提供准确可靠的结果。
–校准和质控样品:定期进行校准和质控样品的使用,以确保结果的准确性和可靠性。
–数据管理:建立完善的数据管理系统,确保数据的安全性和完整性。
3. 结论仿制药BE试验备案的技术要求涉及设计方案、受试者选择、给药和采样、分析方法、数据分析与报告以及质量控制等方面。
严格按照这些技术要求进行BE试验,能够确保仿制药与原研药在生物等效性方面的相似性,并为仿制药上市提供科学依据。
化学药BE试验备案信息表
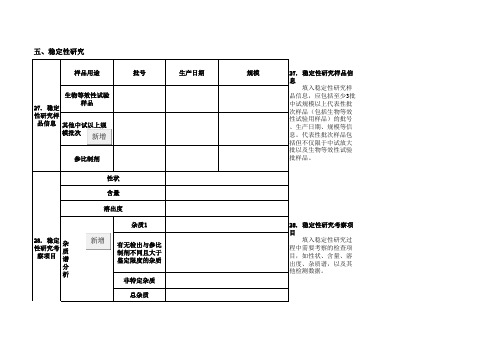
生物等效性试验 样品
新增
参比制剂 性状 含量 溶出度 杂质1 28. 稳定 杂 性研究考 质 察项目 谱 分 析
新增
有无检出与参比 制剂不同且大于 鉴定限度的杂质 非特定杂质 总杂质
28. 稳定性研究考察项 目 填入稳定性研究过 程中需要考察的检查项 目,如性状、含量、溶 出度、杂质谱,以及其 他检测数据。
五、稳定性研究
样品用途 批号 生产日期 规模 27. 稳定性研究样品信 息 填入稳定性研究样 品信息,应包括至少3批 中试规模以上代表性批 次样品(包括生物等效 性试验用样品)的批号 、生产日期、规模等信 息。代表性批次样品包 括但不仅限于中试放大 批以及生物等效性试验 批样品。
27. 稳定 性研究样 品信息 其他中试以上规 模批次
加速试验 31. 稳定 性研究数 据
新增
批号 杂质1(%) 批号 批号
考察项目
新增
杂 质 谱
有无检出与参比 制剂不同且大于 鉴定限度的杂质 (%) 非特定杂质 (%) 总杂质(%) 其他
长期试验
新增
31. 稳定性研究数据 填入各批次样品在不同时间点的稳定性实际考察数据。对于杂质谱检查项,不可仅填入单个杂质和总杂质的数据,必须列出所有大于鉴定 限度的杂质,并按结构已知的特定杂质、结构未定的特定杂质以及总杂质进行填写,非特定杂质指未超过指导原则中鉴定限度的杂质。 (批号:新增按钮,为添加批号 ; 杂质谱:新增按钮,为了增加杂质)
32. 稳定 性研究总 结 32. 填入稳定性研究总结,说明样品与参比制剂稳定性是否一致。 技术要求: 应提供三批中试以上规模自制样品的稳定性研究数据,试验样品包括但不仅限于中试放大批以及生物等效性试验批样品。应提供至少三个 月加速与长期留样研究数据。在生物等效性试验期间继续进行以上样品进行加速与长期留样稳定性研究。 如无充分的依据,拟定的贮藏条件应与参比制剂以及药典规定的贮藏条件相同。如果降低贮藏条件,提示在参比制剂以及药典规定的贮藏 条件下,样品可能不如参比制剂稳定。 稳定性研究期间,如果发现样品的质量(如含量、溶出度、杂质等)不符合质量标准要求,应立即终止生物等效性试验,并以备案形式报 告。如果稳定性研究期间样品在多种溶出介质中的溶出行为发生显著变化的,或者发现大于鉴定限度的杂质谱与参比制剂不同,或杂质种类和/ 或含量明显多于/高于参比制剂,且无法按照杂质研究技术指导原则进行充分解释的,应立即终止生物等效性试验,并以备案形式报告。
仿制药质量和疗效一致性评价参比制剂与BE备案-zheng

总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的 公告(食品药品监管总局公告2016年第106号)
总局药品审评中心正在拟定一致性评价临床技术指南
参比制剂遴选
备案程序
药品生产企业
行业协会 原研企业 国际公认的同种 药物生产企业
备案 推荐
申报
提交
电子版资料 纸质版资料
用户注册
打开网址后进入用户登录页面;
如果用户未注册账号,须首先在申请人之窗进行用户注册,操作如下图;
点击注册
用户登录 化学药BE试验备案信息平台简介
用户登录
企业用户须使用药审中心的申请人之窗账户进行登录。企业用户登录方式有两种:
账号密码登录和UKEY证书登录。
方式一:账号密码登录(管理人员同样使用此方式登录)
圳信立泰药业股份有限公司的替格瑞洛片,亚宝药业集团股
份有限公司的苯甲酸阿格列汀片,上海安必生制药技术有限
公司的孟鲁司特钠咀嚼片)。
现平台为非结构化的,预计从今年7月起全采用结构化的
平台。
化学药BE试验备案信息平台简介 非结构化备案流程
企业使用申请人之窗账户进行登录
新建备案
终止备案
撤销备案
用户注册 化学药BE试验备案信息平台简介
备案范围
2.属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理 和审评审批。
4)不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品; 5)注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。
BE试验备案和程序
备案资料
注册申请人信息 产品基本信息 处方工艺 质量研究和质量标准 参比制剂基本信息 稳定性研究 原料药 试验方案设计 伦理委员会批准证明文件等
国家食品药品监督管理总局关于化学药生物等效性试验实行备案管理的公告(2015年第257号)附件

附件化学药生物等效性试验备案范围和程序一、备案范围(一)属于下列情形的化学药,应当进行BE试验备案:1.仿制已上市的参比制剂,其活性成分、给药途径、剂型、规格应与参比制剂相一致。
参比制剂应为原研药品。
2.已批准在境内上市,需通过BE试验开展相应变更研究的药品。
3.已在境内上市,需通过BE试验与参比制剂进行质量和疗效一致性评价的药品。
参比制剂应为原研药或国际公认的仿制药。
(二)属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批。
1.放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品;2.细胞毒类药品;3.不适用BE试验方法验证与参比制剂质量和疗效一致的药品;4.不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品;5.注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。
二、备案程序(一)注册申请人向具有资质的药物临床试验机构提出申请,获得该机构伦理委员会的批准,并签署BE试验合同。
(二)注册申请人开展生物等效性试验前30天,应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,按要求提交备案资料。
(三)备案资料主要包括注册申请人信息、产品基本信息、处方工艺、质量研究和质量标准、参比制剂基本信息、稳定性研究、原料药、试验方案设计、伦理委员会批准证明文件等。
(四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。
(五)注册申请人在获得备案号后,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,注册申请人须说明情况;2年内未提交受试者入组试验信息的,所获得备案号自行失效。
(六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),按照试验方案开展BE试验。
化学药BE试验备案平台介绍

化学药BE试验备案平台介绍化学药BE试验备案平台是一个为化学药品BE试验提供备案申请和管理服务的平台。
BE试验,即生物等效性试验,是一种用于比较不同药品之间的生物等效性的试验方法,以评估药物在体内的药代动力学和药效学等方面的相似性。
化学药BE试验备案平台的主要功能是协助申请人提交和管理BE试验备案申请。
申请人可以通过平台在线填写申请表格,并上传相关证明材料。
平台会对申请材料进行初步审核,并在一定时间内完成审批。
通过平台,申请人可以随时查看申请状态和审批结果,以及补充或修改申请材料。
化学药BE试验备案平台也提供了一系列辅助功能,以帮助申请人更好地管理BE试验项目。
例如,平台可以协助管理不同批次的BE试验,包括提交批次信息、药品样品的存放和出库等。
此外,平台还提供了试验计划、数据分析和报告生成等功能,以支持BE试验的设计和结果分析。
化学药BE试验备案平台的优势在于提高了BE试验备案流程的效率和透明度。
传统的备案申请通常需要纸质材料的邮寄和人工审核,需要耗费大量的时间和人力。
而通过化学药BE试验备案平台,申请人可以在线提交申请,减少了繁琐的邮寄流程。
平台的自动化审核也能够快速完成初步审批,提高了备案的办理速度。
此外,通过平台,申请人可以实时查看申请状态和进度,避免了传统备案申请中信息不透明的问题。
另外,化学药BE试验备案平台也提供了一些质量管理的功能,提高了BE试验的可靠性和数据的准确性。
例如,平台可以设定数据录入的规范和标准,避免数据录入的错误和混乱。
平台还可以保存试验数据和相关信息,便于日后的查阅和验证。
通过这些质量管理的功能,化学药BE试验备案平台可以更好地保障试验的科学性和可信度。
总体而言,化学药BE试验备案平台是一个为化学药品BE试验提供便捷、高效和可靠服务的在线平台。
通过平台,申请人可以简化备案流程,加快备案审批进度,并提高试验的质量和可信度。
化学药BE试验备案平台在推动化学药品BE试验的规范化和标准化方面发挥着重要作用,促进了药物研发和生产的科学性和可持续发展。
BE审评工作-20181211-来源药审中心

Guideline on the investigation of bioequivalence (2010)
EMA
使申请人的研究有章可循 使BE试验技术审评有据可依 申请人对结果具有可预见性
10
BE审评参考——国外指导原则
仿制药BE指导原则(2013年, draft) 新药的BE指导原则(2014年,draft)
FDA Bioanalytical Method Validation Guidance for Industry(May 2018)
Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System(December 2017)
0
5
10
15
20
一致性评价(前5批)
方法验证及 其他:23% 样品检
测:23%
样品检测&其他:54%
方法验证及样品检测
样品检测&其他
其他 12
BE审评的总体考虑
•PK-BE?PD-BE? •交叉?平行?重复? •采血点设计? •空腹?餐后?
• ……
•剔除数据? •数据集? •随机化? •敏感性分析? •……
适用范围
• 无全身吸收的局部作用 药物 • 生物样本中活性成分浓 度过低,不能可靠测定 • 活性成分的浓度与药品 的有效性和安全性无关
15-药品快检技术平台介绍

药品快检技术平台介绍
车载近红外软件和模型下载 可以及时的更新SFDA_Ident软件和近红外通用性模型, 工作效率高、服务范围广,从而克服模型更新不及时 的问题。各地药品检测车管理人员通过登录“近红外 模型下载”网页,下载各自所需的近红外模型以及升 级说明。2010年度的近红外药品鉴别系统更新工作共 维护了17个通用性定性模型、并新增了31个通用性定 量模型和120个一致性检验模型。
药品快检技术平台介绍
药品检测车数据网络管理平台 通过本平台,国家局可对各地药品监督检查情况进行 实时监控和数据管理,及时传递基层药品质量信息, 逐步实现了对药品质量的动态监管和监督检验相结合 的新模式。通过信息系统软件的数据统计功能,将现 场快检数据统计分析,获知各快检方法的使用情况, 根据实际情况对快检方法进行相应的修改、调整;通 过数据信息网络平台,实时了解各地ห้องสมุดไป่ตู้载移动实验室 的运行检测实际情况,针对各地用户上报的数据进行 统计分析获知当前市场上假药的流行趋势,并可根据 这个信息有针对性的开发相应的快检方法。
药品快检技术平台介绍
中国药品生物制品检定所
药品快检技术平台介绍
网址:/CL0107/
药品快检技术平台介绍
药品检测车介绍 药品快速检测系统以车为载体,将近红外无损伤检测 技术、经典化学分析技术及现代计算机信息技术等融 于一体,可在监管现场进行快速筛查。中央及国家局 多位领导多次视察药品检测车,并给予了高度评价。 美国、俄罗斯、泰国等国家药品监管部门专门派员以 及BBC等国际媒体来中国参观药品检测车,进行交流。 截止2009年4月底,全国29个省(自治区、直辖市) 已配备药品检测车389台。
药品快检技术平台介绍
快检技术网络论坛 本论坛作为一个沟通效率高、交流范围广的技术服务 平台,用于药检系统内部各种快检技术的交流与协作, 尤其是近红外光谱技术的讨论和提高。论坛栏目中分 为模型交流、资料下载、案例分析、学术前沿、技术 答疑、最新动态六个板块,每个板块聘请两名教员负 责提供帮助、答疑和技术服务。在“最新动态”板块 中也会将最新的工作动态予以公告。
化学药BE试验备案平台介绍共21页

2020/1/3
4
非结构化备案流程
企业使用申请人之窗账户进行登录 新建备案 终止备案 撤销备案 下面将按照整个流程简述每个流程的具体操作
2020/1/3
5
用户注册
打开网址后进入用户 登录页面;
2020/1/3
7
企业用户备案
新建备案
–前期准备:在网站首页或者备案信息列表页 面下载备案信息表模板,按模板要求填写, 并准备需要上传的相关附件。
–整个备案流程分为签署承诺书、上传伦理批 件、填写备案申请表信息、上传备案信息表 及相关附件、生成备案号等五个阶段。
2020/1/3
8
企业用户备案
1.承诺书
首先需要签署承诺书:查看相应的承诺书条款, 清楚了解要求后,在同意签署以上承诺的地方选 择“是”,点击下一步进入上传伦理委员会批件 页面。
2020/1/3
9
企业用户备案
2.上传伦理委员会批件
填写伦理委员会批件号,并上传批件的PDF文 件。上传成功后点击“下一步”进入备案申请表页 面。此时备案记录已经生成,备案状态为“待提交 申请表” 。
2020/1/3
2
2019年12月1日,总局发布了“关于化学 药生物等效性试验实行备案管理的公告 (2019年第257号)”,提出了具体的要 求与程序。
注册申请人如需进行化学药BE试验,可 登陆国家食品药品监督管理总局“化学药 BE试验备案信息平台”(网址: ),按要求填写备案 信息,提交备案资料,获取备案号。
2020/1/3
10
企业用户备案
3.填写备案信息申请表
备案申请表共分为四部分,包括基本信 息、申请人信息、参比制剂信息和生物等效 性试验信息。依次填写并检查无误后点击 “保存”将进入备案资料上传页面。保存之 后备案信息状态由“待提交申请表”变为 “待提交资料” 。
化学药BE试验备案平台介绍-PPT精选文档

点击注册
2019/3/17
6
用户登录
企业用户须使用药审中心的申请人之窗账 户进行登录。企业用户登录方式有两种: 账号密码登录和UKEY证书登录。 方式一:账号密码登录(管理人员同样使 用此方式登录) 方式二: UKEY证书登录
2019/3/17
7
企业用户备案
新建备案
–前期准备:在网站首页或者备案信息列表页 面下载备案信息表模板,按模板要求填写, 并准备需要上传的相关附件。 –整个备案流程分为签署承诺书、上传伦理批 件、填写备案申请表信息、上传备案信息表 及相关附件、生成备案号等五个阶段。
2019/3/17 15
企业用户备案
中止备案
–上传完成后点击确定,将提交保存报告文件,
备案信息状态由“已备案”变为“备案中止”。 企业用户将可重新备案。
2019/3/17
16
企业用户备案
撤销备案
–BE试验过程中,发现存在缺陷和风险的,申
请人应撤销该BE试验,并上传总结报告。
–点击“撤销备案”按钮,弹出对话框:上传
2
2019/3/17
2019年12月1日,总局发布了“关于化学 药生物等效性试验实行备案管理的公告 (2019年第257号)”,提出了具体的要 求与程序。 注册申请人如需进行化学药BE试验,可 登陆国家食品药品监督管理总局“化学药 BE试验备案信息平台”(网址: ),按要求填写备案 信息,提交备案资料,获取备案号。
2019/3/17
10
企业用户备案
3.填写备案信息申请表
备案申请表共分为四部分,包括基本信 息、申请人信息、参比制剂信息和生物等效 性试验信息。依次填写并检查无误后点击 “保存”将进入备案资料上传页面。保存之 后备案信息状态由“待提交申请表”变为 “待提交资料” 。
药物临床试验机构备案信息平台操作手册及填报详情

药物临床试验机构备案管理信息平台系统操作手册(V1.2 版)厦门市南方科宇科技有限公司2019 年12 月精品文档文档信息版本记录目录第一章系统总体说明 (1)1. 系统功能综述 (1)1.2 系统角色划分 (1)1.3 系统主要流程 (2)第二章注册与登录 (3)2.1 注册 (3)2.1.1 统一注册平台 (3)2.1.2 新用户 (5)2.1.3 老用户 (15)2.2 登录 (20)2.2.1 系统登录 (20)2.2.2 首页 (25)第三章机构注册管理 (27)3.1 待核对 (27)3.2 已核对 (29)第四章药物机构备案管理 (31)4.1 机构信息维护(机构填报人) (31)4.1.1 基本信息 (32)4.1.2 组织管理机构 (34)4.1.3 备案专业 (36)4.1.4 伦理委员会 (39)4.1.5 年度总结 (40)4.16 接受境外药监部门检查情况报告表 (40)4.17 提交备案内部审核 (42)第五章咨询单位联系方式 (44)第一章系统总体说明1.1系统功能综述本系统采用JAVA 开发语言,使用B/S 架构。
为了达到更好的使用效果,建议使用Windows7 及以上版本的操作系统;IE9.0 及以上的浏览器,如使用360 浏览器,请使用极速模式;屏幕分辨率建议设置为1366*768 及以上。
表 1.1-1 功能模块表1.2系统角色划分表 1.2-1 角色划分表1.3系统主要流程图 1.3-1 系统主要流程图第二章注册与登录通过https:///web/index 网址,进入国家药品监督管理局网上办事大厅,至少完成法人账号和一个个人账号的注册。
1. 新用户:首次使用本系统的临床试验机构,账号注册成功后,需要在本系统中补充资料,并等待监管部门核对,补充资料核对通过后,才能登录系统。
2. 老用户:之前已在本系统成功注册的临床试验机构,需要完成新注册的法人账号和一个个人账号,与本系统之前注册的内审和填报账户之间的绑定。
化学药生物等效性(BE)试验填报说明

化学药⽣物等效性(BE)试验填报说明填报须知《化学药⽣物等效性(BE)试验备案信息平台》主要⾯向新申报的化学药。
按照《国家⾷品药品监督管理总局关于化学药⽣物等效性试验实⾏备案管理的公告》(2015年第257号)中的规定,对符合备案情形的化学药,应当进⾏⽣物等效性(BE)试验备案。
本填报说明仅针对化学药⽣物等效性(BE)试验备案的操作步骤,仿制药⼀致性评价试验备案操作步骤请参见《仿制药⼀致性评价试验备案填报说明》。
⽬录1 ⽤户注册 (1)2 ⽤户登录 (3)3 新建备案 (4)3.1 承诺书 (5)3.2 伦理委员会批件 (5)3.3 备案信息申请表 (6)3.3.1 基本信息 (6)3.3.2 申请⼈信息 (7)3.3.3 参⽐制剂信息 (8)3.3.4 ⽣物等效性试验信息 (8)3.4 备案信息表及相关附件 (9)3.5 ⽣成备案号 (10)4 中⽌备案 (10)5 撤销备案 (11)1 ⽤户注册图1.1⽤户注册图1.2注册信息填写在上图1.2中填写⽤户信息后,点击⽴即注册。
2 ⽤户登录企业⽤户须使⽤药审中⼼的申请⼈之窗账户需要进⾏登录。
登录⽅式有两种:账号密码登录和UKEY证书登录。
如下图:图2.1普通登录图2.2证书登陆登录成功之后进⼊个⼈⾸页,此页⾯包含两部分:⽤户信息、功能模块。
根据药品备案需求选择不同的备份⼊⼝。
本⽂档主要介绍化学药⽣物等效性(BE)试验备案(⾯向新申报的化学药)的操作步骤。
图2.3个⼈⾸页3 新建备案备案前期准备:在⽹站⾸页或者备案信息列表页⾯下载备案信息表模板,按模板要求填写,并了解需要准备的相关材料附件。
模板须启⽤宏功能,具体操作见《2007启⽤宏步骤》、《2010启⽤宏步骤》⽂档。
整个备案流程分为签署承诺书、上传伦理批件、填写备案申请表信息、上传备案信息表及相关附件、⽣成备案号。
平台登录成功后进⼊备案记录列表,如下图:图3.1备案记录列表点击“新建备案”按钮进⼊备案流程。
BE备案资料目录

BE备案资料目录
(一)综述资料
2.证明性文件。
3.立题目的与依据。
4.对主要研究结果的总结及评价。
5.药品说明书、起草说明及相关参考文献。
(二)药学研究资料
7.药学研究资料综述。
8.原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文
献资料。
9.确证化学结构或者组份的试验资料及文献资料。
10.质量研究工作的试验资料及文献资料。
11.药品标准及起草说明,并提供标准品或者对照品。
12.样品的检验报告书。
13.原料药、辅料的来源及质量标准、检验报告书。
14.药物稳定性研究的试验资料及文献资料。
(三)药理毒理研究资料
16.药理毒理研究资料综述。
(四)临床试验资料
28.国内外相关的临床试验资料综述。
中南大学湘雅医院
I期临床试验研究室BE备案审查表
承诺书
中南大学湘雅医院:
××××年×月×日,我单位(公司)向贵院提交了开展项目生物等效性研究(备案)相关前期药学研究资料。
现郑重承诺:申请材料中所涉及的文件、证件及有关附件是真实、原始的,复印件与原件是一致的,并对因申请材料虚假所引发的一切后果承担全部法律责任。
承诺人:
(印章)××××年×月×日。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2019/7/22
2
2019年12月1日,总局发布了“关于化学 药生物等效性试验实行备案管理的公告 (2019年第257号)”,提出了具体的要 求与程序。
注册申请人如需进行化学药BE试验,可 登陆国家食品药品监督管理总局“化学药 BE试验备案信息平台”(网址: ),按要求填写备案 信息,提交备案资料,获取备案号。
4.上传备案信息表及相关附件 备案信息表及相关附件上传后点击“保存”
进入确认生成备案号页面,保存之后备案信息 状态由“待提交资料”变为“待生成备案 号” 。
若使用了尚未在国内批准上市的原料药,需 要填写与上传原料药的相关资料与附件。
2019/7/22
12
企业用户备案
5.生成备案号
生成备案号必须使用ukey验证身份信息,点击 “确认并生成备案号”将弹出证书登录页面。
化学药BE试验 备案信息平台简介
总局药品审评中心
2019/7/22
1
背景
• 根据国务院发布的《关于改革药品医疗器械审评审 批制度的意见》,总局于2019年11月11日发布了 “关于药品注册审评审批若干政策的公告(2019年第 230号) ”,在“三、优化临床试验申请的审评审批 中:自2019年12月1日起,仿制药生物等效性试验由 审批制改为备案制。申请人应按照总局发布的相关 指导原则和国际通行技术要求与原研药进行全面的 质量对比研究,保证与原研药质量的一致性;生物 等效性试验用样品的处方、工艺、生产线应与商业 化生产保持一致。申请人开展生物等效性试验前, 应按总局制定的管理规定与技术要求于试验前30天 向国家食品药品监督管理总局提交备案资料。”
2019/7/22
13
企业用户备案
点击登录将确认备案人身份,确认成功后直接生 成备案号。备案信息状态由“待生成备案号”变 为“已备案”。至此整个备案流程完成。 备案号生成后企业将无法修改备案信息,如修改
须中止该备案记录再重新备案。
2019/7/22
14
企业用户备案
中止备案
–BE试验过程中,需要对原备案资料中原研药、 原料药、制剂处方、工艺等进行变更,申请 人应停止该BE试验,中止备案记录并上传总 结报告,并提交备案变更资料重新备案,生 成新的备案号后重新开展BE试验。
现平台为非结构化的,预计从今年7月起全 采用结构化的平台。
2019/7/22
4
非结构化备案流程
企业使用申请人之窗账户进行登录 新建备案 终止备案 撤销备案 下面将按照整个流程简述每个流程的具体操作
2019/7/22
5
用户注册
打开网址后进入用户 登录页面;
1.承诺书
首先需要签署承诺书:查看相应的承诺书条款, 清楚了解要求后,在同意签署以上承诺的地方选 择“是”,点击下一步进入上传伦理委员会批件 页面。
2019/7/22
9
企业用户备案
2.上传伦理委员会批件
填写伦理委员会批件号,并上传批件的PDF文 件。上传成功后点击“下一步”进入备案申请表页 面。此时备案记录已经生成,备案状态为“待提交 申请表” 。
2019/7/22
7
企业用户备案
新建备案
–前期准备:在网站首页或者备案信息列表页 面下载备案信息表模板,按模板要求填写, 并准备需要上传的相关附件。
–整个备案流程分为签署承诺书、上传伦理批 件、填写备案申请表信息、上传备案信息表 及相关附件、生成备案号等五个阶段。
2019/7/22
8
企业用户备案
–点击“撤销备案”按钮,弹出对话框:上传 撤销总结报告文件。
2019/7/22
17
企业用户备案
撤销备案 上传完成后点击确定,将提交保存报告文件, 备案信息状态由“已备案”变为“备案撤销”。 企业用户将不可重新备案。
2019/7/22
18
备案记录维护
管理员登录之后进入备案信息管理列表, 可以在此查看所有已备案完成信息。管 理员可以对不符合要求的试验进行叫停 或者撤销操作。
2019/7/22
10
企业用户备案
3.填写备案信息申请表
备案申请表共分为四部分,包括基本信 息、申请人信息、参比制剂信息和生物等效 性试验信息。依次填写并检查无误后点击 “保存”将进入备案资料上传页面。保存之 后备案信息状态由“待提交申请表”变为 “待提交资料” 。
Байду номын сангаас
2019/7/22
11
企业用户备案
如果用户未注册账号,须首先在申请人之窗进 行用户注册,操作如下图;
点击注册
2019/7/22
6
用户登录
企业用户须使用药审中心的申请人之窗账 户进行登录。企业用户登录方式有两种: 账号密码登录和UKEY证书登录。
方式一:账号密码登录(管理人员同样使 用此方式登录)
方式二: UKEY证书登录
2019/7/22
19
敬请批评指正! 谢谢各位领导!!
2019/7/22
20
–点击“中止备案”按钮,弹出对话框:上传 中止总结报告文件。
2019/7/22
15
企业用户备案
中止备案
–上传完成后点击确定,将提交保存报告文件, 备案信息状态由“已备案”变为“备案中止”。 企业用户将可重新备案。
2019/7/22
16
企业用户备案
撤销备案
–BE试验过程中,发现存在缺陷和风险的,申 请人应撤销该BE试验,并上传总结报告。
2019/7/22
3
平台进展情况
该平台由药审中心负责开发、维护。
2019年11月项目进入试运行,2019年12月 化学药BE试验备案网站正式运行。
截至目前平台中共有54条记录,已有3个品 种备案成功(深圳信立泰药业股份有限公 司的替格瑞洛片,亚宝药业集团股份有限 公司的苯甲酸阿格列汀片,上海安必生制 药技术有限公司的孟鲁司特钠咀嚼片)。