阿托伐他汀合成
阿托伐他汀合成图解
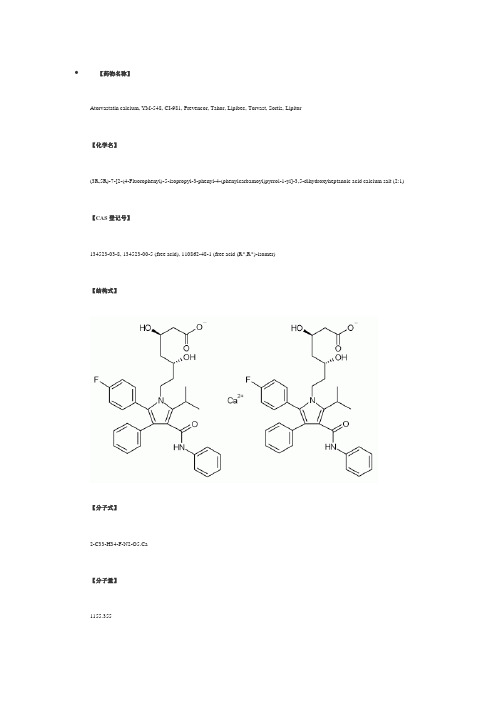
【药物名称】Atorvastatin calcium, YM-548, CI-981, Prevencor, Tahor, Lipibec, Torvast, Sortis, Lipitor【化学名】(3R,5R)-7-[2-(4-Fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxyheptanoic acid calcium salt (2:1) 【CAS登记号】134523-03-8, 134523-00-5 (free acid), 110862-48-1 (free acid (R*,R*)-isomer)【结构式】【分子式】2-C33-H34-F-N2-O5.Ca【分子量】1155.355【原研厂家】Jouveinal (Originator), Pfizer (Originator), Almirall Prodesfarma (Licensee), Syncro (Licensee), Yamanouchi (Licensee), Stanford University (Codevelopment)【作用类别】Alzheimer's Dementia, Treatment of , CARDIOVASCULAR DRUGS, Cognition Disorders, Treatment of, Immunologic Neuromuscular Disorders, Treatment of, Lipoprotein Disorders, Treatment of , METABOLIC DRUGS, Multiple Sclerosis, Agents for, NEUROLOGIC DRUGS, Treatment of Disorders of the Coronary Arteries and Atherosclerosis, HMG-CoA Reductase Inhibitors, TNFSF6 Expression Inhibitors【研发状态】Launched-1997【合成情况】NB2〖来源〗Drugs Fut〖合成路线〗〖标题〗Atorvastatin Calcium〖合成方法〗1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl 2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 C in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4-diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX), which is finally treated first with NaOH in methanol/water, and then with CaCl2 or calcium acetate.〖作者〗Graul, A.; Casta馿r, J.〖参考〗Graul, A.; Casta馿r, J.; Atorvastatin Calcium. Drugs Fut 1997, 22, 9, 956〖出处〗Drugs Fut1997,22,(9):956〖备注〗Synthesis Atorvastatin calcium has been obtained by several different ways: 1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 癈in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4 -diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI) (1, 2), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX) (2, 3), which is finally treated first with NaOH inmethanol/water, and then with CaCl2 or calcium acetate (3, 4). 2) The condensation of the already described aldehyde (XI) with(S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S) (2, 3). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2 (2-4). 3) The condensation of4-cyano-3(R)-hydroxybutyric acid ethyl ester (XXV) with N,N-diphenylacetamide (R1 = R2 = Ph in XXVI) by means of LDA in THF gives 6-cyano-5(R)-hydroxy-3-oxo-N,N-diphenylhexanamide (XXVII), which is reduced with diethylmethoxyborane and NaBH4 in THF yielding 6-cyano-3(R),5(R)-dihydroxy-N,N-diphenylhexanamide (XXVIII). The protection of the two OH groups of (XXVIII) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XXX), which by reduction of its CN group by hydrogenation with H2 over RaNi in methanol/liquid ammonia gives (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]-N,N -diphenylacetamide (XXXI). The cyclization of (XXXI) with 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) (its synthesis is in section 6, Scheme 4) in refluxing toluene yields the protected dihydroxyheptanamide (XXXIII), which is deprotected with HCl in methanol to afford (3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N -phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxy-N,N-diphenylheptanamide (XXXIV). Finally, this compound is hydrolyzed with NaOH and treated with calcium acetate in water (5). Scheme 18007203a. 4) The preceding reaction pathway can be repeated using other substituents for R1 and R2 in acetamide (XXVI) such as R1 = R2 = CH2Ph; R1 = R2 = Et; R1 = Bu, R2 = Me; R1 = t-Bu, R2 = CH2Ph; R1,R2 = -(CH2)5- (5). 5) The hydrolysis of methyl (Et or Bu)3(R)-(tert-butyldimethylsilyloxy)-4-cyanobutyrate (XXV) with NaOH gives the corresponding free acid (XXXVI), which is condensed with malonic acid mono-tert-butyl ester magnesium salt (XXXVII) by means of carbonyldiimidazole (CDI) yielding tert-butyl5(R)-(tert-butyldimethylsilyloxy)-6-cyano-3-oxohexanoate (XXXVIII). The desilylation of (XXXVIII) with tetrabutylammonium fluoride in acetic acid affords the expected hydroxylated ketoester (XXXIX), which is reduced with diethylmethoxyborane and NaBH4 in methanol giving tert-butyl 6-cyano-3(R),5(R)-dihydroxyhexanoate (XL). The protection of the two OH groups of (XL) with acetone dimethylketal(XXIX) and methanesulfonic acid affords the 1,3-dioxane (XLI) (6), which by reduction of its CN group by hydrogenation with H2 overPd/C gives intermediate (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLII). The cyclization of (XLII) with 4-(4-fluorophenyl-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) in refluxing toluene yields the protected dihydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water (7). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 6) The (4R,6R)-2-[6-(cyanomethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLI) can also be obtained by reaction of (4R,6R)-2-[6-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLVIII) with tosyl chloride to give the corresponding tosylate (XVIX), which is then treated with NaCN (6). 7) The tert-butyl6-cyano-5(R)-hydroxy-3-oxohexanoate (XXXIX) can also be obtained by condensation of methyl 4-cyano-3(R)-hydroxybutyrate (L) with tert-butyl acetate (XXIII) by means of LDA in THF (6). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 9) The cyclization of (XXXII) with intermediate (XLII) (preceding synthesis) in refluxing toluene yields the protected dehydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water. References 1. Roth, B.D. (Warner-Lambert Co.). Trans-6-[2-(3- or 4-carboxamido-substd.pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis. EP 247633, US 4681893. 2. Roth, B.D., Blankley, C.J., Chucholowski, A.W., Ferguson, E., Hoefle, M.L., Ortwine, D.F., Newton, R.S., Sekerke, C.S., Sliskovic, D.R., Stratton, C.D., Wilson, M.W. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem 1991, 34: 357-66. 3. Roth, B.D. (Warner-Lambert Co.). (R-(R*R*)-2-(4-Fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl-3-phenyl-4-[(phenylamino)-carbonyl]-1H-pyrrole-1-heptanoic acid, its lactone form and salts thereof. EP 409281, JP 91058967, US 5273995.4. Milb, N., Muhammad, N.A., Weiss, J., Nesbitt, R.U. (Warner-Lambert Co.). Stable oral CI-981 formulation and process for preparing same. EP 680320, JP 96505640, WO 9416693.5. Butler, D.E., Le, T.V., Nanninga, T.N. (Warner-Lambert Co.). Process fortrans-6-[2-(substd.-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis. US 5298627. 6. Brower, P.L., Butler, D.E., Deering, C.F., Le, T.V., Millar, A., Nanninga, T.N., Roth, B.D. The synthesis of (4R-cis)-1,1-dimethylethyl6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate, a key intermediate for the preparation of CI-981, a highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2279-82. 7. Baumann, K.L., Butler, D.E., Deering, C.F., Mennen, K.E., Millar, A., Nanninga, T.N., Palmer, C.W., Roth, B.D. The convergent synthesis of CI-981, an optically active, highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2283-4. 8. McKenzie, A.T. (Warner-Lambert Co.). Form III crystalline(R-(R*,R*))-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methyl-ethyl) -3-phenyl-4-((phenylamino)carbonyl)-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703958. 9. Lin, M., Schweiss, D. (Warner-Lambert Co.). Novel process for the production of amorphous [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid calcium salt (2:1). WO 9703960. 10. Briggs, C.A., Jennings, R.A., Wade, R.A., Harasawa, K., Ichikawa, S., Minohara, K., Nakagawa, S. (Warner-Lambert Co.). Crystalline[R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl) -3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703959.〖来源〗J Med Chem〖合成路线〗〖标题〗Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus〖合成方法〗1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl 2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 C in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4-diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolane group of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX), which is finally treated first with NaOH in methanol/water, and then with CaCl2 or calcium acetate.〖作者〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.〖参考〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.; Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)eth IĶ 셈睋서睋쓰睎ℐ ul> li>a href=쓜睎 Ʈ 0 ๙ιᇐ ꀀÑ밸-00AA004CLSID\{0E59F1D5-1FBE-11D0-8FF2-00A0D10038BC}e Ǔ〖出处〗J Med Chem1991,34,(1):357-66〖备注〗〖来源〗Drugs Fut〖合成路线〗〖标题〗Atorvastatin Calcium〖合成方法〗2) The condensation of the previously described aldehyde (XI) with (S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2.〖作者〗Graul, A.; Casta馿r, J.〖参考〗Graul, A.; Casta馿r, J.; Atorvastatin Calcium. Drugs Fut 1997, 22, 9, 956〖出处〗Drugs Fut1997,22,(9):956〖备注〗Synthesis Atorvastatin calcium has been obtained by several different ways: 1) The condensation of 2-(1,3-dixolan-2-yl)ethylamine (I) with ethyl 2-bromo-2-(4-fluorophenyl)acetate (II) by means of triethylamine in acetonitrile gives ethyl2-[2-(1,3-dioxolan-2-yl)ethylamino]-2-(4-fluorophenyl)acetate (III), which is acylated with isobutyryl chloride (IV) and triethylamine in dichloromethane yielding the corresponding amide (V). Saponification of the ester (V) with NaOH in methanol/water affords the free acid (VI), which is cyclized with N,3-diphenylpropynamide (VII) [obtained in the reaction of 3-phenylpropynoic acid (VIII) with aniline (IX) by means of dicyclohexylcarbodiimide (DCC)] by heating at 90 癈in acetic anhydride giving1-[2-(1,3-dioxolan-2-yl)ethyl]-5-(4-fluorophenyl)-2-isopropyl-N,4 -diphenylpyrrole-3-carboxamide (X). The hydrolysis of the dioxolanegroup of (X) with HCl yields the corresponding aldehyde (XI), which is condensed with methyl acetoacetate (XII) by means of NaH in THF affording 7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-5-hydroxy-3-oxoheptanoic acid methyl ester (XIII). The reduction of the carbonyl group of (XIII) with tributylborane and NaBH4 in THF gives the (3R*,5R*)-dihydroxy ester (XIV), which is saponified with NaOH in water yielding the corresponding free acid (XV). The lactonization of (XV) by heating in refluxing toluene affords the (R*,R*)-lactone (XVI) (1, 2), which is submitted to optical resolution by reaction with (R)-1-phenylethylamine (XVII) followed by fractional crystallization thus obtaining the amide (XVII) as the pure (R,R,R)-enantiomer. The hydrolysis of the amide (XVIII) with NaOH, followed by heating in refluxing toluene gives the (R,R)-lactone (XIX) (2, 3), which is finally treated first with NaOH inmethanol/water, and then with CaCl2 or calcium acetate (3, 4). 2) The condensation of the already described aldehyde (XI) with(S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol -1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S) (2, 3). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2 (2-4). 3) The condensation of4-cyano-3(R)-hydroxybutyric acid ethyl ester (XXV) with N,N-diphenylacetamide (R1 = R2 = Ph in XXVI) by means of LDA in THF gives 6-cyano-5(R)-hydroxy-3-oxo-N,N-diphenylhexanamide (XXVII), which is reduced with diethylmethoxyborane and NaBH4 in THF yielding 6-cyano-3(R),5(R)-dihydroxy-N,N-diphenylhexanamide (XXVIII). The protection of the two OH groups of (XXVIII) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XXX), which by reduction of its CN group by hydrogenation with H2 over RaNi in methanol/liquid ammonia gives (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]-N,N -diphenylacetamide (XXXI). The cyclization of (XXXI) with 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) (its synthesis is in section 6, Scheme 4) in refluxing toluene yields the protected dihydroxyheptanamide (XXXIII), which is deprotected with HCl in methanol to afford(3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N -phenylcarbamoyl)pyrrol-1-yl]-3,5-dihydroxy-N,N-diphenylheptanamide (XXXIV). Finally, this compound is hydrolyzed with NaOH and treated with calcium acetate in water (5). Scheme 18007203a. 4) The preceding reaction pathway can be repeated using other substituents for R1 and R2 in acetamide (XXVI) such as R1 = R2 = CH2Ph; R1 = R2 = Et; R1 = Bu, R2 = Me; R1 = t-Bu, R2 = CH2Ph; R1,R2 = -(CH2)5- (5). 5) The hydrolysis of methyl (Et or Bu)3(R)-(tert-butyldimethylsilyloxy)-4-cyanobutyrate (XXV) with NaOH gives the corresponding free acid (XXXVI), which is condensed with malonic acid mono-tert-butyl ester magnesium salt (XXXVII) by means of carbonyldiimidazole (CDI) yielding tert-butyl5(R)-(tert-butyldimethylsilyloxy)-6-cyano-3-oxohexanoate (XXXVIII). The desilylation of (XXXVIII) with tetrabutylammonium fluoride in acetic acid affords the expected hydroxylated ketoester (XXXIX), which is reduced with diethylmethoxyborane and NaBH4 in methanol giving tert-butyl 6-cyano-3(R),5(R)-dihydroxyhexanoate (XL). The protection of the two OH groups of (XL) with acetone dimethylketal (XXIX) and methanesulfonic acid affords the 1,3-dioxane (XLI) (6), which by reduction of its CN group by hydrogenation with H2 overPd/C gives intermediate (4R,6R)-2-[6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLII). The cyclization of (XLII) with 4-(4-fluorophenyl-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) in refluxing toluene yields the protected dihydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water (7). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means of triethylamine in hot ethanol (7). 6) The (4R,6R)-2-[6-(cyanomethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLI) can also be obtained by reaction of (4R,6R)-2-[6-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetic acid tert-butyl ester (XLVIII) with tosyl chloride to give the corresponding tosylate (XVIX), which is then treated with NaCN (6). 7) The tert-butyl6-cyano-5(R)-hydroxy-3-oxohexanoate (XXXIX) can also be obtained by condensation of methyl 4-cyano-3(R)-hydroxybutyrate (L) with tert-butyl acetate (XXIII) by means of LDA in THF (6). 8) The synthesis of the 4-(4-fluorophenyl)-2-isobutyryl-4-oxo-N-phenylbutyramide (XXXII) is carried out as follows: The condensation of 4-methyl-3-oxo-N-phenylpentanamide (XLIV) with benzaldehyde (XLV) gives2-benzylidene-4-methyl-3-oxo-N-phenylpentanamide (XLVI), which is then condensed with 4-fluorobenzaldehyde (XLVII) by means oftriethylamine in hot ethanol (7). 9) The cyclization of (XXXII) with intermediate (XLII) (preceding synthesis) in refluxing toluene yields the protected dehydroxyheptanoate (XLIII), which is deprotected with HCl in methanol and finally hydrolyzed with NaOH and treated with calcium acetate in water. References 1. Roth, B.D. (Warner-Lambert Co.). Trans-6-[2-(3- or 4-carboxamido-substd.pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis. EP 247633, US 4681893. 2. Roth, B.D., Blankley, C.J., Chucholowski, A.W., Ferguson, E., Hoefle, M.L., Ortwine, D.F., Newton, R.S., Sekerke, C.S., Sliskovic, D.R., Stratton, C.D., Wilson, M.W. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem 1991, 34: 357-66. 3. Roth, B.D. (Warner-Lambert Co.). (R-(R*R*)-2-(4-Fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl-3-phenyl-4-[(phenylamino)-carbonyl]-1H-pyrrole-1-heptanoic acid, its lactone form and salts thereof. EP 409281, JP 91058967, US 5273995.4. Milb, N., Muhammad, N.A., Weiss, J., Nesbitt, R.U. (Warner-Lambert Co.). Stable oral CI-981 formulation and process for preparing same. EP 680320, JP 96505640, WO 9416693.5. Butler, D.E., Le, T.V., Nanninga, T.N. (Warner-Lambert Co.). Process fortrans-6-[2-(substd.-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis. US 5298627. 6. Brower, P.L., Butler, D.E., Deering, C.F., Le, T.V., Millar, A., Nanninga, T.N., Roth, B.D. The synthesis of (4R-cis)-1,1-dimethylethyl6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate, a key intermediate for the preparation of CI-981, a highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2279-82. 7. Baumann, K.L., Butler, D.E., Deering, C.F., Mennen, K.E., Millar, A., Nanninga, T.N., Palmer, C.W., Roth, B.D. The convergent synthesis of CI-981, an optically active, highly potent, tissue selective inhibitor of HMG-CoA reductase. Tetrahedron Lett 1992, 33: 2283-4. 8. McKenzie, A.T. (Warner-Lambert Co.). Form III crystalline(R-(R*,R*))-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methyl-ethyl) -3-phenyl-4-((phenylamino)carbonyl)-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703958. 9. Lin, M., Schweiss, D. (Warner-Lambert Co.). Novel process for the production of amorphous [R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid calcium salt (2:1). WO 9703960. 10. Briggs, C.A., Jennings, R.A., Wade, R.A., Harasawa, K., Ichikawa, S., Minohara, K., Nakagawa, S. (Warner-Lambert Co.). Crystalline[R-(R*,R*)]-2-(4-fluorophenyl)-beta,delta-dihydroxy-5-(1-methylethyl) -3-phenyl-4-[(phenylamino)carbonyl]-1H-pyrrole-1-heptanoic acid hemi calcium salt (atorvastatin). WO 9703959.〖来源〗J Med Chem〖合成路线〗〖标题〗Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus〖合成方法〗2) The condensation of the previously described aldehyde (XI) with (S)-(+)-2-acetoxy-1,1,2-triphenylethanol (XX) by means of lithium diisopropylamide (LDA) in THF gives5-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl)pyrrol-1-yl]-3(R)-hydroxypentanoic acid2-hydroxy-1(S),2,2-triphenylethyl ester (XXI), which is trans-esterified with sodium methoxide in methanol/THF yielding the expected methyl ester (XXII). The condensation of (XXII) with tert-butyl acetate (XXIII) by means of LDA in THF affords(R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(N-phenylcarbamoyl) pyrrol-1-yl]-5-hydroxy-3-oxoheptanoic acid tert-butyl ester (XXIV), which is reduced with triethylborane and NaBH4 in THF, hydrolyzed with NaOH, lactonized by heating in refluxing toluene and finally submitted to fractional crystallization in order to separate the two diastereomers of the obtained lactone, (R,R) and (R,S). The(R,R)-diastereomer (XIX), already obtained, is finally treated with NaOH and then with CaCl2.〖作者〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.〖参考〗Roth, B.D.; Blankley, C.J.; Chucholowski, A.W.; Ferguson, E.; Hoefle, M.L.; Ortwine, D.F.; Newton, R.S.; Sekerke, C.S.; Sliskovic, D.R.; Stratton, C.D.; Wilson, M.W.; Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)eth IĶ 셈睋서睋쓰睎쓜睎 Ʈ 0 ๙ιᇐ ꀀÑ밸-00AA004CLSID\{0E59F1D5-1FBE-11D0-8FF2-00A0D10038BC}e Ǔ ℐ ul> li>a href=〖出处〗。
调血脂药阿托伐他汀钙的合成探究

调血脂药阿托伐他汀钙的合成探究摘要】综合国内外关于阿托伐他汀钙的报道和文献,通过大量的实验研究,对阿托伐他汀钙的合成技术进行优化,探讨适合工业化生产阿托伐他汀钙的新工艺技术,在进行阿托伐他汀钙的合成过程中,考察水解反应时间、水解反应温度及溶剂对阿托伐他汀钙收率的影响,得出阿托伐他汀钙合成技术的优缺点,为大规模、工业化生产阿托伐他汀钙提供依据。
【关键词】阿托伐他汀钙;调血脂药;合成【中图分类号】R97 【文献标识码】A 【文章编号】1007-8231(2015)11-0218-02阿托伐他汀钙是临床上常见的用于治疗高胆固醇血症等胆固醇升高患者,为临床安全有效的降脂药物。
其是还原酶的选择性及竞争性抑制剂,主要通过抑制肝脏内HMG-CoA还原酶及胆固醇的合成从而起到降低脂蛋白及胆固醇水平的作用[1]。
该药是美国Warner-Lambert公司的Paker-Dvais药厂和Pfizer公司共同研制出来的,是第三代他汀类的调脂药,在临床上被广泛的运用[2]。
本文详细论述了阿托伐他汀钙的合成方法。
1.阿托伐他汀钙的合成1.1 线性合成方法仪器:选用核磁共振波谱仪、元素分析仪器、高效液相色谱仪,红外分光光度仪。
1.1.1中间体合成在圆底烧瓶中加入(4R,6R)一6氨乙基一2,2一二甲基一1,3一二氧己环一4一乙酸叔丁酯,氟一d一[2一甲基一1一氧丙基]一7一氧代一N,13一二苯基苯丁混合,加入甲苯200ml及对甲苯磺酸,对其进行加热,回流,采用分流器将反应生成的水分去除,6小时后,进行降温,随后进行减压蒸发掉甲苯,加入MTBE100ml分散,将固体过滤。
1.1.2阿托伐他汀钙的合成方法在圆底烧瓶中加入上述中间体合成的产物,并加入40毫升甲醇、200ml氢氧化钠的水溶液,加热,待水解后溶液变清,进行降温,采用叔丁基甲基醚80毫升重复洗涤;洗涤完成后,将水溶液重新加热,搅拌均匀,加入水溶液醋酸钙搅拌2小时后,冷却至20摄氏度,过滤,将产物水洗,进行减压干燥,直至白色固体出现。
阿托伐他汀侧链结构式

阿托伐他汀侧链结构式
阿托伐他汀(Atorvastatin)是一种降脂药物,属于他汀类药物,用于治疗高胆固醇血症和预防心血管疾病。
其侧链结构是该分子中与羟基苯甲酸酯基团相连的部分,对于药物的吸收、分布、代谢和排泄具有重要作用。
阿托伐他汀的侧链结构包括一个2-[4-氟苯基]-β,δ-二羟基戊酸酯基团。
这个侧链通过酯键与阿托伐他汀分子的主体部分相连。
具体的化学结构如下:
OH
│
HOOC-CH=CH-CH2-CH2-O-CO-R
│
OCH2CH2NHCO
│
R'-COOH
其中,R代表阿托伐他汀分子的主体部分,包含一个内酯环和一个噻唑烷酮环;R'则是指与酯基相连的烃基,在阿托伐他汀中,R'为一个氢原子。
请注意,这个侧链结构式是一个简化的表示,实际的分子结构可能更为复杂,并且需要考虑立体化学因素。
在药物设计和合成中,侧链的精确结构对药物的活性、稳定性和生物利用度都至关重要。
阿托伐他汀钙的合成工艺

摘 要 :对阿托伐他汀钙合成工 艺进行分析 。通过 大量 实验 ,全 面优化 阿托伐他 汀钙的合成路线 ,选择与工业化生产需求 相符合 的新 工艺,构建 阿托伐他 汀钙 的生 产工艺。结果表 明 :利用元素分析、核磁共振氢谱 、红外光谱等分析手段 ,分析 阿托 伐他 汀钙 的结构表征 ,该合 成工艺具有有机 溶剂残 留量低、产品纯度 高、产率 高、操作 简单 、原料 易得 、反应条件温和 等基本 特征 ,与 药用标准相符合 ,适合 用于工业化生产 中。
明显 降 低 ,使 社 会 效 益 、 经 济 效 益 明 显 提 高 。本 文 对 阿 托 伐 收率为 70.00% ;水解反应 时 间为 i.2h时,收率为 81.00% ;水
他 汀 钙 的 合 成 工 艺进 行全 面 分 析 ,现 总 结 如 下 。
解 反 应 时 间 为 1.5h时 ,收 率 为 85.00%;水 解 反 应 时 间 为 1.8h时 ,
为 美 国 辉 瑞 公 司 ,可 用 于 高 胆 固 醇 血 症 的 临 床 治 疗 以及 预 防 行 2h搅 拌 , 并 给 予 抽 滤 ,在 70℃ 温 度 下 将 滤 饼 放 置 在 真 空 状
中 【1]。 阿 托 伐 他 汀 钙 主 要 是 采 用 对 血 脂 异 常 患 者 和 载 脂 蛋 白 态 下 30h,测 量产 品质 量 。
阿 托 伐 他 汀 钙 在 最 后 一 步 合 成 时 , 在 改 变 水 解 反 应 时 间
最近几 年, 国内外开始重视 研究 阿托 伐他 汀钙药物 的制作 工 的 情 况 下 , 维 持 其 他 条 件 不 变 ,可 以得 知 以下 数 据 :水 解 反
艺 ,通 过对其 合成工 艺进行优 化 ,促进 用药成 本、生产 成本 应 时 间 为 0.5h时 ,收 率 为 45.00% ;水 解 反 应 时 间 为 1.0h时 ,
阿托伐他汀说明书

商品名:立普妥英文名:Lipitor通用名:阿托伐他汀钙外文名:Lipitor,AtorvastatinCalcium汉语拼音:liputuo性质:立普妥是白色、椭圆、薄膜衣的阿托伐他汀钙盐片,是一种合成的选择性、竞争性HMG-CoA还原酶抑制剂(他汀类),其分子式为(C33H34FN2O5)2Ca·3H2O,分子量为1209.42。
片剂的一面凹刻“PD155”,另一面有“10”的字样。
药理毒理:立普妥为HMG-CoA还原酶选择性抑制剂,通过抑制HMG-CoA还原酶和胆固醇在肝脏的生物合成而降低血浆胆固醇和脂蛋白水平,并能通过增加肝细胞表面低密度脂蛋白(LDL)受体数目而增加LDL的摄取和分解代谢。
立普妥也能减少LDL的生成和其颗粒数。
立普妥还能降低某些纯合子型家族性高胆固醇血症(FH)的低密度脂蛋白胆固醇(LDL-C)水平,而一类型的人群对其他类型的降脂药物治疗很少有应答。
立普妥能降低纯合子和杂合子家族性高胆固醇血症、非家族性高胆固醇血症以及混合性脂类代谢障碍患者的血浆总胆固醇(TC)、LDL-C和载脂蛋白B(ApoB),还能降低极低密度脂蛋白胆固醇(VLDL-C)和三酰甘油(TG)的水平,并能不同程度地提高血浆高密度脂蛋白胆固醇(HDL-C)和载脂蛋白A1(ApoA1)的水平。
药代动力学:吸收:口服后迅速吸收,1-2小时内达到最大血浆浓度,吸收程度随口服剂量的增加而成正比例地增加。
绝对生物利用度约为12%,抑制HMG-CoA还原酶的全身利用度约为30%。
无论是否与食物同时服用或在一天中无论何时服用,其降低血浆LDL-C的效果都相似。
分布:平均分布容积是381升,其中98%以上与血浆蛋白结合。
代谢:阿托伐他汀在体内被代谢成为邻羟基化和对羟基化代谢产物,以及各种β-氧化产物。
其对循环HMG-CoA还原酶抑制活性大约70%源于活性代谢产物。
消除:阿托伐他汀及其代谢产物通过肝脏和/或肝外途径代谢后主要经胆汁排除。
调血脂药阿托伐他汀钙的合成研究进展

随着 对 高 脂 血 症 的 不 断 研 究 和综 合 治 疗 经 验 的 积 累 , 疗 高 治
几 得 到 关 键 中 间 体 2一( 3一[ 4一氟 苯 基 ) 一异 丙 基 一 苯 基 一 一5 3一
+
F
脂血症的药物品种及其使用情况也在不断地发生变化…。 羟基 一 3一
3一甲基 戊 二 酰 辅 酶 A H ( MG— o 还 原 酶 抑 制 剂 , 称 他 汀 类 C A) 简 (a n) 物 , s t s药 ti 由于 作 用 机 制 新 颖 , 用 范 围 较为 是 目前 最 为 经典 和有 效 的 调血 脂 药 - 。 耐 被 3 ]
a i s u t e i o te r g 1 er snh s ) T e oh ro e i t pe ae ci l3 5一 i —d y r y h pa o c rg e tte f m te c t c r n h i ( n a y tei . h te n s o rp r hr , es i do e t i ai f m n,h o h d r u t n i s a h x n c d a r
线提 供 参 考 。 关键 词 : 血 脂 药 : 调 阿托 伐 他 汀钙 : 成 合
中图 分 类号 : 7 6 T 6 . 1 R92 . ; Q40 3 文 献标 识 码 : A 文章 编 号 :0 6— 9 1 2 1 )1— 0 4 3 1 0 4 3 (0 0 2 0 0 —0
药学专论
21 年第 l 卷第 2 期 00 9 1
调血 脂 药 阿托伐 他 汀 钙 的合成 研 究 进展
张宜凡 虞心红 ,
(.上 海 医药高等 专科 学校 药学系 , 海 1 上 2 11 ; 2 0 3 8 .华东理 工 大学 药学院 , 上海 20 3 ) 0 1 7
阿托伐他汀钙中间体合成研究

阿托伐他汀钙中间体合成研究作者:刘艳玲乔向勇来源:《卷宗》2016年第06期摘要:本文以2-[2-(4-氟苯基)-2-氧代-1-苯基乙基]-4-甲基-3-氧代-N-苯基戊酰胺和(4R,6R)-6-氨乙基-2,2-二甲基-1,3-二氧六环-4-乙酸特戊酯通过Paal–Knorr反应合成出了阿托伐他汀钙中间体缩合物,分析得出了合成阿托伐他汀钙缩合物的最佳反应时间为28h,且收率高达85%。
这对阿托伐他汀钙工业化生产具有重要意义。
关键词:阿托伐他汀钙;Paal–Knorr反应;中间体;合成Abstract:The Intermediate of Atorvastatin Calcium was synthesized from 2-(2-(4-fluorophenyl)-2-oxo-1-phenylethyl)-4-methyl-3-oxo-N-phenylpentanamide and 1-((4R,6R)-6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl)-3-(neopentyloxy)propan-2-one by Paal-Knorr reaction with yield of about 85%. Also, the best reaction condition is made certain: if the resulting mixtureis heated under reflux for 28h. The present method is advantageous for the large-scale synthesis.Key words:Atorvastatin Calcium, Paal-Knorr reaction, Intermediate, Synthesis7-[2-(4-氟苯基)-3-苯基-4-(苯胺基甲酰基)-5-(2-丙基)吡咯-1-基]-3,5-二羟基庚酸钙(阿托伐他汀钙)可治疗其总胆固醇升高,低密度脂蛋白胆固醇升高,载脂蛋白B升高和甘油三酯升高。
阿托伐他汀

主治:
阿托伐他汀的主要用途是治疗血脂异常 和预防心血管疾病。 • 血脂异常:高胆固醇血症、混合性高血 脂和高甘油三酯血症等 • 心血管疾病:预防二级冠状动脉心脏病 和多种危险因素,如心肌梗塞,中风,不 稳定心绞痛和血运重建。 •
禁忌:
•
1、活动性肝病:胆汁淤积,肝性脑 病,肝炎,黄疸 2、AST或ALT水平 3、原因不明的海拔 4、怀孕 5、母乳喂养
阿托伐他汀(Atorvastatin, INN、Lipitor/Pfizer)是治疗高 胆固醇血症和冠心病的常见药 物。由辉瑞公司装造,销售药 品名为胆固清或立普妥(Lipitor)。
它的结构式与立体结构式
药物作用机理 :•阿托伐他汀是HMG-CoA还原酶的竞争性抑制剂。 然而,不同于大多数人来说,它是一个完全人工 合成的化合物。 HMG-CoA还原酶催化还原3 - 羟 基-3 - 甲基 - 辅酶A(HMG-COA)甲羟戊酸,这 是在肝脏胆固醇合成的限速步骤。抑制酶降低从 头合成胆固醇,提高肝细胞表达的低密度脂蛋白 受体(LDL受体)。增加肝细胞低密度脂蛋白的吸 收,降低血液中的低密度脂蛋白胆固醇量。像其 他他汀类药物,阿托伐他汀也降低血甘油三酯水 平,并略有增加高密度脂蛋白胆固醇水平。在临 床试验中,阿托伐他汀阻止胆固醇的吸收。
后记:
阿托伐他汀于1985年由布鲁斯·罗斯-帕克-戴 维斯华纳-兰伯特公司首次合成(现为辉瑞公司 /Pfizer),是制药历史上销售最好的药物。它自 1996年被美国食品药品监督管理局批准以来,累 计销售额超过1250亿美元。并连续保持此销售冠 军纪录达十年。通用阿托伐他汀,由沃森制药公司 和兰伯西实验室制造,并于2011年11月30日开始 于美国上市。
阿托伐他汀侧链中间体的合成

阿托伐他汀侧链中间体的合成阿托伐他汀侧链中间体的合成摘要:阿托伐他汀是HMG-CoA还原酶的选择性、竞争性抑制剂,能有效地降低血脂。
本文着重对其活性中心侧链中间体的合成进行了专题分析,以寻求较好的合成路线。
关键词:阿托伐他汀侧链中间体合成中图分类号:文献标志码:文章编号:阿托伐他汀是目前全球处方量最多的降胆固醇药物。
由于其含有(3R,5S)-双羟基的侧链,并且要求e.e.≥99.5%,因此合成此侧链的中间体很有挑战性,人们通过各种方法合成,得到的侧链中间体也就有所差别,本文就介绍一下几种中间体的合成路线。
1 中间体TBIA(tert-Butyl isopropylidene amine)的合成主要是通过DERA(EC4.1.2.4)突变体Ser238Asp催化的醛醇缩合反应得到内酯化合物,再经过一系列的反应得到TBIA,具体的合成步骤如下:N31O+O+ODERA6d2OHBr2,BaCO3N3OOMeONa,MeOH,83%t-BuOK,t-BuOH,72%[1]OHN3OHOORcamphorsulfonic acid2,2-dimethoxypropane,76%N3OOOOMeMeOH-H2OLiOH,83: R=Me3b R=t-BuON35O4OOHBoc2ODMPA,86%N37536OOPh3P3d,72%OOOH2NOOOTBIA2 阿托伐他汀钙中间体ATS-8的合成ATS-8的中文名称为6-氰甲基-2,2-二甲基-1,3-二氧戊环-乙酸叔丁酯,英文名称为(4R,6R)-1,1-dimethylethyl-6-cyanomethyl-2,2-dimethyl-1,3-dioxane-4-acetate,其合成以高烯丙醇为原料生成碘代的内酯,具体合成步骤如下:OOHBuLi/THF,CO2,I291%1I2ONC4t-BuOH,DCC,DMAP/CH2Cl2,rtOH6NCATS-8[2]OOp-TsOH,acetone,rt90%IOO3ONC5OCH O°KCN/DMSO,40 C75~80%OOsO4-NaIO4/dioxane-H2O orO3,Me2S65~70%OO°CrO3-H2SO4/acetone,0 C70%NCOOOOO3 阿托伐他汀侧链的1,3-二醇中间体的合成该方法主要是通过L-脯氨酸催化正丁醛发生α-氨基氧化以及碘发生的分子内亲电子[3]环化反应,具体的合成路线如下:OHBnO1CHOaBnO2 R=H3 R=MsORbBnO3OcOOOI9ORBnO4 R=H5 R=BocdXOBoceN36 X=OH7 X=OMs8 X=N3OHOgN310OHOHCN11 (1,3-diol)fN3°Reagents and conditions:(a)(i)PhNO,L-proline(25mol%),CH3CN,-20 C,24h then MeOH,NaBH4;(ii)CuSO4(30mol%),°MeOH,0 C,10h,87%(qver twosteps);(b)(i)MsCl,Et°N,CHCl,0C,15min,92%;(ii)KCO,MeOH,rt,1h,95%;(c)vinylma32223gnesium bromide,THF,CuI,-40 C,1h,92%;(d)(i)(Boc)2O,DMAP,CH3CN,rt,5h,95%;(ii)DDQ,CH2Cl2:H2O(2:1),rt,20h,°°85%,(e)(i)MsCl,Et3N,CH2Cl2,0 C,30min,94%;(ii)NaN°3,DMF,60C,2h,83%;(iii)NIS,CH3CN,-40 to 0 C,20h,87%;(f)°K2CO3,MeOH,0 C tort,2h,96%;(g)NaCN,Ti(OPr)°4,n-Bu4NI,DMSO,70 C,6h,80%.4 中间体R(-)-4-氰基-3-羟基丁酸乙酯的合成以环氧氯丙烷为原料得到2-羟基-1,3-二氰基丙烷,此合成的关键步骤就是利用腈水解酶催化该化合物发生不对称反应,从而得到目的产物。
阿托伐他汀合成

题目:阿托伐他汀中间体及其合成工艺研究姓名:髙晔学号:201121030219指导老师:温艳珍时间:2014-06-14摘要:阿托伐他汀是一种羟甲戊二酰辅酶A还原酶抑制剂,通过抑制胆固醇生物合成的限速酶羟甲戊二酰辅酶A还原酶,从而起到抗高血脂症的作用。
大量临床研究表明,他汀类药物高效、安全,为调脂药物中的首选药物。
本文对近年来阿托伐他汀中间体,及阿托伐他汀合成工艺进行总结。
希望找到一种更好的合成阿托伐他汀的方法,应用于实践,帮助更多的患者。
关键词:阿托伐他汀中间体合成目录摘要 (I)第一章他汀类药物的研究背景及意义 (1)1.1他汀类药物简介 (1)1.2阿托伐他汀的简介 (2)第二章阿托伐他汀中间体的合成 (5)2.1 4-氟-α-(2-甲基-1-氧丙基)-γ-氧-N,β-二苯基苯丁酰胺的合成52.2异丁酰乙酸酯的合成 (5)2.3 4-甲基-3-氧-N-苯戊酰胺的合成 (6)2.3 4-甲基-3羰基-N-苯-2-(苯亚甲基)戊酰胺的合成 (6)2.4 4-氟-a-(2-甲基-卜氧丙基)-Y-氧-N,β-二苯基苯丁酰胺的合成6 第三章阿托伐他汀的合成路线 (7)3.1路线一:以2-(4-氟苯基)-2-溴-乙酸乙酯为起始原料 (7)3.2路线二;以异丁酰乙酰苯胺为起始原料 (8)3.3路线三 (8)3.4路线四:以阿托伐他汀醛为起始原料 (9)3.5路线五;以苯乙酸为起始原料 (10)3.6路线六:以L一缬氨酸为起始原料 (11)3.7路线七: (11)第四章总结 (13)参考文献 (14)第一章他汀类药物的研究背景及意义1.1他汀类药物简介心血管疾病(包括冠心病和动脉粥样硬化)是一类严重威胁人类健康的疾病。
近年来,随着人们生活水平的提高和工作节奏的加快,心血管疾病的发病率和死亡率都呈明显的上升态势。
研究表明,血浆胆固醇(CH)水平增高,特别是低密度脂蛋白胆固醇(LDL-CH)水平的增高,是早发性动脉粥样硬化和冠心病的重要因素。
浅谈阿托伐他汀的作用机理及合成

2018年第18期广东化工第45卷总第380期 ·97 ·浅谈阿托伐他汀的作用机理及合成陶溪,王蕙,岳伟杰(盐城工学院化学化工学院,江苏盐城224051)[摘要]阿托伐他汀是一种他汀类的药物,在降低心血管疾病和死亡风险方面,有着高效、安全的长期临床效益。
本文对阿托伐他汀的药动力学机理进行分析,在此基础上,对阿托伐他汀合成路径研究综述。
研究表明:阿托伐他汀主要有线性、收敛性两种合成策略,Paal-Knorr吡咯法通常被认为是最广泛的使用方法。
[关键词]阿托伐他汀;机制;合成路径[中图分类号]TQ [文献标识码]A [文章编号]1007-1865(2018)18-0097-02Mechanism and Synthesis of AtorvastatinTao Xi, Wang Hui, Yue Weijie(Yancheng Institute of Technology, School of Chemistry and Chemical Engineering, Yancheng 224051, China) Abstract: Atorvastatin is a statin drug with high-efficiency, safe long-term clinical benefits in reducing the risk of cardiovascular disease and death. In this paper, the pharmacokinetic mechanism of atorvastatin was analyzed, and on this basis, the research on the synthesis pathway of atorvastatin was reviewed. Studies have shown that atorvastatin has two linear and astringent synthetic strategies, and the Paal-Knorr pyrrole method is generally considered to be the most widely used method.Keywords: atorvastatin;mechanism;synthetic pathway1 引言1.1 背景简介阿托伐他汀,开始作为一种钙盐在辉瑞公司以“立普妥”的商品名而销售,并于1997年在美国获得了FDA的批准。
调血脂药阿托伐他汀钙的合成研究进展

调血脂药阿托伐他汀钙的合成研究进展张宜凡;虞心红【摘要】综述治疗高脂血症的他汀类药物阿托伐他汀钙的合成方法,根据文献报道归纳出阿托伐他汀钙的合成路线有两种,一种是先合成出取代的吡咯环,然后在环上引入手性的3,5-顺式双羟基庚酸结构(线性合成),一种是先制备手性的3,5-顺式二羟基庚酸片断,然后与1,4-二羰基化合物环合得到吡咯环结构(汇聚合成).还比较了各合成工艺方法的优缺点,为寻找一条适合大规模、工业化生产的工艺路线提供参考.【期刊名称】《中国药业》【年(卷),期】2010(019)021【总页数】3页(P4-6)【关键词】调血脂药;阿托伐他汀钙;合成【作者】张宜凡;虞心红【作者单位】上海医药高等专科学校药学系,上海,201318;华东理工大学药学院,上海,200137【正文语种】中文【中图分类】R972+.6%TQ460.31随着对高脂血症的不断研究和综合治疗经验的积累,治疗高脂血症的药物品种及其使用情况也在不断地发生变化[1]。
3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,简称他汀类(statins)药物,由于作用机制新颖,应用范围较广,疗效显著,毒副反应小,耐受性好,被认为是目前最为经典和有效的调血脂药[2-3]。
阿托伐他汀是第3代HMG-CoA还原酶的选择性、竞争性抑制剂,对原发性高胆固醇血症、高甘油三酯血症、家族性高胆固醇血症和混和性高脂血症等各种类型的血脂异常均有效。
阿托伐他汀钙(atorvastatin calcium)的商品名为立普妥(Lipitor),化学名为(R,R)-2-(4-氟苯基)-β,γ-二羟基-5-(1-甲基乙基)-3-苯基-4-苯胺基酰基-1H-吡咯-1-庚酸钙盐(2∶1)三水化合物,分子式C66H68CaF2N4O10·3H2O,相对分子质量为1 209.42。
其化学结构由两部分组成,即有取代的吡咯环结构(主环)和手性3,5-顺式二羟基庚酸结构(侧链)。
阿托伐他汀钙杂质研究

阿托伐他汀钙杂质研究一、基本情况⏹阿托伐他汀钙(Atorvastatin Calcium)是一种人工合成的HMG-CoA还原酶抑制剂,是临床常用降血脂药⏹原研发企业为辉瑞公司(Pfizer)⏹目前国内有原研发企业产品及仿制药上市⏹原料药及其制剂质量标准收载于国家新药转正标准、USP33、EP7.1收载原料药二、杂质限度要求⏹最大日剂量:口服80mg/天四、杂质谱分析⏹工艺杂质:杂质A(去氟阿托伐他汀)⏹杂质C(双氟阿托伐他汀)、杂质F、杂质G⏹工艺杂质及降解产物⏹杂质B(非对映异构体)⏹杂质E(对映异构体)⏹降解产物:杂质D(环氧化物)⏹杂质H(内酯化降解产物)⏹EP中8个已知杂质结构五、本品常用制备工艺为先分别合成主环和侧链,然后缩合、脱保护、成盐。
在常规条件下前述8个杂质均可能出现可作为杂质研究的重要参考依据⏹ 需要注意存在的杂质并不限于前述8个已知杂质六、原料药杂质研究思路⏹ 以EP 标准中的检验方法及限度为参考依据,进行必要的方法验证⏹ 试制样品杂质检查结果符合EP 标准要求,无超过鉴定限度的其它杂质——达到研究目标 ⏹ 若杂质谱与EP 标准一致,但杂质量超过限度要求——完善工艺(精制:优化工艺参数;控制起始原料及中间体质量)⏹ 出现超过鉴定限度(0.1%)的新杂质——鉴定结构,分析原因,修改完善工艺,降低杂质量至0.1%以下。
⏹ 若新杂质不超过质控限度(0.15%),且经鉴定结构明确不是毒性 杂质,可订入质量标准中进行控制七、制剂杂质研究思路⏹ 应选择质量符合要求的原料药⏹ 若原料药质量符合要求,制剂杂质研究重点考察降解产物⏹ 关注降解途径及降解产物⏹ 阿托伐他汀主要降解途径为内酯化降解及氧化降解,氧化降解产物除环氧化物(杂质D )外,尚有其它降解产物。
⏹ EP 、USP 等未收载制剂。
与原研厂产品的对比研究对评价杂质检查方法及限度合理性的尤其重要八、申报品种的杂质研究情况⏹ 已有多家企业申报,研究工作的系统性和深入性参差不齐,有的品种差距较大 ⏹ 有以下几种情况⏹ 仅按照国家标准进行有关物质检查⏹ 在国家标准基础上,增加了对映异构体检查⏹ 在分析杂质谱的基础上,对各已知杂质、光学异构体均进行了研究与控制,并与上市产品进行了质量对比。
阿托伐他汀钙合成工艺

阿托伐他汀钙合成工艺
阿托伐他汀钙合成工艺反应原理:
N
F
O O
O
H
2
O O O
N
F
O
N
O O O +
AT-9M-4AT-10
N
F
O N
O O O
N
F
O
N
OH OH O
N
F
O N
OH OH O
N
F
O
N O
OH OH O
2
Ca++
AT-10AT-11
AT-11
AT
精
制
精品阿托伐他汀钙
AT-10的合成
在有回流冷凝器的园底烧瓶中,加入41.7克(0.1mol)M-4,28.7克(0.1mol)AT-9,0。
002mol 的对甲苯磺酸,200ml甲苯,加热回流反应,并用分水器将反应生成的水分掉,6小时后,降温,
减压整除甲苯,加入100ml叔丁基甲基醚(MTBE)分散,过滤出固体,水洗,直接用于下一步反应。
AT的合成。
园底烧瓶中,加入上一步物料,甲醇40ml,氢氧化钠(0。
105mol)的水溶液200毫升,加热到55度水解1.5小时至溶液彻底变澄清,降至室温,用80*2叔丁基甲基醚洗涤;水溶液(即AT-11)重新加热到55度,搅拌滴加一水合醋酸钙(0.53mol)的水溶液,并在此温搅拌两个小时,冷却至20度,过滤,水洗得产物,70度减压干燥,得白色固体。
总收率85%。
AT 的精制:
用甲醇水加热回流脱色,冷却析晶,过滤得白色晶体。
70度以下减压干燥。
收率94% .。
阿托伐他汀钙合成路线

阿托伐他汀钙合成路线ChineseJournalofNewDrugs2006.V o1.15No.22阿托伐他汀钙合成路线中国新药杂志2006年第15卷第22期徐颂一(天津药物研究院医药信息中心,天津300193),【摘要】对调血脂药阿托伐他汀钙的合成路线进行了归纳和分析.根据经过的中间体的不同,将合成路线分为两大类.经过中间体G的路线有一定的参考价值.为进一步工业化生产和技术创新打下基础.[关键词】阿托伐他汀钙;合成路线;中间体[中圈分类号】R972.6;R916.1[文献标识码]A[文章纳号】1003—3734(2006)22—1913—05GraphicalsyntheticroutesofatorvastatincalciumXUSong(CenterofPharmaceuticalInformation,TianjinInstituteofPharmaceuticalResearch,Tianji n300193,China)[Abstract]Thisreviewsummarizedthevarioussyntheticprocessesofatorvastatincalcium.T he syntheticroutemaybecategorizedintotwoclassesaccordingtotheintermediatesinvolved.T heroutewith theintermediateGshowsmorefavorablecommercialvalueinthemanufacturingscale. [Keywords]atorvastatincalcium;syntheticroutes;intermediate阿托伐他汀钙(atorvastatincalcium,1),为一种新型3一羟基一3-甲基戊二酰辅酶A(HMGCoA)还原酶抑制剂,化学名为:[R.(R,R)]_2-(4.氟苯基)-B,8-二羟基一5一(1一甲基乙基)-3苯基一4一[(苯胺基)-羰基].1日一吡咯.1-庚酸钙盐(2:1.)…,临床用其三水合物,具有同时降低血滤胆固醇和三酰甘油的作用,调脂作用高于其他HMG—CoA还原酶抑制剂,不良反应小,属于第三代全合成的他汀类调血脂药.笔者对近年来有关阿托伐他汀钙的合成方法进行了分析和总结,按其经过的合成中间体的不同分Hl广CHOAcOH为两大类:~类是经Paal-Knott反应进行的环合过程;另一类是经过中间体5-(4-氟一苯基).1(3-丙醛)-2一异丙基-4?苯基-1日-吡咯一3.羧酸苯胺(3)的过程.1经Paal-Knorr反应的合成路线由4-甲基-3-氧代.Ⅳ-苯基戊酰胺(4)与苯甲醛反应,再在三乙胺中以3-乙基-5-(2-羟乙基).4*甲基溴化噻唑错为催化剂与对氟苯甲醛合成得到4-(4-氟苯基)-2一异丁酰基-4-氧代-3一苯基-N?苯基丁酰胺(2).合成路线见图1J.cH..图12的合成将(R).4氰基.3一羟基-丁酸乙酯(6)与Ⅳ,Ⅳ一二硅烷基保护的中间体,再经酯缩合,脱硅烷基,选择苯基乙酰胺在二异丙氨基锂(LDA)作用,经过性还原,羟基保护,氢化还原得中间体B.也可以化Claisen缩合得到(R)-l5一氰基-5一羟基-3一氧代一N,N一二合物l6为原料,在碱性条件下,与4一氯苯磺酰苯基己酰氨(7),还原羟基得到[-(,)]-氰氯(较好的离去基团)酯化得l7,氰化,氢化还原得基'3,5.二羟基.N,N二苯基己酰氨(8),以.2,2一二甲化合物B,后两步收率85%一90%.以氯乙醛氧丙烷保护羟基得到(4.顺)-l5-氰甲基-2,2.二甲(is)为起始原料¨.',氰基直接取代氯得化合物2l,基-Ⅳ,Ⅳ.二苯基-1,3.二嗯烷-4-乙酰氨(9),还原氰在2,2-二甲基甲丙醚和对甲苯磺酸作用下得到化基得到(4R.顺)-l5.(2氨乙基)-2,2-二甲基一Ⅳ,Ⅳ-二合物22,成酰氯,叔丁基醇酯化亦可得到中间体B.苯基1,3.二嗯烷一4.乙酰胺,即中间体A].或将乙酸叔丁基酯与化合物27反应得到化合物以异抗坏血酸为原料],经过一系列反应得到l5,此步收率75%一80%,还原氰基得中间体B,是———1913———比较简单且经济的一种方法.用l,6一庚二烯一4一醇(33)与正丁基锂,二氧化碳和碘作用,在P—TsOH/无水丙酮条件下由碘化物(34)制得缩酮(35),收率90%,与KCN反应125h得到烯(36),收率75%~80%,再经问氯过氧苯甲酸(mCP.BA)和高碘酸氧化得醛(38)12.此路线中每步收率都较高,且省去了以往使用臭氧和有毒的四氧化锇的过程一.经氧化,还原氰基得中间体C.由(4R一顺)一6一(2一氰甲基)一4一(2一羟乙基)一2,2一二甲基一l,3一二烷一4一乙酸叔丁基酯(15)经氢化铝锂在无水乙醚中反应得到(4R一顺)-6一(2一氨基乙醇)一4一(2-羟乙基)一2,2-二甲基一l,3一二烷(40),收率92.1%,与甲基叔丁基醚(MtBE)和三甲基乙酸生垦堑药杂志2006年第l5卷第22期加热回流得(4R一顺)-6一(2-氨乙基)一4一(2-羟乙基)一2,2一二甲基一l,3一二烷三甲基乙酸盐,即中间体D.中间体B与37%盐酸室温反应30min脱去异亚丙基,所得盐酸盐(32)进一步与氯代叔丁基二甲基硅烷(TBDMSC1),三乙胺反应得7一氨基一(3R,5R)一二(叔丁基二甲基硅烷)庚酸叔丁基酯,即中间体E.由6一氰基一3,5一二羟基庚酸叔丁基酯(29)在二氯甲烷溶液中与二氯二异丙基硅烷,三乙胺Ⅳ一羟基苯并三唑(HOBT)中回流得到二异丙基硅保护的化合物(30),收率84.8%.该化合物经氢气还原氰基,三甲基乙酸成盐得6一(2一氨乙基)一2,2-二异丙基一l,3一二氧一2一硅环己烷一4一乙酸叔丁基酯三甲基乙酸盐,即中间体F.合成路线如图2.:::HOptlO--∥:=≥●.如cc一CrOW~SO34c.3335*\一一翮.cl2\v/..1●●●●●●●●●J由化合物18经2次催化剂2一脱氧核糖一5.磷酸醛缩酶(DERA)作用下与乙醛反应得中间体19,甲基化得到手性内酯化合物(2S,4R)一2-氯甲基一6一甲氧四氢吡喃一4一醇(51),经一系列反应得2一[(2尺,4R)一4一苯甲酰基一6一甲氧四氢吡喃一2一醇]乙胺,即中间体G.以氰基乙酸甲酯(56)为起始原料的路线,避免.\,,.S1DMSO/KCNI~.一,,<Ru(Ⅱ)/二膦H~HBr/CH3oHPh/NH¨IONaO.中国新药杂志2006年第l5卷第22期了使用昂贵的手性原料(R)一4-氰基一3一羟基丁酸乙酯(6)和低温非对应选择性硼烷还原.首先与Mt—BE和吗啉在55qC反应12~18h,得到3吗啉一3一氧代一丙腈(57),加氢还原得盐(58),进一步在苯乙酸作用下得3一氨基一1一吗啉一1.丙酮苯乙酸盐,即中间体H.'.合成路线如图3.1)PhCH(oMe)z,H2)KOtBuTHF一一Y,r.,.一Ru(11)I:~Ph,NH1)MeONa,MeOHoHCLo警2)PhCH,CooH—……图3经过中间体G,含芳基的1,4-二羰基物(2)与含氨基和2个手性碳原子的侧链中间体A,B,C,D,E,F,G,H缩合,经脱保护,水解,成盐等反应可制得1.合成路线如图2,3所示.2经中间体5一(4.氟-苯基)-1-(3-丙醛)-2-异丙基-4.苯基.1H-吡咯-3-羧酸苯胺(3)的路线Roth等研究了一条由化合物66通过1,3一偶极环加成反应合成5一(4-氟一苯基)一1一(3-丙醛)一2一异丙基一4一苯基一1H一吡咯一3一羧酸苯胺(3)的方法, 或直接由中间体2与氨基化合物经Paa1.Knorr反应,再脱保护基得到醛中间体(3).合成路线见图4.3.氧代一丁酸甲酯在碱性条件下与醛中间体(3)反应得到的外消旋体8O,经三丁基甲硼烷和硼氢化钠作用,脱保护基,引入(R)一(+)一d一甲基苯胺可以经过拆分成盐得1..,1,.ph~NH62Oc\/5677H的合成路线Roth等以醛中间体与手性试剂(S)一(+)一2一乙酰氧基一1,1,2一三苯基乙醇和溴化镁反应得到化合物81,对映选择性为97%,即经过重结晶纯化可得几乎单一的异构体,经酯交换,与乙酸叔丁酯缩合得到8一羟基一B一酮酯(83).硼氢化钠还原,水解,成盐可得到1.醛中间体(3)与(+)一二乙基酒石酸盐在四氢呋喃中与丙二烯硼酸(allenylboronicacid)不对称合成得到异构体74_2,再经Lindlar催化剂还原得到烯中间体75,也可直接与烯丙基三丁基锡反应直接得到75,手性路易斯酸可以增加产物的对应选择性,这里使用的是三溴化硼和(s,S)一1,2-二胺一1,2一二苯乙烷.顺.甲磺酰胺,使对映选择性达94.4%,再经丙烯酰氯和二甲氨基吡啶(DMAP)作用得到酯中间体76,Grubb's催化剂和丙烯酸酯成环得化合物~1915一.浠一^■●●●●●●●●●●●.●●●●AoaHC12n.]卜2.oChineseJournalofNewDrugs2006,V o1.15No.2277,经1,4-加成,氢解,成盐得到1.合成路线如图4. Ph.三CooH+PhNH——+Ph三—lLNHPh中国新药杂志2006年第l5卷第22期1】THF21AcOH晶FPhNHPhNH图4经过中间体3的合成路线3经过其他中间体的路线Pandey等发展了另一条经过1,3-偶极环加成反应的路线,合成了关键中间体Ⅳ3,4-二苯基-5- (4-氟苯)-2-异丙基-1H-3-吡咯酰胺(91),见图5.此路线以天然,J一缬氨酸(85)为起始原料,研究发现H1)HCl,CH30H2)NH3.町83%,∞№/HN--CH2PhEt3NPhNHNaBH4/Et3BF2)H202化合物89和90并不是以1:1的比例得到,而是化合物89更多一些,而且很容易由重结晶分离而得, 这就提高了中间体91的收率.此方法方便经济,进一步研究正在进行中,有可能会成为合成阿托伐他汀钙的新路线.2)NaOH,CH3OH95%FnNa.NH383%OPh—c;ij,一NH—Ph8880%89PhHN0C,./Ph人CH2Ph90图5经过冥他中1日J体的合成路线综上所述,可以看出,经过中间体G的合成路[3]BUTLERDE,DEERNGCF,MILLARA,.Pm...线步骤短,收率高,所用试剂方便经济,已广泛用于rans?(2-(im..pyrrol-1-y)y1)PYY".2.inhi.有机合成反应中,是一条有参考价值的合成路线."....synthesis:u'00..[P]?一.一? [作者简介]徐颂(190一)'女,硕士,主要从事药物化学Pr....f0th.reprali..fHMG.c.Aredt..ihd及医药信息研究.联系电话:(022)23006829,E-mail:xs_tjiprintemediatec.mp.undsemployedtherein:US,4611067[P].@.1986-09-09[参考文献][1]卢海儒.立普妥[J].中国新药杂志,2001,10(12):943—945[2]BAUMANNKL,BUTLERDE,DEERINGCF,eta1.Thecon—vergcntsynthesisofCI~81,anopticallyactive,highlypotent[5]tissBeselectiveinhibitorofHMG.CoAreduetase[J].Tetrahedron[6] Lett.1992.33(17):2283—2284—1916一BROWERPL,BUTLERDE,DEERINGCF,eta1.Thesynthe—sisof(4R.cis).1,1.dimethylethyl6-cyanomethyl-2,2-dimethyl-1,3.dioxane.4.acetate,akeyintermediateforthepreparationofCI-981,ahighlypotent.tissueselectiveinhibitorofHMG-CoAre- ductasc[J].Tetrahedron",1992,33(17):2279—2282.MILLARA.BUTLERDE.Processforthesynthesisof(4R—cis)-1,1.dimethylethyl-6一cyanomethyl-2,2-dimethyl一1,3-dioxane'4- 皇一莲n荽..C《Chine~JournalofNewDrugs2006.V o1.15No+22acetate:US,5103024[P].1992—04—07.[7]MILLARA.BUTLERDE.Processforthesynthesisof(4R?sis)?1.1-dimethylethyl-6-iodomethylor6-(phenyl-substituted)sulfo- nyloxymethyl?2,2-dimethyl?1,3-dioxane?4?acetate:US.5248793 [P].1993—09—28.[8]蔡正艳.宁奇.周伟撩.HMG-CoA还原酶抑制剂的合成(上) [J].中国医药工业杂志,2004,35(10):621—626.[9]蔡正艳,宁奇.周伟撩.HMG-CoA还原酶抑制剂的合成(下)'[J].中国医药工业杂志,2004,35(11):687—691.[1O]DANIELM,MICHAELW,WILHELMUSB,eta1.Processfor thepreparationof(4-hydroxy-6-oxo-tetrahydropyran-2-y1)aceto-nitrileandderivativesthereof:we,2004/096788[P].2004一●1l—l1.[11]BULTERDE.LETV.NANNINGA TN.Processforthesynthe- Bieof(5R)-I.I-dimethylethyl-6-eyano-5-hydrexy-3-OXO-hexan0- ate:US,5155251[P].1992—10—13.[12]RADLS,STACHJ,HAJ1CELJ,eta1.Animprovedsynthesisof1,1一dimethylethyl6-cyanomethyl-2,2-dimetlIyl-1,3-dioxane- 4-acetate.8keyintermediateforatorvactatinsynthesis[J].m- hsdronLett,2002,43(11):2087—2090.[13]BUTLERDE,DEERINGCF,MILLARA,eta1.Processfor trans-6_[2-(substituted-pyrrol-1-y1)alky1]pyran-2-oneinhibitors ofeholestemlsynthesis:US,5097045[P].1992—03—17.[14]BUTLERDE,DEERINGCF,MILLARA,etu1.Processfor~ans..6-C2-(substituted-pyrrel-1-y1)alky1]pyran-2-oneinh!bitors ofcholesterolsynthesis:US,5124482[P].1992-06—23_[15]BUTLERDE,DEERINGCF,MILLARA.eta1.Processfor trans-6_[2-(substituted-pyrrol-1-y1)alky1]pymn-2-oneinhibitore ofcholesterolsynthesis:US,5149837[P].1992—09—22.[16]BUTLERDE,LARA.eta1.Improved processfortrans-6_f2-(substituted-pyrrol-1-y1)alky1]pyran-2- oneinhibitomofcholesterolsynthesis:we,1989/007598[P]. 1989—08—24[17]ORENJ,DOLITZKYBZ,HARELZ,eta1.Synthesisof3,5-di- hydroxy-7-pyrrol-1-ylheptanoicacid:WO,2004/046105[P]. 2004—06—03.[18]GREENBERGWA,VARV AKA,HANSONsR,eta1.Develop? Imentofanefficient,scalable,aldolase-catalyzedprocessforen? antioseleetivesyhthesisofstatinintermediates[¨.PNAS,2004,101(16):5788—5793.1[19][20][21][22][23][24][25][26][27][28]中国新药杂志20o6年第15卷第22期MOODYDJ.WIFFENm.Processandintermediatecompounds usefulinthepreparationofstatins,particularlyatorvastatin:WO.2005/012246[P].2005—02—10.BUTLERDE.DEJONGRL.NELSONJD,eta1.Novelprocess forthesynthesisof5?(4-fluoropheny1)-1-C2-((2R,4R)-4-hy?droxy-6-OXO-te~ahydro?pyran-2-y1)-ethy1]-2-isopropyl?4?phenyl? 1H-pyrrole-3-carboxylieacidphenylamide:we.2002/055519 [P].2002—07—18.NELSONJD.PAMMENTMG.Processforpreparing5?(4-flu? oropheny1)-1_[(2R.4R)-4-hydroxy-6-OXO-tetrahydro-pyran-2-y1)ethy1]-2,isopropyl?4-pheuyl-1H-pyrrole-3?carboxylicacid phenyla-mide:we,2004/014896[P].2004一o2—19.ROTHBD.BLANKLEYCJ.CHUCHOLOWSKIAW,eta1.In- hibitoreofcholesterolbiosynthesis.3.Tetrahydre-4-hydroxy-6?[2-(1H-pyrrol-1?y1)ethy1]-2H-pyran-2-oneinhibitorsofHMG? CoAreduetase.2.Effectsofintroducingsubstituentsatpositions threeandfourofthepyrrolenucleus[J].JMedChem.1991.34(1):357—366.BJORGESM.BLACKAE.ROTHBD.eta1.[(Hydroxypheny-lamino)earbony1]pyrroles:US,5385929[P].1995—01—31.GRAULA,CASTANERJ.Atorvastatincalcium[J].DrugsFut.1997,22(9):956—968.ROTHBD,ARBORA.Trans-6_[2-(3-or4-earboxamido-sub-stituedpyrrol1,y1)alky1]?4-hydroxypyran-2-oneinhibitorsof cholesterolSynthesis:us,4681893[P].1987—07—21.ROTHBD,ARBORA.MICH.[R一(R'R')]_2一(4一fluorophe-ny1)?8.-,/-dihydroxy-5?(1-methylethyl-3-phenyl-4_[(phenylami?no)carbony1]-1H-pyrrole-I-heptanoieacid,itslactoneformandBaltBthereof:US,5273995[P].1993一l2—28.NELSONJD,PAMMENTMG.Processforpreparing5?(4-flu?ompheny1)-1-[(2R,4R)-4-hydroxy-6-OXO-tetrahydr0-pyran-2?y1)ethy1]-2-isoprepyl-4-phenyl?1H-pyrrole-3-carboxylicacidphenylamide:W0.2004/089894[P].2004—1o一21.PANDEYPS,RAOTS.AnefficientsynthesisofN3,4?diphenyl?5?(4-fluorepheny1)_2?isopropyl?1H?3?pyrrolecarboxamide,akey intermediateforatorv~tatinsynthesis[J],BiolMedChemLett.2OO4.14(1):129—131.编辑:王字梅/接受日期:2006—07—06★薨图治糖尿病新药通过审批美国FDA近日宣布,新药加努维亚(音译)在治疗糖尿病方面确实有效.加努维亚是一种口服的抑制剂,属非胰岛京治疗药物.这种药是第一种能通过改善患者自身能力来控制血糖的糖尿病药物.FDA研究证实,加努维亚主要用于治疗2型糖尿病,无论是单独服用还是作为其他药物的佐剂一起服用,都能起到很好的控制血糖的效果.加努维亚能让患者在进食期间从胃和肠道产生更多激素,从而在抑制肝脏产生糖的同时,让胰脏生成更多的胰岛素.加努维亚是由美国默克公司研制的.通过对2719例患者长达1年的临床观察中发辫.加努维亚不会造成肥胖,低血糖等不良反应,最常见的不良反应是困倦,流鼻涕,咽喉疼痛,头疼,腹泻和关节痛等. 一l9l7一。
阿托伐他汀的全合成研究进展

阿托伐他汀的全合成研究进展王继宇;沈健芬;王立新;王文;蔡泽贵;杜振军【摘要】介绍了近20年来利用消旋体拆分法、非对映选择性醇醛缩合法、Paal-Knorr反应、环加成法、双羰基不对称还原法等,完成阿托伐他汀全合成的研究进展.分析了各条路线的关键步骤,讨论了各方法的特点.参考文献27篇.【期刊名称】《合成化学》【年(卷),期】2007(015)005【总页数】10页(P519-527,594)【关键词】阿托伐他汀;全合成;综述【作者】王继宇;沈健芬;王立新;王文;蔡泽贵;杜振军【作者单位】中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041;中国科学院,研究生院,北京,100039;中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041;中国科学院,研究生院,北京,100039;中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041;中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041;中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041;中国科学院,成都有机化学研究所,四川省不对称合成重点实验室,四川,成都,610041【正文语种】中文【中图分类】R914.5;O621.3阿托伐他汀(1, Chart 1)的化学名为(3R,5R)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-(苯氨基甲酰基)吡咯-1-基]-3,5-二羟基庚酸钙盐,是由美国Warner-Lambert公司和辉瑞(Pfizer)公司共同开发的他汀类血脂调节药,1997年在英国率先上市。
1能强力抑制HMG-CoA 还原酶的活性,阻断HMG-CoA 还原成羟甲戊酸,大大降低总胆固醇和低密度脂蛋白的含量[1]。
由于1的活性优于在它之前的所有他汀类药物,且毒副作用小,因此一经上市就表现出不同凡响的上升势头, 2000年后一跃成为全球销售额最高的药物, 2004年全球销售收入高达120亿美元。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
题目:阿托伐他汀中间体及其合成工艺研究姓名:髙晔学号:************指导老师:***时间:2014-06-14摘要:阿托伐他汀是一种羟甲戊二酰辅酶A还原酶抑制剂,通过抑制胆固醇生物合成的限速酶羟甲戊二酰辅酶A还原酶,从而起到抗高血脂症的作用。
大量临床研究表明,他汀类药物高效、安全,为调脂药物中的首选药物。
本文对近年来阿托伐他汀中间体,及阿托伐他汀合成工艺进行总结。
希望找到一种更好的合成阿托伐他汀的方法,应用于实践,帮助更多的患者。
关键词:阿托伐他汀中间体合成目录摘要 (I)第一章他汀类药物的研究背景及意义 (1)1.1他汀类药物简介 (1)1.2阿托伐他汀的简介 (2)第二章阿托伐他汀中间体的合成 (5)2.1 4-氟-α-(2-甲基-1-氧丙基)-γ-氧-N,β-二苯基苯丁酰胺的合成52.2异丁酰乙酸酯的合成 (5)2.3 4-甲基-3-氧-N-苯戊酰胺的合成 (6)2.3 4-甲基-3羰基-N-苯-2-(苯亚甲基)戊酰胺的合成 (6)2.4 4-氟-a-(2-甲基-卜氧丙基)-Y-氧-N,β-二苯基苯丁酰胺的合成6 第三章阿托伐他汀的合成路线 (7)3.1路线一:以2-(4-氟苯基)-2-溴-乙酸乙酯为起始原料 (7)3.2路线二;以异丁酰乙酰苯胺为起始原料 (8)3.3路线三 (8)3.4路线四:以阿托伐他汀醛为起始原料 (9)3.5路线五;以苯乙酸为起始原料 (10)3.6路线六:以L一缬氨酸为起始原料 (11)3.7路线七: (11)第四章总结 (13)参考文献 (14)第一章他汀类药物的研究背景及意义1.1他汀类药物简介心血管疾病(包括冠心病和动脉粥样硬化)是一类严重威胁人类健康的疾病。
近年来,随着人们生活水平的提高和工作节奏的加快,心血管疾病的发病率和死亡率都呈明显的上升态势。
研究表明,血浆胆固醇(CH)水平增高,特别是低密度脂蛋白胆固醇(LDL-CH)水平的增高,是早发性动脉粥样硬化和冠心病的重要因素。
抑制CH合成,特别是控制LDL-CH可有效防治动脉粥样硬化和冠心病。
胆固醇合成过程复杂,有近三十步酶促反应,可划分三个阶段:①由B-羟基-B-甲基戊二酰酶A(HMG-CoA)经羟甲基戊二酰酶A还原酶(HMG-CoAreductase)催化生成甲羟戊酸(mevflonicacid,MV A);②由甲羟戊酸经一系列反应生成鲨烯;③鲨烯进入微粒体环化为胆固醇。
理论上选择性抑制胆固醇合成中的某一部反应都可以达到控制胆固醇过高的目的。
然而,若抑制后面的步骤往往会导致甾醇中间体的积累而引起严重的副反应,这一发现使人们放弃了对抑制后续步骤的研究。
HMG-CoA还原酶作用于胆固醇合成的初始步骤且为重要的限速酶,寻找该酶的抑制剂用作调节高血脂症的有效药物成为近年来人们关注的热点。
他汀类药物,(3R,5S,6E)-3,5-二羟基-6-庚烯酸(酯或盐),就是一种HMG-CoA还原酶抑制剂,它可竞争性抑制胆固醇合成过程中的跟速酶HMG-CoA还原酶,而降低体内的内源性合成的胆固醇水平,选择性强,疗效确切,能显著降低LDL中胆固醇水平,并能提高HDL中胆固醇水平,使胆同醇形式从有害到无害。
这对冠心病的防治非常有益,是目前治疗高胆固醇血症中疗效良好的药物。
他汀类药物上市。
就表现出了强大的市场竞争力。
第一个上市的他汀类药物是美国默克公司(MerckCo.)研制开发的洛伐他汀(Lovastatin),于1987年在美国上市,该药上市后仅数年,销售额即突破10亿美元大关,90年代初期就已经成为世界十大畅销药物之一。
随后,疗效更好的辛伐他汀(Simvastatin)及普伐他汀钠(PavastatinSodium)也分别在1988年和1989年上市,获得了更大的成功。
90年代后,氟伐他汀(Fluvastatin),阿伐他汀(Atorvastatin)和赛伐他(Cerivastatin)又相继上市,90年代他汀类药物的年销售额以20%的年他汀类新药物的合成平均增长率增长。
1995年,他汀类药物的全球总售额为53亿美元,到1999年总销售额已超过了100亿美元。
在预测2005年全球最畅销的15种药物中,他汀类HMG-CoA还原酶抑制剂就占了三席,总金额超过200亿美元。
1.2阿托伐他汀的简介1.2.1阿托伐他汀的背景阿托伐他汀的化学名为( 3R, 5R ) -7-[ 2-( 4-氟苯基) -5-异丙基-3-苯基-4-(苯氨基甲酰基)吡咯-1-基] -3,5-二羟基庚酸钙盐,它是由美国Warner-Lambert公司和辉瑞( Pfizer)公司共同开发的他汀类血脂调节药, 1997年在英国率先上市。
由于可以大大降低总胆固醇和低密度脂蛋白的含量,具有高活性、低毒低副作用等优点。
活性优于在它之前的所有他汀类药物,且毒副作用小,因此一经上市就表现出不同凡响的上升势头, 2000年后一跃成为全球销售额最高的药物, 2004年全球销售收入高达120亿美元,成为全球首个销量额破百亿美元的药品,并连续七年全球销售总额过百亿。
其研究愈发的引起人们的关注,巨大的经济效益刺激着人们长期关注其合成方法的研究,有关其全合成的研究不断被报道。
1.2.2阿托伐他汀的物化性质分子式:C33H33CaFNO5分子量:582.6947熔点 176-178℃熔点:176-178℃结构式及立体结构式:结构式立体结构式1.2.3阿托伐他汀的作用机理是一种新型羟甲基戊二酶辅酶A(HMG—CoA)还原酶抑制剂,阻断HMG-CoA 还原成羟甲戊酸,它通过长时间持续竞争性抑制作用,降低血浆中胆固醇合成总量,从而抑制了VLDL的生成,降低甘油三酯水平;另外,阿托伐他汀可增加产生肝细胞表面LDL受体的mRNA表达,使LDL受体数量显著增加并使其活性增强,增加血浆LDL-胆固醇的清除,保持细胞内胆固醇内环境的稳定,同时也降低载脂蛋白的水平。
适用于杂合子家族性或非家族性高胆固醇血症和混合性高脂血症,也用于纯合子高胆固醇血症。
(1)抑制HMGu-CoA还原酶HMG—CoA还原酶是胆固醇合成酶系中的限速酶,通过对其的抑制作用,从而使胆固醇合成减少。
当前所使用的几种HMG-CoA丕原酶抑制剂都具有相同的作用机制。
然而,动物实验表明,这类药物并非完全一致胆固醇的合成,给药后仍有足够的胆固醇以完成生理功能,例如胆固醇的合成和细胞的生长,阿托伐他汀吸收后即有生物活性。
(2)增加低密度脂蛋白(LDL)受体阿托伐他汀通过对VLDL必需的。
阿托伐他汀通过使血浆胆固醇浓度降低,使VLDL合成分泌减少。
VLDL是携带和转用三酰甘油所必需的,VLDL-C又是LDL-C的前提,故阿托伐他汀可使三酰甘油、VLDL-C、LDL-C均降低。
(3)抗动脉粥样硬化作用阿托伐他汀通过降低血脂、减少脂质浸润和泡沫的形成,对延迟动脉粥样硬化病有利。
阿托伐他汀可防止动脉粥样硬化瓣破裂。
体外实验表明可抑制平滑肌增殖和转移。
1.2.4阿托伐他汀国内外研制情况1991年美国华纳-兰伯特公司发明了立普妥,于1997年首次上市,由辉瑞公司代理销售。
2001年辉瑞公司完成对华纳-兰伯特的收购,完全将立普妥归于帐下。
2000年,立普妥进入中国。
在国内,北京红惠制药有限公司与1999年9月获得了阿托伐他汀钙及片剂的新药证书和生产批件;2001年2月,广州南新制药有限公司、天然药物及仿生药物国家重点实验室、湖南迪诺制药有限公司也获得了阿托伐他汀钙及片剂的生产许可。
1.2.5国内外市场情况在国际上,阿托伐他汀品牌药全球销售一路飙升,2002年销售额创历史新高,也创下了重磅炸弹药全球销售的新记录,达到了79亿美元。
在国内,阿托伐他汀钙国内由红惠制药有限公司于1999年在辉瑞公司的利普妥获得行政保护前获得了生产批文。
2001年红惠公司开始向医院推广阿托伐他汀钙(商品名阿乐)。
2001年16大典型城市样本医院的购药金额为101.6万元,购药量为15.0万片,2002年购药金额增至771.4万元,增加了6.6倍,购药量为114.0万片,增加了6.6倍,可以说,无论是购药金额,还是购药量均呈快速递增态势。
2003年一季度购药金额为321.2万元,较2002年同期增长了260.5%;购药量为48.1万片,较2002年同期增长了265.0%。
根据ATPIII,由于高脂血症诊断标准的改变,预见会有更多的美国人因高胆固醇而接受治疗:接受膳食治疗者将会从5200万增至6500万,接受药物治疗者将会从1300万增至3600万。
在我国,心脑血管疾病发病率、死亡率近年来逐年上升,发病率高达8%,死亡率接近总死亡率的50%;平均每20分钟就有一人因心脑血管疾病而死亡。
近年来国内学者进行了一些调查研究,得出的结论是中国城镇居民的血脂水平要高于农民,成年人血TC随年龄而增高,50岁以前男性血TC高于女性,但50岁以后女性血TC增高的幅度超过男性;平均值亦比男性为高,LDL-C的变化趋势与TC相同。
TG亦随年龄而增高。
TC与TG均在60~80岁时达到最高水平,尔后便下降。
而HDL-C在男性各年龄组相对稳定而无明显差异,但较美国的同年龄人群为高,而中美两国中年女性的HDL-C水平无明显差别。
在地区分布上呈现北高南低,西高东低的特点。
由高脂血症发病的这些特点可以预见,随着我国人口老龄化进程的加速,城市化发展的加速以及生活方式的日渐西化,未来我国高脂血症的发病率和发病人数将会继续增长。
1.2.6知识产权情况阿托伐他汀具有欧洲专利,专利号EP0409281,2011年到期;美国专利,专利号US4681893。
在中国有行政保护,与1999年9月30日授权,授权号:B-US99093016,到期日:2004年7月21日。
1.2.7阿托伐他汀的研究意义阿托伐他汀(Astorvastatin)作为一种合适的一线降脂药物,它能有效降低冠心病的发生率和死亡率。
该药为最新合成药物,医学界公认比天然微生物发酵,半合成的他汀类药物好,患者使用时,起始剂量即有优异的调脂达标率。
2005年一季度销售数据山显示,增长率比上一年四季度有了较大增长,正在以大幅增加的速率逐渐吞噬着国内调血脂药物的市场。
目前阿托伐他汀为在保护期内的新药品种,但是在专利保护期终止后,必将出现群雄逐鹿的市场局面。
因此,对于我国研制开发阿托伐他汀降脂药具有巨大重要意义。
第二章阿托伐他汀中间体的合成2.1 4-氟-α-(2-甲基-1-氧丙基)-γ-氧-N,β-二苯基苯丁酰胺的合成4-氟a(2-甲基-1-氧丙基)-γ-氧-N,β-二苯基苯丁酰胺是合成阿托伐他汀所需要的重要中间体。
2.2异丁酰乙酸酯的合成文献报道了两种合成异丁酰乙酸乙酯的方法:一种是乙酰乙酸乙酯在乙醇镁的作用下,和异丁酰氯在0~5。