临床试验项目培训_图文
合集下载
clinicaltrials临床试验方案培训课件

o Ex: Every 3 gets higher dose
• Nonrandomized: All with Hep. C = cases; others = controls
• Protocol: Study design - instructions
• Blinded: Participants do not know if in experimental or control group
clinicaltrials临床试验方案
5
Another Classification System
• rected Data Capture
• Ex: Vital Statistics
• Directed Data Capture & Hypothesis Testing
• Ex: Cohort Studies, Case Control Studies
developed a systematic methodology for studying & evaluating therapeutic interventions
clinicaltrials临床试验方案
11
Core Components of Clinical Trials
• Involve human subjects • Move forward in time • Most have a comparison CONTROL group • Must have method to measure intervention • Focus on unknowns: effect of medication • Must be done before medication is part of
• Nonrandomized: All with Hep. C = cases; others = controls
• Protocol: Study design - instructions
• Blinded: Participants do not know if in experimental or control group
clinicaltrials临床试验方案
5
Another Classification System
• rected Data Capture
• Ex: Vital Statistics
• Directed Data Capture & Hypothesis Testing
• Ex: Cohort Studies, Case Control Studies
developed a systematic methodology for studying & evaluating therapeutic interventions
clinicaltrials临床试验方案
11
Core Components of Clinical Trials
• Involve human subjects • Move forward in time • Most have a comparison CONTROL group • Must have method to measure intervention • Focus on unknowns: effect of medication • Must be done before medication is part of
临床试验培训课件

参照系统选择
对比试剂 已上市同类产品; 临床普遍认为质量较好的产品; 具有可比性的产品:方法学、预期用途、性能指标、校准品 溯源、推荐的阳性判断值/参考区间、样本类型等。
18
IVD
临床试验设计—举例
参照系统选择
病原体IgM、TORCH 类指导原则: 除同类产品对比试验外,还应对急性期患者样本进行考核试 剂与参考方法的一致性研究,不少于30例,或采用血清转化盘 验证。 药物滥用类指导原则: 除同类产品对比试验外,还需完成至少30例参考方法对比试 验。 流感病毒核酸检测: 除同类产品对比试验外,还需完成不少于30例病毒培养阳性 样本验证。
27
IVD
临床试验设计--注册变更
试 验 方 法
对照试验 对照试验:变更后产品 vs. 变更前产品/ 已上市同类产品; 证明两者等效
样 本 量
最低样本量要求: 检测条件优化、增加与原样本类型具有可比性的其他样本类 型等:三类至少200例,二类至少100例。(至少两家机构) 主要原材料供应商变更、阳性判断值或参考区间变化、增加 临床适应症:根据具体情况,酌情增加总样本数。 (至少两家 机构)
统 计 方 法
定量检测 线性回归( 最小二乘法、Passing-Bablok 、Deming等) Bland-Altman分析 简单相关性分析(Pearson、 Spearman 等) 配对t检验 结果应依据产品的检测性能选择回归分析等适宜的统计 分析方法,在合理的置信区间,考察两种试剂结果是否呈显著 相关性,定量值结果应无显著统计学差异。如有可能,建议应 考虑到在不同的样本浓度区间试剂的性能可能存在的差异,对 总体浓度范围进行区间分层统计。 实验数据离群点的剔除须有依据
对比试剂 已上市同类产品; 临床普遍认为质量较好的产品; 具有可比性的产品:方法学、预期用途、性能指标、校准品 溯源、推荐的阳性判断值/参考区间、样本类型等。
18
IVD
临床试验设计—举例
参照系统选择
病原体IgM、TORCH 类指导原则: 除同类产品对比试验外,还应对急性期患者样本进行考核试 剂与参考方法的一致性研究,不少于30例,或采用血清转化盘 验证。 药物滥用类指导原则: 除同类产品对比试验外,还需完成至少30例参考方法对比试 验。 流感病毒核酸检测: 除同类产品对比试验外,还需完成不少于30例病毒培养阳性 样本验证。
27
IVD
临床试验设计--注册变更
试 验 方 法
对照试验 对照试验:变更后产品 vs. 变更前产品/ 已上市同类产品; 证明两者等效
样 本 量
最低样本量要求: 检测条件优化、增加与原样本类型具有可比性的其他样本类 型等:三类至少200例,二类至少100例。(至少两家机构) 主要原材料供应商变更、阳性判断值或参考区间变化、增加 临床适应症:根据具体情况,酌情增加总样本数。 (至少两家 机构)
统 计 方 法
定量检测 线性回归( 最小二乘法、Passing-Bablok 、Deming等) Bland-Altman分析 简单相关性分析(Pearson、 Spearman 等) 配对t检验 结果应依据产品的检测性能选择回归分析等适宜的统计 分析方法,在合理的置信区间,考察两种试剂结果是否呈显著 相关性,定量值结果应无显著统计学差异。如有可能,建议应 考虑到在不同的样本浓度区间试剂的性能可能存在的差异,对 总体浓度范围进行区间分层统计。 实验数据离群点的剔除须有依据
药物临床试验与GCP培训课件ppt

对试验过程进行实时监控,确保试验操作规 范、数据记录准确。
安全性评估
对试验过程中出现的不良事件进行及时处理 和记录,确保受试者的安全和权益。
04 伦理审查与知情同意书
伦理审查的流程与内容
伦理审查流程
提出申请、递交资料、组织评审、作 出决定、结果通知
审查内容
试验方案的科学性、可行性;受试者 的权益保障;知情同意书的签署情况 等
规范原则
制定明确的试验方案、操 作规程和标准操作流程, 确保试验过程的规范性和 一致性。
GCP原则在临床试验中的应用
试验设计
根据GCP原则,制定科学的试验设计 ,包括试验目的、样本量、随机分组 、对照设置、给药方案等。
统计分析
遵循统计学原理和方法,对试验数据 进行统计分析,得出科学结论。
01
02
知情同意
对研究结果进行深入 讨论,阐述结论,并 指出研究的局限性和 未来研究方向。
报告撰写的基本要求与技巧
01
清晰明了
确保报告内容清晰易懂,避免使用 过于专业或复杂的术语。
详实准确
报告中的数据和信息应详实准确, 并注明来源。
03
02
客观公正
报告应客观公正,避免主观臆断或 偏见。
逻辑严谨
报告的结构应严谨,各部分内容之 间逻辑关系应清晰。
。
保密性原则
必须对受试者的个人信息和试 验数据保密,防止泄露和滥用
。
02 GCP原则及其在临床试验 中的应用
GCP原则概述
01
02
03
伦理原则
尊重受试者权利、利益和 尊严,遵循知情同意、自 愿参加、不损害受试者利 益等原则。
科学原则
遵循科学方法和统计学原 理,确保试验设计、数据 收集、分析和报告的科学 性。
安全性评估
对试验过程中出现的不良事件进行及时处理 和记录,确保受试者的安全和权益。
04 伦理审查与知情同意书
伦理审查的流程与内容
伦理审查流程
提出申请、递交资料、组织评审、作 出决定、结果通知
审查内容
试验方案的科学性、可行性;受试者 的权益保障;知情同意书的签署情况 等
规范原则
制定明确的试验方案、操 作规程和标准操作流程, 确保试验过程的规范性和 一致性。
GCP原则在临床试验中的应用
试验设计
根据GCP原则,制定科学的试验设计 ,包括试验目的、样本量、随机分组 、对照设置、给药方案等。
统计分析
遵循统计学原理和方法,对试验数据 进行统计分析,得出科学结论。
01
02
知情同意
对研究结果进行深入 讨论,阐述结论,并 指出研究的局限性和 未来研究方向。
报告撰写的基本要求与技巧
01
清晰明了
确保报告内容清晰易懂,避免使用 过于专业或复杂的术语。
详实准确
报告中的数据和信息应详实准确, 并注明来源。
03
02
客观公正
报告应客观公正,避免主观臆断或 偏见。
逻辑严谨
报告的结构应严谨,各部分内容之 间逻辑关系应清晰。
。
保密性原则
必须对受试者的个人信息和试 验数据保密,防止泄露和滥用
。
02 GCP原则及其在临床试验 中的应用
GCP原则概述
01
02
03
伦理原则
尊重受试者权利、利益和 尊严,遵循知情同意、自 愿参加、不损害受试者利 益等原则。
科学原则
遵循科学方法和统计学原 理,确保试验设计、数据 收集、分析和报告的科学 性。
临床试验项目管理培训课件
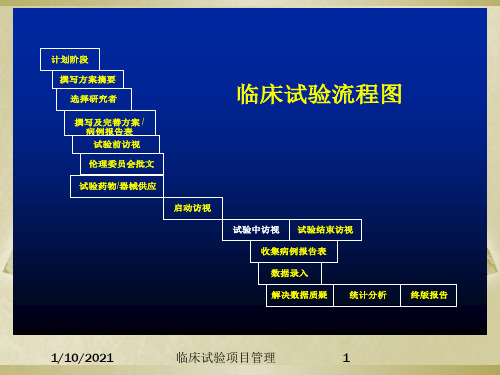
1/10/2021
临床试验1项3 目管理
文件管理(3)
试验结束阶段
试验用药的回收和销毁 CRF的回收及数据的质疑 试验文件完整性的确认 伦理委员会的通报 临床试验的报告
1/10/2021
临床试验1项4 目管理
临床试验项目管理要素
文件管理
进度管理
质量管理 风险管理 1/10/2021
临床试验1项5 目管理
Have blood pressure and heart/pulse rate been taken at this visit?
No
Yes
Please record the results in the Vital Signs Log at the
end
of the CRF.
Is there any clinically significant abnormality?
病史(患病时间等) 目前身体状况、伴随疾病和用药 近期停止用药时间(满足方案要求) 受试者参加临床试验号/签署知情同意书 访视日期 实验室/X-线等结果 试验用药数量及伴随治疗记录
1/10/2021
临床试验9项目管理
1/10/2021
临床试验1项0 目管理
Protocol 265805/249
批文(CFDA批文、伦理委员会批件及其成员) 签字合同(研究者合同、财务规定) 试验器械药品器械接收单和检测报告 实验室文件(正常值范围、质控证明) 研究者简历、研究者授权表及相关文件
1/10/2021
临床试验5项目管理
临床试验方案:
方案的形成过程中需申办者 与研究者讨论,并严格遵守 中国注册法规要求。
计划阶段 撰写方案摘要 选择研究者
GCP培训PPT课件

GCP在全球范围内的推广与普及
各国药品监管机构将积极推广和实施GCP标准,加强国际合作和交流,促进全球 临床试验的规范化和标准化。
GCP标准的普及将有助于提高药物研发的整体水平,加速新药上市进程,为患者 提供更加安全、有效、质量可控的药品。
THANKS
谢谢
01
ICH-GCP是国际协调会议发布的GCP指南,与各国GCP标准相
比更加国际化。
GCP与美国GCP的比较
02
美国GCP更加注重实用性和灵活性,强调对受试者的保护和数
据质量。
GCP与中国GCP的比较
03
中国GCP更加注重与本国法律法规和实际情况相结合,强调规
范化和标准化。
03
CHAPTER
GCP认证与考试
GCP常见问题及解决方案
伦理问题
确保伦理审查严格把关 ,遵循伦理原则,保护
受试者权益。
数据质量问题
建立数据核查机制,确 保数据准确性、可靠性
和完整性。
试验操作问题
加强培训和监管,确保 试验操作符合规范要求
。
受试者权益问题
加强受试者保护措施, 确保受试者知情同意和
隐私保护。
GCP与其他标准的比较
GCP与ICH-GCP的比较
02
GCP标准将与国际药品监管机构 合作,推动全球药品监管的协调 和统一,促进国际临床试验的互 认和药品上市许可的便利化。
GCP在数字化转型中的作用与影响
随着数字化技术的发展,GCP标准将 更加注重临床试验数据的数字化采集 、存储和管理,提高数据质量和可追 溯性。
GCP标准将推动临床试验过程的智能 化和自动化,利用人工智能等技术手 段提高试验效率和质量,降低人为错 误和偏差。
药物临床试验与GCP培训课件ppt

伦理审查委员会
负责对药物临床试验的伦理进行审查,确保符合伦理原则和法规要 求,保护受试者的权益和安全。
01
药物临床试验的设 计与实施
临床试验的设计原则
科学性原则
确保试验设计科学合理 ,能够得出可靠的结论
。
伦理原则
遵循伦理原则,保护受 试者的权益和安全。
随机原则
保证试验结果的客观性 和公正性。
对照原则
01
02
03
04
试验方案设计
遵循GCP原则,制定科学的试 验方案,明确研究目的、方法 、样本量、统计学分析等。
伦理审查
确保临床试验符合伦理原则, 保护受试者的权益和安全,通
过伦理委员会的审查。
知情同意
确保受试者充分了解试验内容 、风险和权益,自愿参与并签
署知情同意书。
质量控制
实施严格的质量控制措施,确 保试验数据的可靠性、完整性
药物临床试验的法规要求
阐述药物临床试验必须遵循的法规要求,包括伦理要 求、GCP要求等。
药物临床试验必须遵循一系列法规要求,以确保研究 过程的规范性和研究结果的可信度。伦理要求是其中 最为重要的,必须保障受试者的权益、安全和福祉, 遵循伦理审查和知情同意原则。此外,药物临床试验 还需遵循GCP(药品生产质量管理规范)的要求,确 保试验数据的真实、完整、可靠和可追溯。这些法规 要求旨在保护受试者的权益,同时为新药研发提供科 学依据,促进医药行业的健康发展。
通过设置对照组,排除 其他因素的干扰。
临床试验的实施流程
试验准备
确定试验目的、方案设计、伦 理审查等。
受试者招募
筛选符合条件的受试者,签署 知情同意书。
试验实施
按照方案进行试验,确保数据 准确可靠。
负责对药物临床试验的伦理进行审查,确保符合伦理原则和法规要 求,保护受试者的权益和安全。
01
药物临床试验的设 计与实施
临床试验的设计原则
科学性原则
确保试验设计科学合理 ,能够得出可靠的结论
。
伦理原则
遵循伦理原则,保护受 试者的权益和安全。
随机原则
保证试验结果的客观性 和公正性。
对照原则
01
02
03
04
试验方案设计
遵循GCP原则,制定科学的试 验方案,明确研究目的、方法 、样本量、统计学分析等。
伦理审查
确保临床试验符合伦理原则, 保护受试者的权益和安全,通
过伦理委员会的审查。
知情同意
确保受试者充分了解试验内容 、风险和权益,自愿参与并签
署知情同意书。
质量控制
实施严格的质量控制措施,确 保试验数据的可靠性、完整性
药物临床试验的法规要求
阐述药物临床试验必须遵循的法规要求,包括伦理要 求、GCP要求等。
药物临床试验必须遵循一系列法规要求,以确保研究 过程的规范性和研究结果的可信度。伦理要求是其中 最为重要的,必须保障受试者的权益、安全和福祉, 遵循伦理审查和知情同意原则。此外,药物临床试验 还需遵循GCP(药品生产质量管理规范)的要求,确 保试验数据的真实、完整、可靠和可追溯。这些法规 要求旨在保护受试者的权益,同时为新药研发提供科 学依据,促进医药行业的健康发展。
通过设置对照组,排除 其他因素的干扰。
临床试验的实施流程
试验准备
确定试验目的、方案设计、伦 理审查等。
受试者招募
筛选符合条件的受试者,签署 知情同意书。
试验实施
按照方案进行试验,确保数据 准确可靠。
药物临床试验与GCP培训课件ppt

药物临床试验的监管机构
国际药物临床试验监管机构
介绍国际药物临床试验监管机构,如世界卫生组织、国际药品监管机构协会等 及其职能。
中国药物临床试验监管机构
分析中国国家药品监督管理局及其地方药品监管部门在药物临床试验监管方面 的职责、监管措施和成效。
04
药物临床试验的流程与操作
药物临床试验的方案设计
总结词
知情同意是药物临床试验的伦理要求,需要确保受试者充分了解试验内容、风险和权益保障等信息。
详细描述
在知情同意过程中,研究者应向受试者充分说明试验内容、风险和权益保障等信息,并回答受试者的 问题。知情同意书应包含必要的信息,并由受试者自愿签署。同时,研究者应关注受试者的心理和生 理状KS
试验过程中的数据收集与记录
总结词
数据收集与记录是药物临床试验的重要 环节,需要确保数据的准确性和完整性 。
VS
详细描述
在试验过程中,研究者应按照试验方案的 要求,准确、完整地记录数据,包括受试 者的基本信息、观察指标、不良反应和意 外事件等。数据记录应采用适当的工具和 方法,确保数据的可追溯性和可靠性。同 时,研究者应定期对数据进行核查和整理 ,确保数据的准确性和完整性。
行为的处理措施。
中国的药物临床试验法规
中国药物临床试验法规概述
介绍中国药物临床试验法规的制定背景、主要内容和目的。
中国药物临床试验法规的核心条款
阐述中国药物临床试验法规中规定的受试者权益保护、伦理审查、数据真实性和完整性等 核心条款。
中国药物临床试验法规的执行现状与改进方向
分析中国药物临床试验法规的执行情况,探讨存在的问题和改进方向。
案例二:失败的药物临床试验案例分析
案例名称
[PPT]-临床试验项目培训(第一部分)---药物临床试验质量管理
![[PPT]-临床试验项目培训(第一部分)---药物临床试验质量管理](https://img.taocdn.com/s3/m/1de1dcb2915f804d2b16c1bb.png)
*** 临床试验项目培训
(第一部分)
---药物临床试验质量管理规范(GCP)
GCP的发展、概念、原则与组织实施
医学的进步是以研究为基础的,这些研究在一定程度上 最终有赖于以人作为受试者的试验 。
--《赫尔辛基宣言》
相关国际法规的发展历程
相关国际法规的发展历程-1
20 世纪初叶的医学
在美国,药品在一个个城市的 “药品表演会 ”上做广告和销售;
申办者及所在国的义务来自不同国家的150名代表审核了这份文件包括卫生部长科学家医生伦理学家哲学家律师等为发展中国家如何进行临床研究提出建议相关国际法规的发展历程14国际协调会议ich临床试验规范的国际性指导原则为欧盟日本和美国的临床研究提供了统一的标准以促进相互间接受临床研究的结果其制定考虑到了不同组织和地区的现行标准包括欧盟日本美国澳大利亚北欧国家和世界卫生组织whoichgcp涵盖了以下三个方面的内容
受试者知情同意 选择研究对象 资料的保密 意外伤害的补偿 伦理委员会的工作程序 申办者及所在国的义务
来自不同国家的 150 名代表审核了这份文件(包括卫生部长、 科学家、医生、伦理学家、哲学家、律师等) 为发展中国家如何进行临床研究提出建议
相关国际法规的发展历程-14
国际协调会议 (ICH) – 1996 年
相关国际法规的发展历程-4
第二次世界大战期间纳粹的实验
数以千计的犹太人被强迫参加非人道的实验; 在儿童受害者身上进行了实验性烧伤和实验性创伤实验; 实施饥饿实验以观察饥饿的症状 。
相关国际法规的发展历程-5
纳粹实验的特征
实施前未获得参加者的同意 导致了不必要的疼痛、痛苦和死亡 对参加者未带来任何益处 缺乏足够的科学依据
这些药物来自植物、动物、矿物等天然物质,根据经验选择; 对安全性或疗效没有控制; 上市前不需要验证; 只有少数几种药物在后来被证明有效 (如 吗啡、洋地黄和奎宁
(第一部分)
---药物临床试验质量管理规范(GCP)
GCP的发展、概念、原则与组织实施
医学的进步是以研究为基础的,这些研究在一定程度上 最终有赖于以人作为受试者的试验 。
--《赫尔辛基宣言》
相关国际法规的发展历程
相关国际法规的发展历程-1
20 世纪初叶的医学
在美国,药品在一个个城市的 “药品表演会 ”上做广告和销售;
申办者及所在国的义务来自不同国家的150名代表审核了这份文件包括卫生部长科学家医生伦理学家哲学家律师等为发展中国家如何进行临床研究提出建议相关国际法规的发展历程14国际协调会议ich临床试验规范的国际性指导原则为欧盟日本和美国的临床研究提供了统一的标准以促进相互间接受临床研究的结果其制定考虑到了不同组织和地区的现行标准包括欧盟日本美国澳大利亚北欧国家和世界卫生组织whoichgcp涵盖了以下三个方面的内容
受试者知情同意 选择研究对象 资料的保密 意外伤害的补偿 伦理委员会的工作程序 申办者及所在国的义务
来自不同国家的 150 名代表审核了这份文件(包括卫生部长、 科学家、医生、伦理学家、哲学家、律师等) 为发展中国家如何进行临床研究提出建议
相关国际法规的发展历程-14
国际协调会议 (ICH) – 1996 年
相关国际法规的发展历程-4
第二次世界大战期间纳粹的实验
数以千计的犹太人被强迫参加非人道的实验; 在儿童受害者身上进行了实验性烧伤和实验性创伤实验; 实施饥饿实验以观察饥饿的症状 。
相关国际法规的发展历程-5
纳粹实验的特征
实施前未获得参加者的同意 导致了不必要的疼痛、痛苦和死亡 对参加者未带来任何益处 缺乏足够的科学依据
这些药物来自植物、动物、矿物等天然物质,根据经验选择; 对安全性或疗效没有控制; 上市前不需要验证; 只有少数几种药物在后来被证明有效 (如 吗啡、洋地黄和奎宁
药物临床试验与GCP培训课件

GCP的基本原则
伦理原则
尊重受试者的权益和安全 ,遵循伦理准则,确保受 试者知情同意。
科学原则
保证试验设计科学合理, 数据采集、分析和报告准 确可靠。
公正原则
确保所有参与试验的人员 都遵循试验方案,不受任 何不正当的影响。
GCP的适用范围
药物临床试验全过程
01
从试验设计、实施、监查、记录到数据整理、分析、报告等各
目的
药物临床试验的主要目的是为药物注 册审批提供科学依据,以证明药物在 特定适应症上的安全性和有效性,从 而为患者提供安全有效的治疗选择。
药物临床试验的阶段
01
02
03
04
I期临床试验
初步评估药物在人体上的安全 性和耐受性,以及药物代谢和
药效动力学特征。
II期临床试验
评估药物的疗效和安全性,通 常涉及一定数量的患者。
试验数据收集与记录
数据收集
按照数据收集表的要求,及时收 集受试者的相关数据和观察结果
。
数据记录
将收集的数据准确无误地记录在 数据表中,确保数据的可追溯性
和可靠性。
数据核查
对记录的数据进行核查,确保数 据的准确性和完整性,及时发现
并纠正错误或遗漏。
试验结束与总结
数据整理与分析
对收集到的数据进行整理和分析,采用适当的统 计分析方法对数据进行分析和解释。
GCP培训的考核
培训结束后,需要进行考核,考核形式包括笔试、面试和实践操作等。
GCP培训的证书
通过考核的人员可以获得GCP培训证书,证书有效期一般为3年,到期需要进行 复训并重新考核。
CHAPTER 05
药物临床试验的质量管理
质量管理概述
药物临床试验管理规范培训共116页PPT

。 ——德 谟克利 特 67、今天应做的事没有做,明天再早也 是耽误 了。——裴斯 泰洛齐 68、决定一个人的一生,以及整个命运 的,只 是一瞬 之间。 ——歌 德 69、懒人无法享受休息之乐。——拉布 克 70、浪费时间是一桩大罪过。——卢梭
药物临床试验管理规范培训
21、没有人陪你走一辈子,所以你要 适应孤 独,没 有人会 帮你一 辈子, 所以你 要奋斗 一生。 22、当眼泪流尽的时候,留下的应该 是坚强 。 23、要改变命运,首先改变自己。
24、勇气很有理由被当作人类德性之 首,因 为这种 德性保 证了所 有其余 的德性 。--温 斯顿. 丘吉尔 。 25、梯子的梯阶从来不是用来搁脚的 ,它只 是让人 们的脚 放上一 段时间 ,以便 让别一 只脚能 够再往 上登。
药物临床试验管理规范培训
21、没有人陪你走一辈子,所以你要 适应孤 独,没 有人会 帮你一 辈子, 所以你 要奋斗 一生。 22、当眼泪流尽的时候,留下的应该 是坚强 。 23、要改变命运,首先改变自己。
24、勇气很有理由被当作人类德性之 首,因 为这种 德性保 证了所 有其余 的德性 。--温 斯顿. 丘吉尔 。 25、梯子的梯阶从来不是用来搁脚的 ,它只 是让人 们的脚 放上一 段时间 ,以便 让别一 只脚能 够再往 上登。
临床试验基础知识ppt课件

1 研究者必须详细阅读和了解试验方案的内容,并严 格按照方案执行。
2 获得伦理委员会的批准。 3 在未获得伦理委员会及申办者同意时,研究者不可
随意违反方案。除非有突发影响受试者安全的事件 发生。 4 研究者应记录和说明任何违背方案的现象 5 取得受试者知情同意书 6 试验总结报告,签名并注明日期
伦理委员会
研 究 者 的 职 责(一)
负责临床试验的研究者 究者的职责:
1.在合法的医疗机构中具有任职行医的资格。 2.具有试验方案中所要求的专业知识和经验。 3.对临床试验研究方法具有丰富经验或者能得到
本单位有经验的研究者在学术上的指导。
4.熟悉申办者所提供的与临床试验有关的资料与
文献。
研 究 者 的 职 责(二)
与临床试验相关的文件
试验方案 叙述试验的背景、理论基础和目的,试
验设计、方法和组织,包括统计学考虑、 试验执行和完成的条件。方案必须由参加 试验的主要研究者、研究机构和申办者签 章并注明日期。
与临床试验相关的文件
病例报告表 指按试验方案所规定设计的一种
文件,用以记录每一名受试者在试验 过程中的数据。
注意事项
• 将保护受试者利益放在首位: • 1)提供详实的ICF (按我国GCP的要求) • 2)ICF a. 时间:在所有的与试验有关的检查之前签署
b. 签字:本人签字(不会签,不能签一定要有 证人签字)
c. 医师的电话:确认是否可以真的联系上
• 3)及时与研究者沟通药物安全性信息
要求 1)对试验方案要透彻了解
临床试验知识培 训
药物临床试验概念
药物临床试验(Clinical Trial):指任 何在人体(病人或健康志愿者)进行药物 的系统性研究,以证实或揭示试验药物的 作用、不良反应及/或试验药物的吸收、分 布、代谢和排泄,目的是确定试验药物的 疗效与安全性。 临床试验一般分为I、II、 III和IV期临床试验。
医学研究方法与临床试验培训ppt

数据收集
确保数据的准确性和完整性,采用标准化的数据采集方法,减少误差和偏倚。
数据处理
对数据进行清洗、整理和转换,确保数据质量,满足统计分析的要求。
统计分析方法
描述性统计
对数据进行描述性分析,如均值、标准差、频数等,以了解数据的基本特征和分布情况 。
推断性统计
运用合适的统计模型和参数检验方法,对数据进行深入分析,以得出具有统计学意义的 结论。
范操作。
案例二:某流行病学调查研究
要点一
总结词
要点二
详细描述
大样本、代表性、可靠性
该案例探讨了一个大样本的流行病学调查研究,强调了样 本的代表性和可靠性,以及研究方法的科学性和规范性。 该研究对于了解疾病流行情况和制定预防措施具有重要意 义。
案例三:某疾病诊断试验研究
总结词
准确性、特异性、敏感性
目的
改善人类健康状况,提高疾病防 治水平,促进医学科学的发展。
医学研究类型与设计
类型
基础研究、临床研究、流行病学研究 等。
设计
随机对照试验、观察性研究、队列研 究等。
医学研究伦理与法规
伦理
尊重受试者权益、保障受试者安全、 遵循知情同意原则等。
法规
涉及伦理审查、数据保护、知识产权 等方面。
02
临床试验基础
调查研究法
要点一
总结词
调查研究法是一种通过问卷、访谈等 方式收集数据并进行分析的方法。
要点二
详细描述
调查研究法通常用于大规模的流行病 学研究或临床试验的基线调查。通过 设计问卷或访谈提纲,研究者可以收 集到关于疾病、健康状况、生活习惯 等方面的信息,并对数据进行统计分 析,以得出结论。
要点三
确保数据的准确性和完整性,采用标准化的数据采集方法,减少误差和偏倚。
数据处理
对数据进行清洗、整理和转换,确保数据质量,满足统计分析的要求。
统计分析方法
描述性统计
对数据进行描述性分析,如均值、标准差、频数等,以了解数据的基本特征和分布情况 。
推断性统计
运用合适的统计模型和参数检验方法,对数据进行深入分析,以得出具有统计学意义的 结论。
范操作。
案例二:某流行病学调查研究
要点一
总结词
要点二
详细描述
大样本、代表性、可靠性
该案例探讨了一个大样本的流行病学调查研究,强调了样 本的代表性和可靠性,以及研究方法的科学性和规范性。 该研究对于了解疾病流行情况和制定预防措施具有重要意 义。
案例三:某疾病诊断试验研究
总结词
准确性、特异性、敏感性
目的
改善人类健康状况,提高疾病防 治水平,促进医学科学的发展。
医学研究类型与设计
类型
基础研究、临床研究、流行病学研究 等。
设计
随机对照试验、观察性研究、队列研 究等。
医学研究伦理与法规
伦理
尊重受试者权益、保障受试者安全、 遵循知情同意原则等。
法规
涉及伦理审查、数据保护、知识产权 等方面。
02
临床试验基础
调查研究法
要点一
总结词
调查研究法是一种通过问卷、访谈等 方式收集数据并进行分析的方法。
要点二
详细描述
调查研究法通常用于大规模的流行病 学研究或临床试验的基线调查。通过 设计问卷或访谈提纲,研究者可以收 集到关于疾病、健康状况、生活习惯 等方面的信息,并对数据进行统计分 析,以得出结论。
要点三
生物医药行业临床试验培训ppt

研究报告的结构与内容
标题页
包括研究标题、研究人员姓名和单位、研究资助 来源等信息。
摘要
简要概述研究目的、方法、结果和结论。
引言
阐述研究背景、目的和研究问题。
研究报告的结构与内容
方法
详细描述试验设计、研 究对象、数据采集和分
析方法。
结果
客观呈现试验结果,包 括表格、图表等形式。
讨论
对结果进行解释和讨论 ,提出可能的机制和意
持续改进
根据质量控制结果和经验反馈,持续改进临床试验过程和方法,提高试验质量和效率。
05
临床试验总结与报告
试验总结的撰写与审核
总结撰写
按照规定的格式和要求,全面、 准确地记录试验过程、数据分析
和结果。
审核流程
由资深研究人员或专家对总结进 行审核,确保内容的真实性和准
确性。
修改和完善
根据审核意见,对总结进行必要 的修改和完善,提高报告质量。
目的
临床试验旨在为药物或医疗设备 的上市批准提供科学依据,同时 为患者提供新的治疗选择。
临床试验的阶段和类型
阶段
临床试验通常分为四个阶段:I期、II 期、III期和IV期。每个阶段的目的和 试验对象有所不同。
类型
临床试验可根据目的分为治疗性试验 、预防性试验、诊断性试验和流行病 学试验等。
临床试验的法规和伦理要求
内容选择
根据临床试验的要求和参与人员的实际需求,选择合适的培训内容,包括临床试验设计 、伦理审查、数据采集与分析等方面的理论知识。
方法选择
根据培训内容和目标,选择适合的培训方法,如讲座、案例分析、模拟操作等,确保培 训效果的有效传递。
培训效果评估与改进
要点一
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
相关国际法规的发展历程-16
国际管理法规对临床研究的影响
过去 50 年中,在伦理上和科学上提高了临床研究的质量 减少了科学研究中的不当行为; 统一了相关的法规,从而促进了临床研究的全球化进程; 促进了许许多多治疗人类疾病的新药的开发,延长了寿命并 提高了生活质量 。
反应停的悲剧
1950 ~ 1960 年间,在欧洲、加拿大和拉丁美洲的数千名 儿童出生时患先天发育异常; 他们的母亲在妊娠时服用了酞胺哌啶酮(反应停); 在 300 个人中进行了上市前研究,研究者并未发现毒性作用; 消费者对此事件的关注影响到美国国会,在 1962年颁布了 一项新法律, Kefarver-Harris 修正案,对新药物的批准制 订了更严格的安全与疗效方面的要求。
保护受试者 试验的科学性 数据完整真实性
相关国际法规的发展历程-15
在人体中使用研究药物的管理规定
1906 1938 1947 1962 1964 1977 1978 1990 1993 1996 完全食品与药物法 FDA 美国食品、药物和化妆品法 纽伦堡法典 Kefarver-Harris 修正案 赫尔辛基宣言 FDA 联邦管理法典 – GCP Belmont 报告 ICH 和 GCP 的开创 国际生物医学研究伦理指导原则 国际协调会议 – GCP 生效
相关国际法规的发展历程-9
反应停的悲剧
由于在妊娠期服用酞胺哌啶酮导致 的先天畸形
反应停 (酞胺哌啶酮 )在1954年 WHO做为催眠药物注册
相关国际法规的发展历程-10
世界医学协会赫尔辛基宣言 1964 年,芬兰
最著名的生物医学研究的国际管理规范; 规定了在人体进行研究的基本原则和依据; 提出了以下概念:
针对在美国进行的临床研究的管理法规 提出了 " 临床试验质量管理规范 (GCP) " 和 " 数据 完整性 " 的概念
1978 年 , Belmont 报告(美国国家委员会为保护参加生物医学和 行为学研究的人体实验对象而制定的道德原则和准则)
1974 年,美国国会任命了一个国家委员会,以保护参加临 床研究的受试者 审核临床研究的伦理原则 :
自主性 受益性 公正性
相关国际法规的发展历程-13
WHO 生物医学研究国际伦理指导原则 日内瓦, 1993 年 15 项指导原则 ,涉及广泛,包括 :
受试者知情同意 选择研究对象 资料的保密 意外伤害的补偿 伦理委员会的工作程序 申办者及所在国的义务
来自不同国家的 150 名代表审核了这份文件(包括卫生部长、 科学家、医生、伦理学家、哲学家、律师等) 为发展中国家如何进行临床研究提出建议
相关国际法规的发展历程-14
国际协调会议 (ICH) – 1996 年
临床试验规范的国际性指导原则为欧盟、日本 和美国的临床 研究提供了统一的标准,以促进相互间接受临床研究的结果 其制定考虑到了不同组织和地区的现行标准,包括欧盟、日 本、美国、澳大利亚、北欧国家和世界卫生组织 (WHO) ICH-GCP 涵盖了以下三个方面的内容 :
1948 年颁布了纽伦堡法典
目的是为了防止纳粹实验这类暴行的再现; 受试者的参加必须出于自愿 ( 知情同意 ); 实验的进行必须有强有力的科学依据; 不允许对受试者造成肉体或精神上的损害或伤害 (do no harm ); 在试验进行中的任何时间受试者有权退出 。
相关国际法规的发展历程-8
减压实验,杀死了一名囚犯
在一名集中营囚犯身上诱导低温
相关国际法规的发展历程-6
1947 年的纽伦堡审判
骇人听闻的纳粹实验暴露给公众 23 名纳粹医生 受到了战争罪行的审判
一名波兰证人在法庭展示了气性坏 疽实验给腿部留下的伤疤
纳粹医生 Karl Brandt 被 判处死刑
相关国际法规的发展历程-7
GCP的发展、概念、原则与组织实施
医学的进步是以研究为基础的,这些研究在一定程度上 最终有赖于以人作为受试者的试验 。
--《赫尔辛基宣言》
相关国际法规的发展历程
相关国际法规的发展历程-1
20 世纪初叶的医学
在美国,药品来自植物、动物、矿物等天然物质,根据经验选择; 对安全性或疗效没有控制; 上市前不需要验证; 只有少数几种药物在后来被证明有效 (如 吗啡、洋地黄和奎宁 等 )。
相关国际法规的发展历程-2
食品与药品管理局 (FDA) 的诞生
1906 年, Upton Sinclair 出版了一本名为 “ 丛林 ” 的书,揭露了芝加哥肉类加工时恶劣 的卫生环境; 公众到美国国会群起抗议示威; 1906 年,通过 “完全食品与药物法 ”,食品与 药品管理局 (FDA) 诞生; 要求每种药物的标签必须准确; 未要求检测安全性。
相关国际法规的发展历程-4
第二次世界大战期间纳粹的实验
数以千计的犹太人被强迫参加非人道的实验; 在儿童受害者身上进行了实验性烧伤和实验性创伤实验; 实施饥饿实验以观察饥饿的症状 。
相关国际法规的发展历程-5
纳粹实验的特征
实施前未获得参加者的同意 导致了不必要的疼痛、痛苦和死亡 对参加者未带来任何益处 缺乏足够的科学依据
相关国际法规的发展历程-3
1938 年 "美国食品、药物和化妆品法 " 1938 年 "美国食品、药物和化妆品法 " 1937 年,由于使用了一种含有磺胺和乙烯乙二醇的,名为抗链 球菌奇药( strep elixir )的药物,美国有 100 多人死亡; 国会通过了一项名为 “食品、药物和化妆品法” 的法案。这项 法律要求,任何药物在上市前应有安全性的科学证据。
研究方案由独立的伦理委员会批准; 研究者应对受试者的医疗照顾负责; 书面知情同意。
1975 年在东京,1983 年在意大利,1989 年在香 港 ,1996 年在南非,2001 年在爱丁堡曾对内容做 了多次修订。
相关国际法规的发展历程-11
相关国际法规的发展历程-12
1977 年,美国 FDA 颁布了联邦管理法典