液相色谱分析方法的建立
建立液相色谱方法一般步骤
液相色谱仪的使用
必须做!
溶剂和水的前处理:过滤
• 设备和材料:溶剂过滤器、真空泵、0.45mm或更 细的水性滤膜和有机滤膜
• 过滤的方法:用加了滤膜的溶剂过滤器和真空泵 抽滤
• 过滤的目的:除去溶剂中的微小颗粒,避免堵塞 色谱柱,尤其是使用无机盐配制 的缓冲液。
方法
反相 HPLC
离子对 HPLC
正相 HPLC
样品特点/色谱柱
何时应用该方法
流动相:水/有机溶剂
色谱柱:C18(ODS),C8,苯基, 三甲基硅烷,氰基
首选为能溶于水/有机混合物的中性或非 离子化合物。
流动相:水/有机溶剂(一种缓冲 物控制pH和离子对试剂)
色谱柱:C18(ODS),C8,氰基
离子或可电离化合物,尤其是碱性或阳 离子化合物的良好选择
液相色谱分析
液相色谱的检测模式
灵敏度、选择性、适应能力
项目 示差折光 电导
响应 灵敏度 线性范围
通用 毫克 104
选择性 纳克 106
流速敏感 温度敏感 梯度淋洗
是 是 不可
是 是 有限制
紫外 选择性
纳克 105 否 否 可以
荧光 选择性
皮克 105 否 否 可以
电化学 选择性
皮克 106 是 是 脉冲可以
改变容量因子 K'
①
①
②
50/50
② ③
③ 60/40
① 40/60
② ③
液相色谱分析
分析方法建立的实例
改变选择性∶a
• 通过改变流动相改变选择性
– 同样强度的不同溶剂
液相色谱教程液相色谱方法开发

液相色谱教程液相色谱方法开发液相色谱(Liquid Chromatography,简称LC)是一种常用的分离和分析技术,广泛应用于化学、生物、医药等领域。
液相色谱方法的开发是为了解决特定问题和满足特定需求而进行的,本文将介绍液相色谱方法开发的一般步骤和注意事项。
液相色谱方法的开发步骤如下:1.确定分离目标:首先确定需要分离和分析的目标化合物,包括确定化合物的物理化学性质和分离特性等。
2.选择色谱柱:根据分离目标,选择合适的色谱柱。
色谱柱的选择应考虑样品的性质、分离机理、应用要求等因素。
3.选择流动相和梯度条件:根据分离目标,选择合适的流动相(包括溶剂和缓冲剂等)和梯度条件(包括流动相的组成和梯度程序等)。
4.优化色谱条件:通过改变流动相组成、流速、柱温等参数,优化色谱条件,达到最佳分离效果。
5.建立分析方法:根据样品的特点和分析需求,建立分析方法。
包括确定检测器的波长或离子选择器、设置进样量和检测浓度范围等。
6.方法验证:对开发的液相色谱方法进行验证,包括准确度、精密度、线性范围、检出限等指标的确定。
液相色谱方法开发过程中需要注意的事项如下:1.样品制备:样品的制备要充分考虑到样品的性质和分析方法的要求,如需要进行前处理、提取、洗脱、浓缩等。
2.色谱柱保养:液相色谱柱的保养对于保证色谱方法的重复性和稳定性至关重要。
包括定期清洁、适当的保存和使用。
3.流动相准备:流动相的配制要严格按照要求,注意流向的调整、PHA值的调节、气泡和杂质的排除等。
4.柱温控制:柱温对色谱分离的效果有很大影响,需要根据分析需求对柱温进行控制和调节。
5.检测器选择:根据分析的目标和样品的特性,选择合适的检测器,如紫外检测器、荧光检测器、质谱检测器等。
6.数据处理:对色谱结果进行正确的数据处理和解释,包括峰面积计算、峰识别和归一化等。
总结来说,液相色谱方法的开发是一个系统的工程,需要综合考虑样品特性、分析需求和分离机理等因素。
液相色谱串联质谱检测醛固酮的方法学建立和临床应用

液相色谱串联质谱检测醛固酮的方法学建立和临床应用液相色谱串联质谱(LC-MS/MS)是一种先进的分析技术,已经被广泛应用于药物分析、生化分析、环境监测等领域。
本文将重点介绍液相色谱串联质谱在醛固酮检测中的方法学建立和临床应用。
1.方法学建立1.1样品制备对于醛固酮的检测,样品制备是关键的一步。
血液中的醛固酮浓度非常低,常规的前处理方法一般包括固相萃取、液液萃取等。
其中固相萃取是应用较广泛的方法,通过一系列萃取步骤,将醛固酮进行富集,提高检测的灵敏度。
1.2色谱条件优化在液相色谱中,需要选择合适的色谱柱、流动相和梯度条件。
常用的色谱柱包括C18柱、C8柱等,流动相一般为甲醇-水溶液。
通过优化梯度条件,可以实现对醛固酮的高效分离和灵敏检测。
1.3质谱条件优化在质谱条件中,需要选择合适的离子源和工作模式。
常用的离子源包括电喷雾离子源(ESI)和大气压化学电离离子源(APCI),工作模式可以选择正离子模式或负离子模式。
通过调节离子源和工作模式的参数,可以获得稳定的质谱信号,并实现对醛固酮的定量测定。
1.4校准曲线绘制校准曲线是进行定量分析的关键。
通过准备一系列不同浓度的标准品,测定其峰面积或峰高,再进行曲线拟合,得到醛固酮的标准曲线。
校准曲线需要具有良好的线性关系、低限度、高精度和高稳定性。
2.临床应用2.1确诊与鉴别诊断液相色谱串联质谱法能够快速准确地测定血液中的醛固酮浓度,可用于醛固酮增多症的确诊和鉴别诊断。
此外,液相色谱串联质谱还可以与其他指标联合使用,例如血压、血钾等指标,提高对肾上腺皮质功能异常的准确性。
2.2疾病监测醛固酮在机体内起到调节电解质平衡和水盐平衡的功能,与多种疾病的发生发展密切相关。
液相色谱串联质谱法可以定量测定血液中醛固酮的浓度变化,对于高血压、肾上腺异常增生以及一些代谢性疾病的监测具有重要意义,能够及早发现和诊断疾病。
2.3药物治疗监测液相色谱串联质谱法还可以监测针对醛固酮分泌异常的药物治疗效果。
高效液相色谱检测葡萄及葡萄酒中7种有机酸方法的建立

高效液相色谱检测葡萄及葡萄酒中7种有机酸方法的建立沈 燕1,张正文1,刘兴凯1,王福成2,邵学东1*(1.君顶酒庄有限公司,山东烟台 265607;2.烟台市蓬莱区葡萄与葡萄酒产业发展服务中心,山东烟台 265699)摘 要:目的:建立了利用高效液相色谱测定葡萄及葡萄酒中7种有机酸的检测方法。
方法:色谱条件为Hi-Plex H色谱柱(300 mm×7.7 mm,8 μm),PL Hi-Plex H保护柱(50 mm×7.7 mm,8 μm);紫外检测波长210 nm;流动相为0.006 mol·L-1的硫酸;流速0.6 mL·min-1;柱温55 ℃;进样量10 μL。
结果:草酸、柠檬酸、酒石酸、苹果酸、琥珀酸、乳酸和乙酸出峰时间均在20 min以内;标准曲线线性关系良好,相关系数(R2)在0.999 9~1.000 0;检出限在0.031 4~0.722 3 mg·L-1;加标回收率在95.24%~102.04%,其相对标准偏差在0.16%~1.87%。
结论:该方法操作简单,检测时间短,具有较高的准确性和精确性。
关键词:有机酸;葡萄;葡萄酒;高效液相色谱;检测方法Establishment the Method of Seven Kinds of OrganicAcids in Grape and Wine by High Performance LiquidChromatographySHEN Yan1, ZHANG Zhengwen1, LIU Xingkai1, WANG Fucheng2, SHAO Xuedong1*(1.Junding Castle Co., Ltd., Yantai 265607, China; 2.Yantai Penglai District Grape and Wine Industry DevelopmentService Center, Yantai 265699, China)Abstract: Objective: To establish a HPLC method for the determination of 7 organic acids in grape and wine. Method: The chromatographic conditions were Hi-Plex H column (300 mm×7.7 mm, 8 μm) and PL Hi-Plex H protective column (50 mm×7.7 mm, 8 μm). UV detection wavelength 210 nm; sulfuric acid with 0.006 mol·L-1 mobile phase; flow rate 0.6 mL·min-1; column temperature 55 ℃. The sample size was 10 μL. Result: The peak time of oxalic acid, citric acid, tartaric acid, malic acid, succinic acid, lactic acid and acetic acid were all within 20 min. The standard curve has a good linear relationship with the correlation coefficient(R2) ranging from 0.999 9 to 1.000 0. The detection limit was 0.031 4~0.722 3 mg·L-1. The recoveries ranged from 95.24% to 102.04%, and the relative standard deviations ranged from 0.16% to 1.87%. Conclusion: The method is simple to operate, short detection time, and has high accuracy and accuracy.Keywords: organic acid; grape; wine; high performance liquid chromatography; detection method有机酸是葡萄果实及葡萄酒中体现风味的主要物质之一,其含量是衡量葡萄果实成熟度和品质的重要参数,在葡萄酒的酿造中发挥着关键作用,对酒的感官质量有一定的影响,与酒的物理化学以及微生物稳定性有着紧密联系[1-4]。
液相色谱方法开发案例

液相色谱方法开发案例全文共四篇示例,供读者参考第一篇示例:液相色谱是一种常用的分析技术,广泛应用于化学、生物和环境领域。
液相色谱方法的开发通常涉及样品预处理、柱填料选择、流动相优化等方面。
本文将以某种化合物的分析为例,介绍液相色谱方法的开发过程。
为了开发一种液相色谱方法用于分析某种药物残留物,首先需要准备样品。
样品可能包含复杂的基质,需要进行适当的前处理。
通常可以选择提取、浓缩或洗涤样品,以去除干扰成分或减少基质中其他化合物的干扰。
经过前处理后的样品将被注入色谱仪进行分析。
柱填料的选择是液相色谱方法开发中的关键步骤。
柱填料的种类、尺寸和性质将影响色谱分离的效果。
在选择柱填料时,需要考虑到目标分析物的性质、分子大小、亲水性或疏水性等因素。
通常会进行试验性的柱填料筛选,确定最适合的柱填料种类。
流动相的选择和优化也对液相色谱方法的开发至关重要。
流动相的成分、流速、温度等参数将直接影响分析的灵敏度和分离度。
通过对不同流动相条件的尝试和优化,可以找到最佳的流动相组合,以实现对目标化合物的高效分离和检测。
在经过样品预处理、柱填料选择和流动相优化后,可以开始进行基准分析。
通过注入标准品和重复试验,确定色谱方法的准确性、重复性和灵敏度。
也可以进行不同因素对色谱分离的影响研究,以进一步优化色谱方法。
在实际应用中,可能会遇到复杂样品矩阵、稀有目标成分等挑战。
在这种情况下,需要进行更深入的研究和优化,可能需要调整柱填料种类、流动相组合或色谱条件等。
通过持续地改进和优化,可以开发出适用于不同类型样品的高效液相色谱方法。
液相色谱方法的开发是一个复杂而细致的过程,需要多方面的考虑和实验。
通过样品预处理、柱填料选择、流动相优化和基准分析等步骤,可以开发出高效、灵敏和可靠的液相色谱方法,用于不同领域的分析和检测。
在未来的研究中,还需要不断改进和完善液相色谱方法,以应对越来越复杂的样品要求。
第二篇示例:液相色谱方法是一种用于分离和检测化合物的常用技术,它通过利用溶解在液相中的化合物在固定相上的不同吸附性质来实现分离,是现代化学分析中不可或缺的手段之一。
沃特世液相仪器方法建立

沃特世液相仪器方法建立沃特世液相仪器是一种常用的分析仪器,广泛应用于化学、生物、环境等领域的分析实验中。
本文将介绍如何使用沃特世液相仪器建立分析方法,并探讨其在实验中的应用。
一、沃特世液相仪器的基本原理沃特世液相仪器是基于液相色谱法原理设计和研发的,其主要原理包括进样、分离、检测和数据处理等步骤。
在进样过程中,待测样品通过自动进样器被引入色谱柱中;在分离过程中,样品在色谱柱中按照不同的化学性质被分离开来;在检测过程中,利用检测器对分离后的化合物进行测定;最后,将检测到的信号通过数据处理系统进行分析和处理。
二、建立沃特世液相仪器分析方法的步骤1. 样品预处理:根据待测物的特性,选择适当的样品处理方法,如提取、浓缩、净化等,以便得到适合进样的样品。
2. 选择合适的色谱柱:根据待测物的特性和分离要求,选择合适的色谱柱,如反相色谱柱、离子交换色谱柱等。
3. 优化分离条件:确定适当的流动相组成、流速和温度等分离条件,以使待测物在色谱柱上得到良好的分离。
4. 确定检测器和检测条件:根据待测物的特性和分析要求,选择合适的检测器,如紫外检测器、荧光检测器等,并确定合适的检测条件,如波长、增益等。
5. 方法验证:进行方法验证,包括线性范围、灵敏度、重复性、选择性等指标的评价,确保该分析方法的准确性和可靠性。
6. 样品分析:按照建立的分析方法,进行待测样品的分析实验,并记录所得数据。
三、沃特世液相仪器方法的应用沃特世液相仪器广泛应用于各个领域的化学分析实验中,如药物分析、环境监测、食品安全等。
以下是几个常见的应用案例:1. 药物分析:沃特世液相仪器可用于药物的含量测定、成分分析、质量控制等方面。
通过建立合适的分析方法,可以准确测定药物中各种成分的含量,保证药物的质量和安全性。
2. 环境监测:沃特世液相仪器在环境监测领域的应用较为广泛。
例如,可以利用沃特世液相仪器对水体中的有机污染物进行分析,如农药、有机溶剂等,以评估水质的安全性。
液相色谱分析方法建立-系列一基本理论及色谱介绍

硅 胶 的 溶 解 曲 线
1 2 3 4 5 6 7 8 9 p H
硅胶表面键合相的水解(PH<2)
O
OH
Si
O
+
O
“Column Bleed”
H3C
C
C
C
C
Si
C
C
C
CH3
Cl
CH3
O
+ H3C
C
C
C
C
Si
C
C
C
CH
O 硅胶表面未被键合的酸性硅羟基将会与被分析物相互作用,特别是在 中性条件下会对强碱性化合物产生严重的峰形拖尾。
O 封尾工艺使得表面残余的硅羟基被进一步的消除,为小分子所取代。 O 封尾工艺能促使色谱柱对极性和碱性化合物的分离具有良好的峰形。
50
色谱柱的pH值
O 填料的pH稳定性 O 硅胶填料 pH:2-8 O 聚合物填料 pH:2-12 O 杂化硅胶填料 pH:2-12
O
Low pH (hydrolysis of ligand)
O Si
O
O
OH
+
H3C
C
C
C
C
Si
C
C
C
CH3
HO
CH3
硅胶的溶解(PH>8)
Nucleophilic attack
• Complete dissolution of Silica
• Catastrophic column failure
蒸发光散射
任何挥发性低于流动相的样 品均能被 检测
可检测所有物质。
液相色谱常见问题及处理方法(LCMS)资料

常用弱酸的pKa
pK1
醋酸
4.87
柠檬酸
3.13
磷酸
2.16
pK2
4.76 7.2 1
pK3
6.40
12.32
22
6. 不使用时,两头要盖上盖子,避免固定相干枯。
7. 使用预柱。
8. 避免流动相组成及极性的剧烈变化(梯度洗脱)
9、避免压力脉冲的剧烈变化
14
肩峰或分叉峰产生的原因:
1. 柱子劣化 (柱头处产生不均匀的空隙)
2. 样品变化 (生成氧化物、分解物等)
判断标准:
所有的峰都产生有肩峰或分叉时则为柱子劣化。
1. 在使用新柱之前,最好用强洗脱溶剂在低流量下 (0.2-
0.3 ml/min)冲洗 30 min,长时间未用的分析柱也要同
样处理。
2. 定期使用强洗脱溶剂冲洗柱子。
3. 使用缓冲盐作为流动相后,要先用水(含有少量甲醇
5~10%)冲洗,再用有机溶剂(甲醇)冲洗。
4. 净化样品。
5. 分离条件优化(防止鬼峰出现)。
使宝石球与垫片分开 2)拆下单向阀,放入异丙醇或水中,
用超声波清洗
故障2:宝石球或塑料垫片受污导致密封不好。 表现:系统压力波动大 措施:拆下单向阀,放入异丙醇或水中,用超声波清
洗。
7
2. 柱塞密封圈
密封圈
柱塞杆 泵头清洗管路 流动相
故障:密封圈磨损而导致 密封不良
现象:系统压力波动大或 漏夜
措施:更换密封圈 注意点:更换密封圈拆卸
19
2. 脱气: 除去流动相中溶解或因混合而产生的气泡。
气泡对测定的影响:
1)泵中气泡使液流波动,改变保留时间和峰面积; 2)柱中气泡使流动相绕流,峰变形; 3)气泡过多可使泵压力不稳甚至停止工作; 4)检测器中的气泡产生基线波动,
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一. 方法建立的步骤二.开始前应知道1. 样品的性质在开始方法建立之前,我们应该检查自己对样品的了解程度,并明确分离目标。
表 1 有关样品组分和性质的重要信息所含化合物的数目化合物的化学结构(官能团)化合物的分子量化合物的pKa值化合物的UV光谱图化合物在样品中的浓度范围样品的溶解度样品的化学成分能够为选择HPLC分离的最佳初始条件提供有价值的线索根据已知的样品信息,HPLC方法建立有两种不甚相同的模式。
一种模式依据样品的“化学性质”选择最佳初始条件,色谱工作者需很大程度依赖于过去的经验(如类似结构化合物的分离)和/或用文献资料补充现有信息而另一种模式则直接开始色谱分离,而对样品的性质不大注意这两种HPLC的方法建立模式可分别称为理沦型与经验型初始分离一旦开始,可以根据类似的思路(理论的与经验的)选择进一步的实验。
2.分离的目的HPLC分离的目的必须十分明确,下面的问题在建立方法之初就应确定:(1)主要目的是什么?定量或定性,还是定性、定量同时做?;(2)是否有必要解析出样品的所有成分?譬如可能有必要分离出产品中的所有降解物或杂质,以使含量测定结果更加可靠,但却没必要将它们彼此完全分开。
(3)如要求定量分析,准确度与精密度需多大?样品主要成分的精密度通常能达到±1—2%,特别是不需样品预处理的情况。
(4)特殊化合物可能会以不同的样品形式出现(如:原料药,一种或多种形态,环保样品等)。
是否需要一种以上的HPLC方法?单一方法分离不同形态样品是否理想?(5)一次将分析多少样品?当必须同时处理大量样品时,运行时间将变得非常重要。
有时甚至为了缩短运行时间而以牺牲样品分离度作代价,如缩短柱长或加快流速。
当一次分析的样品数目超过10个,运行时间一般应控制在20min以内。
(6)将要使用该方法的实验室中,有哪些HPLC设备?色谱柱能否恒温系统能否做梯度洗脱?该方法是否可在不同设计与生产的设备上运行?方法建立实验开始之前,应明确对方法的这些要求。
三. 样品的预处理和检测1. 预处理:样品来源形式不同,可能以如下形式出现:(1)可直接进样的溶液;:(2)需稀释、pH缓冲、加人内标或其它定容操作的溶液;(3)必须首先溶解或提取处理的固体;。
(4)样品需预处理以除去干扰物和/或保护色谱柱或仪器不受损害。
可直接进样的样品有操作方便、精密度好的优点,然而大多数HPLC分析的样品需称重或稀释定容后才能进样。
当样品溶剂成分接近于流动相时,一般不会产生基线波动等问题,通常能够得到较好的结果。
有些样品在注入HPLC前需进行部分分离(预处理),以除去干扰物、浓缩样品或消除“柱杀手”,因此了解样品和各种被测物的浓度范围非常重要。
2. 检测:HPLC方法建立期间,在第一次进样前,我们必须有把握所选择的检测器能检测到所有的被测样品组分。
通常首选可变波长紫外(UV)检测器,因这种检测器使用方便,且可用于大多数样品。
UV光谱数据可从文献中查找,也可通过所测样品成分的化学结构估算,直接测得(如果可拿到化合物纯品)或在HPLC分离期间用二极管阵列检测器(PDA)得到。
样品对UV检测响应不足时,可考虑使用其它类型检测器(荧光,电化学等),或衍生化样品使检测信号增强。
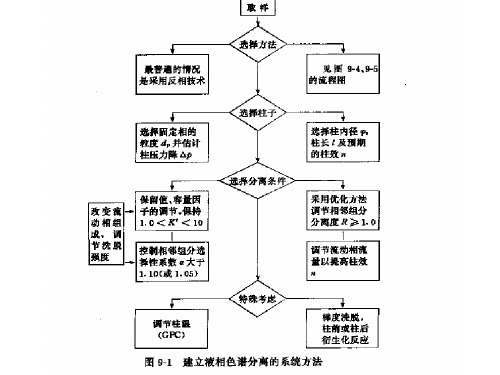
四. 建立分离方法1. 选择HPLC分离模式与起始条件首先,我们要确定使用什么方法最合适;如果我们选定HPLC分离模式,将样品分为一般样品或特殊样品。
一般样品为小分子典型混合物,用近乎标准化的起始条件即可分开。
而样品特殊需用特殊的色谱柱和特定的条件才能很好地分离。
一般样品可进一步分为中性或离子样品。
离子样品包括酸、碱、两性化合物和有机盐类(电离的强酸或强碱)。
如样品为中性,一般不需在流动相中加缓冲液或添加剂,但对于酸或碱性样品,通常需在流动相中加入缓冲剂。
碱性或阳离子样品,推荐用“弱酸性”的反相色谱柱,流动相中加人胺添加剂可能对分离有利。
首次实验完成后,再按照结果系统地加以进一步完善。
样品的性质HPLC GC CE SEC TLC一般样品特殊样品中性的离子的无机离子异构体对映体生物样品等度肽类糖类梯度大分子蛋白质离子对核酸糖类正相合成聚合物图1. 利用样品的有关条件,选择初始实验分离条件表2特殊样品的处理样品要求无机离子检测为主要问题;使用离子色谱异构体有些异构体能用反相HPLC分开,可分为一般样品类中;用(I)正相HPLC或(2)环糊精—硅胶色谱柱的反相分离能较好地分离异构体对映体分离这些化合物需要“手性”条件生物样品多种因素如分子构象,极性官能团和宽范围的疏水性使这种样品很特殊大分子“大”分子需要大孔径的色谱柱填料(≥10.m孔径)表3首次HPLC分离的较好实验条件分离条件较好的首要选择色谱柱尺寸(长度,内径ID)粒度固定相15ⅹ4.6cn 5um a. C8或C18流动相溶剂A和B缓冲液—乙睛%B 80—100% b.缓冲液(化合物、pH、浓度)25mM磷酸钾,2.0<pH<3.0 c.添加剂(如胺改性剂、离子对试剂)开始时不用流速0.5—2.0 ml / min温度35—45℃样品大小体积重量d.<25ul <100ug备注:a. 3.5um微粒为另一选择; b. 用于初始等度分离;初始用梯度实验较好;c. 中性样品不需要缓冲液;d. 小体积色谱柱(如7.5*0.46cm,3.5um)需要的量更小。
表4主要HPLC方法的特性方法/特点/色谱柱何时应该使用该方法反相HPLC流动相:水—有机溶剂色谱柱:C18(ODS)、C8、苯基、三甲基硅烷(TMS)、氰基能溶于水一有机混合液的大多数样品尤其是中性或非离子化合物的首选离子对HPLC流动相:水一有机溶剂,控制PH的缓冲液和离子对试剂色谱柱: C18、C8、氰基离子或可电离的化合物,尤其是碱或阳离子样品的良好选择正相HPLC流动相:有机溶剂混合液色谱柱:氰基、乙醇基、氨基、硅胶当反相或离子对HPLC无效时的第二个良好选择石不溶于水一有机混合液的亲脂样品的首选品异构体混合物和制备规模HPLC的首选(最好用硅胶〕此处推荐的所有色谱柱〔除了未键合硅胶)均用健合相硅胶微粒装填。
当分离目的是为了分离制备纯样品时,优化的最终HPLC方法将不同于用于常规定量分析的方法。
2. 开始建立方法在首次进样之前,唯一需做的选择是流动相中的有机调节剂比例(%B)。
通常不推荐用中等强度的流动相开始方法建立。
更好的选择是先用非常强的流动相(如80%—100%B),然后按需要降低%B。
另一种有别于起始等度分离的方法是梯度洗脱,梯度分离一般使样品分离得更好。
3. 改善分离在HPLC定量分析中,分离开是首要要求。
基线分离是提供精确结果的有效保障。
含有5个组分或更少的样品,使谱峰之间达到基线分离(R>1.5)一般较为容易。
分离度会随色谱柱的使用时间逐步降低,也会由于分离条件的微小波动而在不同时间有所变化,因此在建立简单混合物的分离方法时,最好使R≥2。
这样的分离度将有利于改善测试精度和方法的普适性;含10团种或10种以上组分的样品,分离一般会很困难,目标分离度往往必须降低至R>1.0—1.5。
表5 HPLC方法建立的目标目标评论分离度精密和普适性好的定量分析要求R>1.5分离时间<5—10min时日常工作较理想定量含量测定:≤2%(1S D);要求不高的分析:≤5 %;痕量分析:≤15%压力(如果为新柱)<l50bar最好,<200bar通常是基本的要求峰高大信噪比的窄峰最好溶剂消耗每次运行少用流动相最好注:大约按重要性递减排列,但因分析要求不同而异。
有些HPLC测定不必使全部被测组分达到基线分离,这种情况最常见于所测样品中可能存在数种化合物但仅需检测其中的一种时。
比如在普查水或土壤样品中的某污染物(如定性分析不同的除芳剂)时,仅需要分离出个别除莠剂,确定其特征保留时间进行峰鉴别即可。
尽管大多数HPLC设备能在更高的压力下工作,使用新色谱柱的工作压力不应超过170bar(2500psi , 17MPa),压力上限最好在2000psi以下。
限制该压力基于两点原因:第一. 随着色谱柱的使用,微粒杂质会逐步堵塞柱头,使柱压成倍升高;第二. 较低的压力(< 170bar)适于泵、进样阀、尤其是自动进样装置的稳定工作,密封垫的寿命更长,色谱柱堵塞的情况减少,系统的耐久性明显提高。
因此,最好以小于泵的最大允许压力的50%作为目标压力。
4. 可重现的分离在进行实验时,要能够重现每次实验色谱图。
在方法建立过程中改变实验条件(流动相、色谱柱、温度)时,必须经过足够长的时间使色谱柱在新流动相和温度下达到平衡。
通常以10—20倍柱体积的新流动相冲洗色谱柱后,色谱柱可达到平衡。
五.HPLC方法的完善最后的实验方案应达到方法建立开始时所设定的所有目标。
方法本身在日常工作中应经受住考验,应适用于所有的相关实验室以及人员。
1. 定量与方法论证一旦完成了HPLC方法建立,应对其进行论证,进行全面论证通常需通过简略核查方法的专属性、线性、准确性、精密度、回收率、灵敏度等而实施。
在最终论证方法之前,应准备和检查书面分析方案,使之清晰、严密。
实际的论证程序按方法的重要性不同,需时为1天一2周不等。
理想的情况是,通过方法论证能找出可能因仪器和操作者的不同而导致的潜在问题。
在日常HPLC分析中,柱间重现性可能是主要问题之一。
同种方法在不同批号的色谱柱上可能有不同的结果,任何意外的结果都应进行研究,以找出其原因,避免在日后的工作中出现同类错误。
表6 完善方法1. 应预备数据表明所需方法的性能;2. 应有为其它操作者使用的书面分析步骤;3. 以一个以上的系统或操作者,用包括期望组分和期望被测物浓度的样品系统地论证方法的性能;比较不同时间和不同实验室之间的实验数据;4. 应得到色谱柱的期望寿命以及柱间重现性的数据;5. 研究不正常的结果,以纠正潜在的问题;6. 研究所有影响分离的条件(温度、流动相组成、pH等);规定这些条件的限度;对可能发生的问题(关键谱峰对分离不足;随运行时间延长,最末谱峰保留值增加等)提出建议措施。
备注:主要用于日常工作或质量监控方法上表的要求适用于满足精密准确、稳定可靠并可移植转让的严格HPLC标准方法。
在其它情况下,可能只要求一次成功的分离或能快速、“粗略”地答复某一特定问题,此时可免掉表5和表6中的许多建议,图1中一些步骤也可能是不必要的,仅明白并知道一个方法建立方案的真实目标就足够了。
2. 解决出现的问题随着方法建立的进行,会出现各种问题,其中有些已在表7中列出,其它的问题包括:开始时色谱图中有的谱峰可能比所期望的要宽(塔板数低),或谱峰严重拖尾;在方法应用中还会发现用同一(或不同)厂家的“规格相同”色谱柱替代原柱后,分离情况会变得难以接受,常规实验室无法用(规格相同的)另柱重复此方法;色谱柱寿命也可能很短(如进样不足100次即不能再用);(同一色谱柱)重复进样的色谱图不一样,定量精度较差,保留时间从一系列运行的开始直至结束一直漂移;在后面样品的色谱图中可能出现了其它峰干扰被测物测定等。